
Abstract
Structural class characterizes the overall folding type of a protein or its domain. Most of the existing methods for determining the structural class of a protein are based on a group of features that only possesses a kind of discriminative information for the prediction of protein structure class. However, different types of discriminative information associated with primary sequence have been completely missed, which undoubtedly has reduced the success rate of prediction. We present a novel method for the prediction of protein structure class by coupling the improved genetic algorithm (GA) with the support vector machine (SVM). This improved GA was applied to the selection of an optimized feature subset and the optimization of SVM parameters. Jackknife tests on the working datasets indicated that the prediction accuracies for the different classes were in the range of 97.8–100% with an overall accuracy of 99.5%. The results indicate that the approach has a high potential to become a useful tool in bioinformatics.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The concept of protein structure classes, which was introduced by Levitt and Chothia in 1976, was initially based on a visual inspection of polypeptide chain topologies in a dataset of 31 globular proteins (Levitt and Chothia 1976). According Levitt and Chothia's definition (1976), a protein of known structure can generally be categorized into one of four structural classes: all-α, all-β, α/β, and α + β. Since the implementation of this classification, the structural class has become one of the most important features for characterizing the overall folding type of a protein, and it has played an important role in molecular biology, cell biology, pharmacology, rational drug design, and many other related fields (Chen et al. 2006a; Chou 1992, 2004; Chou 2000; Feng et al. 2005; Kedariseti et al. 2006).
During the past three decades, many methods have been proposed for predicting protein structure class, such as the Mahalanobis distance approach (Chou 1995; Chou and Zhang 1994), covariant discrimination approach (Chou and Maggiora 1998), information theory (Jin et al. 2003), artificial neural network (Cai and Zhou 2000; Metfessel et al. 1993), fuzzy clustering (Shen et al. 2005), support vector machine (SVM) (Cai et al. 2002, 2001; Chen et al. 2006a, b; Sun and Huang 2006) and boosting (Cai et al. 2006; Feng et al. 2005). The successes in predicting protein structural classification have, in particular, greatly stimulated the development of predicting other attributes of proteins (Chou 2005), such as subcellular localization (Cedano et al. 1997; Chou and Elrod 1999; Chou and Shen 2007d, 2008), among many others (Chou and Elrod 2002, 2003; Guo et al. 2006; Kuric 2007; Liu et al. 2005a; Shen and Chou 2007c; Shen et al. 2007a, b; Wang et al. 2004, 2005b, 2006; Zhang SW et al. 2006). Many predictors have been proposed to predict protein structure classes with their amino acid composition (Bahar et al. 1997; Chou 1995; Chou and Zhang 1992; Zhang and Chou 1992; Zhang et al. 1995; Zhou and Assa-Munt 2001; Zhou et al. 1992). However, one of the reasons for the lower successful prediction rate may be the complete lack of sequence-order effects in the primary sequence. To take into account the sequence-order effects, a diverse set of descriptors were proposed for enhancing the prediction quality; these include pair-coupled amino acid composition (Chou 1999a), polypeptide composition (Luo et al. 2002), pseudo-amino acid composition (Chen et al. 2006a, b; Chou 2001; Xiao et al. 2006b), various auto-correlation descriptors (Feng and Zhang 2000; Lin and Pan 2001; Horne 1988), and other composition factors (Du et al. 2003, 2006). Since the concept of Chou’s pseudo-amino acid composition was introduced, various pseudo-amino acid composition approaches have been developed to deal with the varieties of problems encountered in proteins and protein-related systems (Aguero-Chapin et al. 2006; Caballero et al. 2007; Cai and Chou 2006; Chen and Li 2007a, b; Chen et al. 2006a, b; Chou and Shen 2008; Diao et al. 2007; Du and Li 2006; Fang et al. 2008; Gao et al. 2005; Gonzalez-Diaz et al. 2006, 2007a, b, c; Kurgan et al. 2007; Li and Li 2007; Lin and Li 2007a, b; Liu et al. 2005a, b; Mondal et al. 2006; Mundra et al. 2007; Pan et al. 2003; Pu et al. 2007; Shen and Chou 2005a, b, 2006, 2007c; Shen et al. 2006, 2007a, b; Shi et al. 2007, 2008; Wang et al. 2004, 2006; Xiao et al. 2006a, b; Zhang SW et al. 2006, 2007; Zhang TL et al. 2006; Zhang and Ding 2007; Zhou et al. 2007; ). Due to its wide usage, a very flexible pseudo-amino acid composition generator, called “PseAAC” (Shen and Chou 2008), was recently made available at the website http://chou.med.harvard.edu/bioinf/PseAAC/, enabling users ton generate 63 different kinds of PseAA composition. Chou and Cai (2002) proposed a completely different approach, the so-called functional domain composition, to incorporate the information of various function types. The validity of their approach has been tested by numerous previous investigations (Cai and Chou 2005a, b; Chou and Cai 2004). Unfortunately, most of the existing methods are based on a group of features that only possesses a kind of discriminative information for the prediction of protein structure class. However, different types of discriminative information associated with primary sequence have been completely missed, which undoubtedly has reduced the success rate of prediction. Empirical studies have demonstrated that the merging of descriptors should increase the predictive accuracy if the descriptors represent different types of discriminative information. Alternatively, the merging of descriptors will simultaneously increase the information redundancy that could, in turn, decrease the predictive accuracy (Kohavi and John 1997). Therefore, there is a need to explore whether an effective combination of descriptors could help to enhance predictive performance.
The SVM, firstly proposed by Cortes and Vapnik (1995), is an excellent machine learning method. Compared with other machine learning systems, SVM has many attractive features, including the absence of local minima, speed and scalability, and the ability to condense information contained in the training set (Chen et al. 2006b). In recent years, SVMs have performed well in predicting protein secondary structure (Hua and Sum 2001), subcellular localization (Chou and Cai 2002; Kim et al. 2006; Yu et al. 2006; Zhang ZH et al. 2006), membrane protein types (Cai et al. 2004; Wang et al. 2004), among others. When using a SVM for predicting protein diverse attributes, two problems are encountered, namely the choice of the optimal features subset and the set of the kernel parameters. The choice of the optimal features subset is how to choose the optimal feature subset that is relevant to protein attributes. Large numbers of features fed to SVM can increase computational complexity and cost (Shen et al. 2007a, b), suffer from the curse of dimensionality and the risk of overfitting and also impede the identification of some biologically mechanism that describe the relationship between the protein and its attributes. In contrast, when a small feature set that is not relevant to protein attributes is used, the result can be bad generalization performance and accurateness. Consequently, the selection of an optimized feature subset is necessary to speed up computation and to improve the generalization performance of the SVM. The choice of the set of the kernel parameters involves how to set the kernel parameters so that the performance of SVM can be brought into full play. These parameters include the penalty constant C and the parameters in the kernel function (width parameter σ of radial basis function, etc.), and they affect more or less the performance of the SVM (Yuan and Chu 2007). Providing adequate solutions to these two problems is crucial because the feature subset choice influences the appropriate kernel parameters, and vice versa (Huang et al. 2008).
Unfortunately, SVMs do not offer the option of a free choice of the optimal features subset and the set of the kernel parameters. In practice, we usually choose a kernel function and set the kernel parameters by experience when a SVM system is constructed. Furthermore, the grid search algorithm is often utilized to find the best kernel parameters when the radial basis function is used; however, the algorithm is time consuming and does not perform well (Hsu and Lin 2002; LaValle and Branicky 2002). The principal component analysis (PCA) and t test are also applied when choosing the optimal features subset when a SVM is used, and although the efficiency of the filter approach of PCA and the t test is high, the results of these methods are poor. In fact, a number of different heuristic algorithms, such as the particle swarm optimization algorithm (Shen et al. 2007a, b), ant colony optimization algorithm (Sivagaminathan and Ramakrishnan 2007), artificial immunization algorithm (Yuan and Chu 2007), and genetic algorithm (GA), have been applied for feature selection. The basic idea of GA is to imitate life evolution in nature according to Darwinian survival of the fittest principle (Jalali-Heravi and Kyani 2007; Lv et al. 2003). GA can effectively search the interesting space and easily solve complex problems without requiring a priori knowledge about the space and the problem. These characteristics of GA make it possible to simultaneously optimize the feature subset and the SVM parameters.
The objective of the research reported here was to develop an effective approach by combining existing descriptors for protein structure class prediction based on an improved GA and SVM. The improved GA was used to simultaneously optimize the kernel parameters of the SVM and to determine the optimized features subset. The prediction quality evaluated by the jackknife cross-validation test exhibited a significant improvement compared to those obtained with several published methods.
Materials and methods
Data sets
In order to facilitate the comparison, the dataset constructed by Chou (1999b) and other two datasets constructed by Zhou (1998) were used as the working dataset. The dataset constructed by Chou (1999b) contains 204 proteins, of which 52 are all-α, 61 are all-β, 45 are α/β, and 46 are α + β. Of the two datasets constructed by Zhou, one consists of 277 domains (70 all-α domains, 61 all-β domains, 81 α/β domains, and 65 α + β domains); the other consists of 498 domains (107 all-α domains, 126 all-β domains, 136 α/β domains, and 129 α + β domains).
Protein primary sequence representation
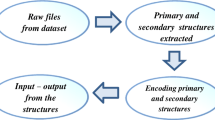
An important issue in the prediction of protein structure class is to represent the primary sequence of proteins with certain encoding scheme. In this work, six feature groups are composed of ten structural and physicochemical features of proteins and peptides from amino acid sequences, and 1447 features were used to represent the protein samples. These features can be easily computed by the PROFEAT web server (Li et al. 2006). PROFEAT is accessible at http://jing.cz3.nus.edu.sg/cgi-bin/prof/prof.cgi. The ten features are summarized in Table 1.
Couple the improved genetic algorithm with SVMs
The publicly available LIBSVM software (Chang and Lin 2001), which can be downloaded freely from http://www.csie.ntu.edu.tw/~cjlin/libsvm, was used to process the SVM classification. The radial basis function was selected as the kernel function. Prediction of protein structure class is a multi-class classification problem. In general, the most commonly used approach for solving multi-class problems is to reduce a single multi-class problem into multiple binary problems known as the one-versus-one and one-versus-rest. However, the one-versus-rest strategy has the well-known ‘false positives’ problem (Ding and Dubchak 2001). Consequently, we adopt here the one-versus-one method to transfer the multi-class problem into a two-class problem. The GA based on chaos (Lv et al. 2003) was used to simultaneously select the feature subset and optimize kernel parameters. In the improved GA, the mutation method based on the chaotic system is used to maintain the population diversity and prevent the incest leading to misleading local optima (Eshelmen and Schaffer 1991). The chromosome representations, fitness function, selection, crossover and mutation operator are described in the following sections.
Chromosome representation
There are three parts to the chromosome: C, γ and the features mask. The chromosome was represented as the binary and decimal coding systems. The hybrid chromosome-encoding method is illustrated in Fig. 1. The hybrid chromosome consists of 1447 binary genes for the selection of features and two decimal genes, C and γ, for the optimization of parameters. If g n = 0, the feature with index n is excluded from the given feature set. Otherwise, the feature with index n is included.

The chromosome coding
Fitness function
A good fitness function is the key to assessing the performance of each chromosome and to obtaining a high classification accuracy. Two objectives must be considered when designing the fitness function. One is to maximize the classification accuracy of fivefold cross-validation, and the other is to minimize the number of selected features. The performances of these two objectives can be evaluated by Eq. (1),
Where SVM_accuracy is the SVM classification accuracy by fivefold cross-validation, and N is the number of selected features.
Selection, crossover and mutation operator
Elitist strategy was used to select the operation. The elitist model guarantees that the chromosome with the highest fitness value is always replicated in the next generation of chromosome. Hence, the function of maximal fitness versus the number of generated chromosome is a monotonous increasing function (Handels et al. 1999). Once a pair of chromosome has been selected for crossover, five random selected positions are assigned to the crossover operator of the binary coding part. The crossover operator was determined according to Eqs. (2) and (3) for the decimal coding part, where p is the random number of (0, 1).
The GA based on the chaotic method was applied to the mutation operator and to the part of decimal coding in the chromosome. Mutation to the part of binary coding in the chromosome is the same as traditional GA.
In the study described here, the population size of improved GA was 30, and the termination condition was that the generation number was 30,000. The whole procedure of GA/SVM-coupled GA with SVM is illustrated in Fig. 2, and the steps were as follows:
-
Step 1. Produce all of the initial chromosome of GA randomly with an appropriate size of the population.
-
Step 2. Run SVM and calculate the fitness values of each chromosome in the population using the fitness function. If the generation number reaches 30,000, stop the process with the output of results, otherwise, go to the next step.
-
Step 3. Select a given percentage of the fittest chromosomes from the current generation based on their fitness value. The selected chromosomes as a part of the next generation are used as parent chromosomes to produce new chromosomes in the next step.
-
Step 4. Produce a given percentage of new chromosomes of the next generation by the mating and mutating operation based on the parents.
-
Step 5. Go back to the second step to run SVM and calculate the fitness values of the renewed population.

The chart of the improved genetic algorithm/support vector machine (GA/SVM) scheme
Results and discussion
Analysis of the convergence processes for current method
Figures 3 and 4 illustrate the convergence processes for the improved GA to optimize kernel parameters and to select the feature subset based on the working datasets constructed by Chou (1999b). The better fitness value, higher classification accuracy, and optimized features subset can be obtained from about 15,000 generations, 8000 generations, and 15,000 generations, respectively. Initially, improved GA selected approximate 700 features into SVM and achieved a predictive accuracy about 90%, which is a distinct symptom of overfitting. Along with the implementation of the process, the number of selected features gradually decreased while fitness value and classification accuracy were improved. Classification accuracy was invariable when the number of generations gradually increased from 7000 to 15,000. Fewer than 100 features were fed to SVM at this time. The results indicate that our method has the ability to overcome the overfitting problem and to achieve a high success rate by searching the optimized features subset and kernel parameters.

Convergence curves for improved GA/SVM. Curve 1 Fitness value was obtained from the most fitted member of each generation, curve 2 classification accuracy was obtained from the most fitted member of each generation)

The relationship between the number of features and the number of generations using improved GA/SVM
Analysis of the optimized features subset
The results of the best features subset are summarized in Table 2. From Table 2 we can see that the optimized features subset contains nine composition, transition, and distribution descriptors; five sequence order descriptors; three amino acid and dipeptide composition descriptors; one autocorrelation descriptors. The results appear to suggest that the order of these feature groups that contributed to the prediction of protein structural class were: composition, transition, and distribution > sequence order descriptors > amino acid and dipeptide composition > autocorrelation descriptors.
In fact, there are two compositions of polarity and polarizability, one transition of charge, three distributions of hydrophobicity, and three distributions of polarity, polarizability, and charge in the group of composition, transition, and distribution descriptors. The five sequence order descriptors include two sequence-order-coupling numbers based on Schneider–Wrede distance and three sequence-order-coupling numbers based on normalized Grantham chemical distance. The three amino acid and dipeptide composition are alanine, leucine, and lysine–arginine composition. One autocorrelation descriptor was the Moran autocorrelation hydrophobicity scale. These results suggest that factors such as hydrophobicity, polarizability, polarity, charge, and composition of alanine, leucine, lysine–arginine are important to protein structure class. We therefore expect that if a new encoding scheme can integrate with (1) composition, transition, and distribution, (2) sequence order, (3) amino acid and dipeptide composition, and (4)autocorrelation information, it would be of great significance in terms of predicting the attributes of protein.
Comparison with different methods
In statistical prediction, the most widely used cross-validation methods for examining the accuracy of a predictor are the sub-sampling test and jackknife test (Chou and Shen 2008; Chou and Zhang 1995). However, as demonstrated by Eq. 50 in a recent comprehensive review by Chou and Shen (2007d), the sub-sampling (e.g., fivefold cross-validation) test cannot avoid arbitrariness even for a very simple benchmark dataset. Accordingly, the jackknife test has been increasingly and widely adopted by investigators (Chen et al. 2006a, b, 2007; Chou and Shen 2006a, b, 2007a, b, c, e; Diao et al. 2008; Ding et al. 2007; Du and Li 2006; Fang et al. 2008; Gao et al. 2005; Guo et al. 2006; Kedarisetti et al. 2006; Li and Li 2007; Lin and Li 2007a, b; Liu et al. 2007; Mondal et al. 2006; Niu et al. 2006; Shen and Chou 2007a, b, c, d; Shen et al. 2007a, b; Shi et al. 2007, 2008; Sun and Huang 2006; Tan et al. 2007; Wang et al. 2005a; Wen et al. 2006; Xiao and Chou 2007; Xiao et al. 2005a, b, 2006a; Zhang and Ding 2007; Zhang SW et al. 2006, 2007; Zhou 1998; Zhou and Doctor 2003; Zhou et al. 2007) to test the power of various predictors. To facilitate a comparison with previous studies for the dataset constructed by Chou (1999b), the optimized kernel parameters and features subsets were utilized to perform jackknife cross-validation test. The success rates by the jackknife test are listed in Table 3 and compared with several published results for the same dataset.
The reliability of the optimized features subset was further evaluated by the two datasets constructed by Zhou (1998). We performed the jackknife cross-validation based on the optimized features subset. Grid searches strategy was adopted to find the best C and γ for obtaining maximal jackknife-tested overall rates. The success rates by the jackknife cross-validation test are listed in Tables 4 and 5.
Table 3 shows that the overall rates by the current approach were 99.5% with the one-versus-one method. The results indicate that our method was about 24 and 14% higher than other two SVMs, which were based on amino acid composition and pseudo-amino acid composition respectively. The results also reveal that our improvements can be attributed to the adoption of the optimized features subset and kernel parameters. It is worth noting that the success rates were improved markedly to 100% for all-α, all-β and—the most difficult case—α + β. Consequently, our proposed method is superior to other methods in identifying the structural classification for the Chou’s (1999b) dataset. Table 4 shows that the overall success rate by the our approach was 84.5% for the 277 dataset, which is about 5% higher than that obtained with the SVM method, which was performed with the conventional amino acid composition as the input. In addition, the result is only 3% lower than the SVM fusion, which had the highest overall predictive rate for the dataset. Table 5 shows that the overall success rate by our method was 94.2% for the 498 dataset, which indicates that our method is superior to other existing methods and comparable to LogitBoost. Accordingly, it can be expected that the current method and the SVM fusion or the LogitBoost, if complemented, may further improve the overall rate for the 277 and 498 dataset, respectively. In short, based on both the rationality of the testing procedure and the present success rates, as shown here by our test results, we believe that the optimized features subset may be used to explore the protein folding mechanism by using abundant discriminative information related to protein structure class.
Conclusion
We have proposed a strategy based on improved GA to simultaneously select the feature subset and optimize the parameters of SVM for predicting protein structure class. The results indicate that the proposed method is very effective for the optimal combination of different features. Moreover, it can be anticipated that this method may also have a great impact by improving the success rates for many other protein attributes, such as subcellular localization, membrane types, enzymes family and subfamily classes, and G-protein-coupled receptor classification.
References
Aguero-Chapin G, Gonzalez-Diaz H, Molina R, Varona-Santos J, Uriarte E, Gonzalez-Diaz Y (2006) Novel 2D maps and coupling numbers for protein sequences. The first QSAR study of polygalacturonases; isolation and prediction of a novel sequence from Psidium guajava L. FEBS Lett 580:723–730
Bahar I, Atilgan AR, Jernigan RL, Erman B (1997) Understanding the recognition of protein structureal classes by amino acid composition. Proteins 29:172–185
Caballero J, Fernandez L, Garriga M, Abreu JI, Collina S, Fernandez M (2007) Proteometric study of ghrelin receptor function variations upon mutations using amino acid sequence autocorrelation vectors and genetic algorithm-based least square support vector machines. J Mol Graph Model 26:166–178
Cai YD, Chou KC (2005a) Using functional domain composition to predict enzyme family classes. J Proteome Res 4:109–111
Cai YD, Chou KC (2005b) Predicting enzyme subclass by functional domain composition and pseudo amino acid composition. J Proteome Res 4:967–971
Cai YD, Chou KC (2006) Predicting membrane protein type by functional domain composition and pseudo amino acid composition. J Theor Bio 238:395–400
Cai YD, Zhou GP (2000) Prediction of protein structural classes by neural network. Biochimie 82:783–785
Cai YD, Liu XJ, Xu XB, Zhou GP (2001) Support vector machines for predicting protein structural class. BMC Bioinformatics 2:1–5
Cai YD, Liu XJ, Xu XB, Chou KC (2002) Prediction of protein structural classes by support vector machines. Comput Chem 26:293–296
Cai YD, Ricardo PW, Jen CH, Chou KC (2004) Application of SVM to predict membrane protein types. J Theor Boil 226:373–376
Cai YD, Feng KY, Lu WC, Chou KC (2006) Using logitboost classifier to predict protein structural classes. J Theor Biol 238:172–176
Cao YF, Liu S, Zhang L, Qin J, Wang J, Tang KX (2006) Prediction of protein structural class with rough sets. BMC Bioinformatics 7:1–6
Cedano J, Aloy P, P’erez-Pons JA, Querol E (1997) Relation between amino acid composition and cellular location of proteins. J Mol Biol 266:594–600
Chang CC, Lin CJ (2001) LIBSVM: a library for support vector machines. Software available at http://www.csie.ntu.edu.tw/~cjlin/libsvm
Chen C, Tian YX, Zou XY, Cai PX, Mo JY (2006a) Using pseudo-amino acid composition and support vector machine to predict protein structural class. J Theor Biol 243:444–448
Chen C, Zhou XB, Tian YX, Zou XY, Cai PX (2006b) Predicting protein structural class with pseudo-amino acid composition and support vector machine fusion network. Anal Biochem 357:116–121
Chen J, Liu H, Yang J, Chou KC (2007) Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids 33:423–428
Chen YL, Li QZ (2007a) Prediction of apoptosis protein subcellular location using improved hybrid approach and pseudo amino acid composition. J Theor Biol 248:377–381
Chen YL, Li QZ (2007b) Prediction of the subcellular location of apoptosis proteins. J Theor Biol 245:775–783
Chou KC (1992) Energy-optimized structure of antifreeze protein and its binding mechanism. J Mol Biol 223:509–517
Chou KC (1995) A novel-approach to predicting protein structural classes in a (20–1)-D amino –acid-composition space. Proteins 21:319–344
Chou KC (1999a) Using pair-coupled amino acid composition to predict protein secondary structure content. J Protein Chem 18:473–480
Chou KC (1999b) A key driving force in determination of protein structural classes. Biochem Biophys Res Commun 264:216–224
Chou KC (2000) Prediction of protein structural classes and subcellular locations. Curr Protein Pept Sc 1:171–208
Chou KC (2001) Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 43:246–255
Chou KC (2004) Review: structural bioinformatics and its impact to biomedical science. Curr Med Chem 11:2105–2134
Chou KC (2005) Review: progress in protein structural class prediction and its impact to bioinformatics and proteomics. Curr Protein Pept Sc 6:423–436
Chou KC, Cai YD (2002) Using functional domain composition and support vector machines for prediction of protein subcellular location. J Biol Chem 277:45765–45769
Chou KC, Cai YD (2004) Predicting protein structural class by functional domain composition. Biochem Biophys Res Commun 321:1007–1009
Chou KC, Elrod DW (1999) Protein subcellular location prediction. Protein Eng 12:107–118
Chou KC, Elrod DW (2002) Bioinformatical analysis of G-protein-coupled receptors. J Proteome Res 1:429–433
Chou KC, Elrod DW (2003) Prediction of enzyme family classes. J Proteome Res 2:183–190
Chou KC, Maggiora GM (1998) Domain structural class prediction. Protein Eng 11:523–538
Chou KC, Shen HB (2006a) Hum-PLoc: a novel ensemble classifier for predicting human protein subcellular localization. Biochem Biophys Res Commun 347:150–157
Chou KC, Shen HB (2006b) Predicting eukaryotic protein subcellular location by fusing optimized evidence-theoretic K-nearest neighbor classifiers. J Proteome Res 5:1888–1897
Chou KC, Shen HB (2007a) Euk-mPLoc: a fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites. J Proteome Res 6:1728–1734
Chou KC, Shen HB (2007b) Large-scale plant protein subcellular location prediction. J Cell Biochem 100:665–678
Chou KC, Shen HB (2007c) MemType-2L: a Web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM. Biochem Biophys Res Comm 360:339–345
Chou KC, Shen HB (2007d) Review: recent progresses in protein subcellular location prediction. Anal Biochem 370:1–16
Chou KC, Shen HB (2007e) Signal-CF: a subsite-coupled and window-fusing approach for predicting signal peptides. Biochem Biophys Res Comm 357:633–640
Chou KC, Shen HB (2008) Cell-PLoc: a package of web-servers for predicting subcellular localization of proteins in various organisms. Nat Protoc 3:153–162
Chou KC, Zhang CT (1992) A correlation-coefficient method to predicting protein-structural classes form amino-acid compositions. Eur J Biochem 207:429–433
Chou KC, Zhang CT (1994) Predicting protein folding types by distance functions that make allowances for amino acid interactions. J Biol Chem 269:22014–22020
Chou KC, Zhang CT (1995) Review: prediction of protein structural classes. Crit Rev Biochem Mol Biol 30:275–349
Cortes C, Vapnik V (1995) Support-vector networks. Mach Learn 20:273–297
Diao Y, Li M, Feng Z, Yin J, Pan Y (2007) The community structure of human cellular signaling network. J Theor Biol 247:608–615
Diao Y, Ma D, Wen Z, Yin J, Xiang J, Li M (2008) Using pseudo amino acid composition to predict transmembrane regions in protein: cellular automata and Lempel-Ziv complexity. Amino Acids. doi:10.1007/s00726-007-0550-z
Ding CHQ, Dubchak I (2001) Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics 17:349–358
Ding YS, Zhang TL, Chou KC (2007) Prediction of protein structure classes with pseudo amino acid composition and fuzzy support vector machine network. Protein Peptide Lett 14:811–815
Du P, Li Y (2006) Prediction of protein submitochondria locations by hybridizing pseudo-amino acid composition with various physicochemical features of segmented sequence. BMC Bioinformatics 7:518
Du QS, Wei DQ, Chou KC (2003) Correlations of amino acids in proteins. Peptides 24:1863–1869
Du QS, Jiang ZQ, He WZ, Li DP, Chou KC (2006) Amino acid principal component analysis (AAPCA) and its applications in protein structural class prediction. J Biomol Struct Dyn 23:635–640
Eshelmen LJ, Schaffer JD (1991) Preventing premature convergence in genetic algorithms by preventing incest. In: Belew RK, Booker LB (eds) Proc 4th Int Conf Genetic Algorithms. Morgan Kaufmann, San Francisco, pp 115–122
Feng KY, Cai YD, Chou KC (2005) Boosting classifier for predicting protein domain structural class. Biochem Biophys Res Commun 334:213–217
Fang Y, Guo Y, Feng Y, Li M (2008) Predicting DNA-binding proteins: approached from Chou’s pseudo amino acid composition and other specific sequence features. Amino Acids. doi:10.1007/s00726-007-0568-2
Feng ZP, Zhang CT (2000) Prediction of membrane protein types based on the hydrophobic index of amino acids. J Protein Chem 19:269–275
Gao Y, Shao SH, Xiao X, Ding YS, Huang YS, Huang ZD, Chou KC (2005) Using pseudo amino acid composition to predict protein subcellular location: approached with Lyapunov index, Bessel function, and Chebyshev filter. Amino Acids 28:373–376
Gonzalez-Diaz H, Perez-Bello A, Uriarte E, Gonzalez-Diaz Y (2006) QSAR study for mycobacterial promoters with low sequence homology. Bioorg Med Chem Lett 16:547–553
Gonzalez-Diaz H, Aguero-Chapin G, Varona J, Molina R, Delogu G, Santana L, Uriarte E, Podda G (2007a) 2D-RNA-coupling numbers: a new computational chemistry approach to link secondary structure topology with biological function. J Comput Chem 28:1049–1056
Gonzalez-Diaz H, Perez-Castillo Y, Podda G, Uriarte E (2007b) Computational chemistry comparison of stable/nonstable protein mutants classification models based on 3D and topological indices. J Comput Chem 28:1990–1995
Gonzalez-Diaz H, Vilar S, Santana L, Uriarte E (2007c) Medicinal chemistry and bioinformatics—current trends in drugs discovery with networks topological indices. Curr Top Med Chem 10:1015–1029
Guo YZ, Li M, Lu M, Wen Z, Wang K, Li G, Wu J (2006) Classifying G protein-coupled receptors and nuclear receptors based on protein power spectrum from fast Fourier transform. Amino Acids 30:397–402
Handels H, Ross T, Kreusch J, Wolff HH, Pöppl SJ (1999) Feature selection for optimized skin tumor recognition using genetic algorithms. Artif Intell Med 16:283–297
Horne DS (1988) Prediction of protein helix content from an autocorrelation analysis of sequence hydrophobicities. Biopolymers 27:451–477
Hua SJ, Sum ZR (2001) A novel method of protein secondary structure prediction with high segment overlap measure: support vector machine approach. J Mol Biol 308:397–407
Huang CL, Liao HC, Chen MC (2008) Prediction model building and feature selection with support vector machines in breast cancer diagnosis. Expert Syst Appl 34:578–587
Hsu CW, Lin CJ (2002) A simple decomposition method for support vector machine. Mach Learn 46:219–314
Jahandideh S, Abdolmaleki P, Jahandideh M, Asadabadi EB (2007a) Novel two-stage hybrid neural discriminant model for predicting proteins structural classes. Biophys Chem 128:87–93
Jahandideh S, Abdolmaleki P, Jahandideh M, Hayatshahi SHS (2007b) Novel hybrid method for the evaluation of parameters contributing in determination of protein structural classes. J Theor Biol 244:275–281
Jalali-Heravi M, Kyani A (2007) Application of genetic algorithm-kernel partial least square as a novel nonlinear feature selection method: activity of carbonic anhydrase II inhibitors. Eur J Med Chem 42:649–659
Jin LX, Fang WW, Tang HW (2003) Prediction of protein structural classes by a new measure of information discrepancy. Comput Biol Chem 23:373–380
Kedarisetti KD, Kurgan L, Dick S (2006) Classifier ensembles for protein structural class prediction with varying homology. Biochem Biophys Res Commun 348:981–988
Kim JK, Raghava GPS, Bang SY, Choi S (2006) Prediction of subcellular localization of proteins using pairwise sequence alignment and support vector machine, Pattern. Recogn Lett 27:996–1001
Kohavi R, John GH (1997) Wrappers for feature subset selection. Artif Intell 97:273–324
Kurgan LA, Stach W, Ruan J (2007) Novel scales based on hydrophobicity indices for secondary protein structure. J Theor Biol 248:354–366
Kuric L (2007). The digital language of amino acids. Amino Acids 33:653–661
LaValle SM, Branicky MS (2002) On the relationship between classical grid search and probabilistic roadmaps. Int J Robot Res 23:673–692
Levitt M, Chothia C (1976) Structural patterns in globular proteins. Nature 261:552–558
Li FM, Li QZ (2007) Using pseudo amino acid composition to predict protein subnuclear location with improved hybrid approach. Amino Acids. doi:10.1007/s00726-007-0545-9
Li ZR, Lin HH, Han LY, Jiang L, Chen X, Chen YZ (2006) PROFEAT: a web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res 34:32–37
Lin H, Li QZ (2007a) Predicting conotoxin superfamily and family by using pseudo amino acid composition and modified Mahalanobis discriminant. Biochem Biophys Res Commun 354:548–551
Lin H, Li QZ (2007b) Using pseudo amino acid composition to predict protein structural class: approached by incorporating 400 dipeptide components. J Comput Chem 28:1463–1466
Lin Z, Pan XM (2001) Accurate prediction of protein secondary structural content. J Protein Chem 20:217–220
Liu DQ, Liu H, Shen HB, Yang J, Chou KC (2007) Predicting secretory protein signal sequence cleavage sites by fusing the marks of global alignments. Amino Acids 32:493–496
Liu H, Wang M, Chou KC (2005a) Low-frequency Fourier spectrum for predicting membrane protein types. Biochem Biophys Res Commun 336:737–739
Liu H, Yang J, Wang M, Xue L, Chou KC (2005b) Using Fourier spectrum analysis and pseudo amino acid composition for prediction of membrane protein types. Protein J 24:385–389
Luo RY, Feng ZP, Liu JK (2002) Prediction of protein structural class by amino acid and polypeptide composition. Eur J Biochem 269:4219–4225
Lv QZ, Shen GL, Yu RQ (2003) A chaotic approach to maintain the population diversity of genetic algorithm in network training. Comput Biol Chem 27:363–371
Metfessel BA, Saurugger PN, Connelly DP, Rich SS (1993) Cross-validation of protein structural class prediction using statistical clustering and neural networks. Protein Sci 2:1170–1182
Mondal S, Bhavna R, Mohan Babu R, Ramakumar S (2006) Pseudo amino acid composition and multi-class support vector machines approach for conotoxin superfamily classification. J Theor Biol 243:252–260
Mundra P, Kumar M, Kumar KK, Jayaraman VK, Kulkarni BD (2007) Using pseudo amino acid composition to predict protein subnuclear localization: approached with PSSM. Pattern Recognit Lett 28:1610–1615
Niu B, Cai YD, Lu WC, Zheng GY, Chou KC (2006) Predicting protein structural class with AdaBoost learner. Protein Peptide Lett 13:489–492
Pan YX, Zhang ZZ, Guo ZM, Feng GY, Huang ZD, He L (2003) Application of pseudo amino acid composition for predicting protein subcellular location: stochastic signal processing approach. J Protein Chem 22:395–402
Pu X, Guo J, Leung H, Lin Y (2007) Prediction of membrane protein types from sequences and position-specific scoring matrices. J Theor Biol 247:259–265
Shen HB, Chou KC (2005a) Predicting protein subnuclear location with optimized evidence-theoretic K-nearest classifier and pseudo amino acid composition. Biochem Biophys Res Comm 337:752–756
Shen HB, Chou KC (2005b) Using optimized evidence-theoretic K-nearest neighbor classifier and pseudo amino acid composition to predict membrane protein types. Biochem Biophys Res Comm 334:288–292
Shen HB, Chou KC (2006) Ensemble classifier for protein fold pattern recognition. Bioinformatics 22:1717–1722
Shen HB, Chou KC (2007a) Gpos-PLoc: an ensemble classifier for predicting subcellular localization of Gram-positive bacterial proteins. Protein Eng Des Sel 20:39–46
Shen HB, Chou KC (2007b) Hum-mPLoc: an ensemble classifier for large-scale human protein subcellular location prediction by incorporating samples with multiple sites. Biochem Biophys Res Commun 355:1006–1011
Shen HB, Chou KC (2007c) Using ensemble classifier to identify membrane protein types. Amino Acids 32:483–488
Shen HB, Chou KC (2007d) Virus-PLoc: a fusion classifier for predicting the subcellular localization of viral proteins within host and virus-infected cells. Biopolymers 85:233–240
Shen HB, Chou KC (2008) PseAAC: a flexible web-server for generating various kinds of protein pseudo amino acid composition. Anal Biochem 373:386–388
Shen HB, Yang J, Liu XJ, Chou KC (2005) Using supervised fuzzy clustering to predict protein structural classes. Biochem Biophys Res Commun 334:577–581
Shen HB, Yang J, Chou KC (2006) Fuzzy KNN for predicting membrane protein types from pseudo amino acid composition. J Theor Biol 240:9–13
Shen HB, Yang J, Chou KC (2007a) Euk-PLoc: an ensemble classifier for large-scale eukaryotic protein subcellular location prediction. Amino Acids 33:57–67
Shen Q, Shi WM, Kong W, Ye BX (2007b) A combination of modified particle swarm optimization algorithm and support vector machine for gen selection and tumor classification. Talanta 71:1679–1683
Shi JY, Zhang SW, Pan Q, Cheng YM, Xie J (2007) Prediction of protein subcellular localization by support vector machines using multi-scale energy and pseudo amino acid composition. Amino Acids 33:69–74
Shi JY, Zhang SW, Pan Q, Zhou GP (2008) Using pseudo amino acid composition to predict protein subcellular location: approached with amino acid composition distribution. Amino Acids. doi:10.1007/s00726-007-0623-z
Sivagaminathan RK, Ramakrishnan S (2007) A hybrid approach for feature subset selection using neural networks and ant colony optimization. Expert Syst Appl 33:49–60
Sun XD, Huang RB (2006) Prediction of protein structural classes using support vector machines. Amino Acids 30:469–475
Tan F, Feng X, Fang Z, Li M, Guo Y, Jiang L (2007) Prediction of mitochondrial proteins based on genetic algorithm – partial least squares and support vector machine. Amino Acids 33:669–675
Wang M, Yang J, Chou KC (2005a) Using string kernel to predict signal peptide cleavage site based on subsite coupling model. Amino Acids 28:395–402 (Erratum, ibid. 2005, 29:301)
Wang M, Yang J, Liu GP, Xu ZJ, Chou KC (2004) Weighted-support vector machines for predicting membrane protein types based on pseudo amino acid composition. Protein Eng Ses Sel 17:509–516
Wang M, Yang J, Xu ZJ, Chou KC (2005b) SLLE for predicting membrane protein types. J Theor Biol 232:7–15
Wang SQ, Yang J, Chou KC (2006) Using stacked generalization to predict membrane protein types based on pseudo amino acid composition. J Theor Biol 242:941–946
Wen Z, Li M, Li Y, Guo Y, Wang K (2006) Delaunay triangulation with partial least squares projection to latent structures: a model for G-protein coupled receptors classification and fast structure recognition. Amino Acids 32:277–283
Xiao X, Chou KC (2007) Digital coding of amino acids based on hydrophobic index. Protein Peptide Lett 14:871–875
Xiao X, Shao S, Ding Y, Huang Z, Chen X, Chou KC (2005a) Using cellular automata to generate Image representation for biological sequences. Amino Acids 28:29–35
Xiao X, Shao S, Ding Y, Huang Z, Huang Y, Chou KC (2005b) Using complexity measure factor to predict protein subcellular location. Amino Acids 28:57–61
Xiao X, Shao SH, Ding YS, Huang ZD, Chou KC (2006a) Using cellular automata images and pseudo amino acid composition to predict protein subcellular location. Amino Acids 30:49–54
Xiao X, Shao SH, Huang ZD, Chou KC (2006b) Using pseudo amino acid composition to predict protein structural classes: approached with complexity measure factor. J Comput Chem 27:478–482
Yu CS, Chen YC, Lu CH, Hwang JK (2006) Prediction of protein subcellular localization. Proteins 64:643–651
Yuan SF, Chu FL (2007) Fault diagnosis based on support vector machines with parameter optimization by artificial immunization algorithm. Mech Syst Signal Pr 21:1318–1330
Zhang CT, Chou KC (1992) An optimization approach to predicting protein structural class form amino-acid-composition. Protein Sci 1:401–408
Zhang CT, Chou KC, Maggiora GM (1995) Predicting protein structural classes from amino-acid-composition—application of fuzzy Clustering. Protein Eng 8:425–435
Zhang SW, Pan Q, Zhang HC, Shao ZC, Shi JY (2006) Prediction protein homo-oligomer types by pseudo amino acid composition: approached with an improved feature extraction and naive Bayes feature fusion. Amino Acids 30:461–468
Zhang SW, Zhang YL, Yang HF, Zhao CH, Pan Q (2007) Using the concept of Chou’s pseudo amino acid composition to predict protein subcellular localization: an approach by incorporating evolutionary information and von Neumann entropies. Amino Acids. doi:10.1007/s00726-007-0010-9
Zhang TL, Ding YS, Chou KC (2006) Prediction of protein subcellular location using hydrophobic patterns of amino acid sequence. Comput Biolo Chem 30:367–371
Zhang TL, Ding YS (2007) Using pseudo amino acid composition and binary-tree support vector machines to predict protein structural classes. Amino Acids 33:623–629
Zhang TL, Ding YS, Chou KC (2008) Prediction protein structural classes with pseudo-amino acid composition: approximate entropy and hydrophobicity pattern. J Theor Biol 250:186–193
Zhang ZH, Wang ZH, Zhang ZR, Wang YX (2006) A novel method for apoptosis protein subcellular localization prediction combining encoding based on grouped weight and support vector machine. FEBS Lett 580:6169–6174
Zhou GF, Xu XH, Zhang CT (1992) A weighting method for predicting protein structural class form amino-acid-composition. Eur J Biochem 210:747–749
Zhou GP (1998) An intriguing controversy over protein structural class prediction. J Protein Chem 17:729–738
Zhou GP, Assa-Munt N (2001) Some insights into protein structural class prediction. Proteins 44:57–59
Zhou GP, Doctor K (2003) Subcellular location prediction of apoptosis proteins. Proteins 50:44–48
Zhou XB, Chen C, Li ZC, Zou XY (2007) Using Chou’s amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J Theor Biol 248:546–551
Acknowledgments
The authors acknowledge financial support from the National Natural Science Foundation of China (Nos. 20475068, 20575082), the Natural Science Foundation of Guangdong Province (No. 031577, 7003714), the Scientific Technology Project of Guangdong Province (No. 2005B30101003) and the Scientific Technology Project of Guangzhou City (No. 2007Z3-E0441).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, ZC., Zhou, XB., Lin, YR. et al. Prediction of protein structure class by coupling improved genetic algorithm and support vector machine. Amino Acids 35, 581–590 (2008). https://doi.org/10.1007/s00726-008-0084-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-008-0084-z