
Abstract
The properties of antimicrobial peptides adsorbed on inorganic or organic surfaces are of interest for their potential applications in intracellular drug delivery. In this work, continuous-wave (CW) electron paramagnetic resonance (EPR) and pulsed electron-electron double resonance (PELDOR) techniques were applied to study adsorption of the short-sequence trichogin GA IV and the medium-length sequence ampullosporin A antimicrobial peptides on the monodisperse colloidal silica nanospheres of 20 nm diameter. The results obtained by CW EPR support the view that the adsorbed peptides form close-packed clusters. PELDOR data show that both trichogin and ampullosporin adsorbed on the silica surface possess a more disordered conformation as compared to that in solution. For ampullosporin, disordering is much more pronounced than for trichogin. After desorption, the peptides restored their conformations; upon adsorption the peptides in some cases may lose partly their biradical character.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Antimicrobial peptides are members of a new class of drugs which are interesting because of their rapid therapeutic effect and reduced immunogenicity. Naturally occurring antimicrobial peptides belonging to the peptaibiotic family are membrane-active polypeptides isolated from fungal sources which are known for their ability to modify the permeability of biological membranes [1–4]. Peptaibiotics are characterized by a large presence of an unusual amino acid, α-aminoisobutyric acid (Aib), typically bear an N-terminal acyl, most often an acetyl (Ac) moiety, and a C-terminal 1,2-amino alcohol. Their sequences show an extremely large molecular diversity. A typical classification of peptaibiotics, exclusively based on the number of constitutive amino acids, involves three sub-classes [4]: long-sequence with 17–21 residues; medium-length sequence (14–16 residues); and short-sequence (4–11 residues). Their amphipathic nature allows them to self-associate into oligomeric ion-channel assemblies which span the width of lipid bilayer membranes. Almost 1300 peptaibiotic sequences have been reported to date [4].
When incorporated into nanocarriers, antimicrobial peptides may be used for targeted drug delivery [5–9]. This insertion can be performed using different methods and utilizing different natural and synthetic materials. When antimicrobial peptides are adsorbed or covalently linked onto the surfaces of the carriers, they are released by simple desorption or breaking the covalent linkages with the particle surfaces.
The properties of peptides adsorbed on surfaces can be studied using circular dichroism [10], molecular dynamics and Monte-Carlo simulations [11, 12], Fourier transform infrared absorption [13, 14], nuclear magnetic resonance [15, 16]. In most cases, these approaches allow one to extract conclusions on the peptide secondary structure (α-helix, β-pleated sheet, 310-helix, etc.) and in some cases on the tertiary structure as well.
Recently, pulsed electron–electron double resonance (PELDOR), which is a pulsed version of electron paramagnetic resonance (EPR) [17, 18], was also applied to study a peptaibiotic adsorbed on an organic surface [19]. For a spin-labeled long-sequence alamethicin F50/5 analog adsorbed on the Oasis HLB sorbent, it was shown that PELDOR allows one to detect aggregation of peptide molecules on the surface and to probe peptide conformation.
In this work, we continue our exploration of the possibilities of continuous-wave (CW) EPR and PELDOR spectroscopies to study the properties of peptaibiotics from other sub-classes, adsorbed on different surfaces. The results obtained show that the spin-labeled, short-sequence (10-amino-acid long) trichogin GA IV (Tri) and the medium-length sequence (14-amino-acid long) ampullosporin A (Amp) adsorbed on the inorganic colloidal monodisperse silica nanospheres tend to form closely-packed aggregates. Their conformations on the surface were found to be remarkably disordered as compared with those in solution. Also, PELDOR is shown to be capable to evaluate the peptide integrity during the adsorption–desorption processes.
The sequences of Tri and Amp and their doubly spin-labeled (Tri 1,8 and Amp 3,13) analogs are given below:
- Tri :
-
nOct-Aib1-Gly-Leu-Aib4-Gly–Gly-Leu-Aib8-Gly-Ile-Lol
- Tri 1,8 :
-
nOct-TOAC-Gly-Leu-Aib-Gly–Gly-Leu-TOAC-Gly-Ile-Leu-OMe
- Amp :
-
Ac-Trp-Ala-Aib3-Aib-Leu-Aib-Gln-Aib–Aib-Aib-Gln-Leu-Aib13-Gln-Lol
- Amp 3,13 :
-
Ac-Trp-Ala-TOAC-Aib-Leu-Aib-Gln-Aib-Aib–Aib-Gln-Leu-TOAC-Gln-Lol
where nOct is n-octanoyl and Lol is the 1,2-amino alcohol leucinol. The chemical structures of the amino acid residues Aib and spin-labeled TOAC (2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4-carboxylic acid) are

As an experimental approach, we employ a three-pulse PELDOR sequence [17]. As compared with a more common four-pulse experimental setup [18], this approach allows to avoid the distortions appearing because of the finite pulselengths [20], and to increase the sensitivity [20, 21].
2 Experimental Section
2.1 Materials and Methods
Syntheses and characterizations of the TOAC-labeled Tri [22] and Amp [23] peptaibiotics used here were already reported. Solvents (methanol, ethanol, methylene chloride) were obtained from Ekros-Analytica (St. Petersburg, Russia).
For adsorption, we used colloidal silica powder of monodisperse nanospheres, commercially produced as a sorbent for medical uses. It was purchased from “ZAO Polisorb” (Chelyabinsk Region, RF), with the reported specific surface area of 300 m2/g. The size and shape of silica nanoparticles were controlled by transmission electron microscopy (TEM). A JEM-2010 microscope (JEOL, Japan) was employed. The accelerating voltage was 200 kV; the resolution was 1.4 Å. Samples for these measurements were prepared by ultrasonic dispersion on a copper base.
The silica powder was placed on the surface of a 0.1-μm Amicon Ultrafree-MC Durapore PVDF centrifugal filter (Millipore), then a 2 mM solution of the spin-labeled peptide either in methanol (containing 5 % of ethanol for glass formation at low temperatures) or in methylene chloride CH2Cl2 was added. The solution was left standing for 30 min, then centrifuged (2000 rpm, 2 min, 20 °C) and the supernatant was removed. The silica powder with the adsorbed peptide deposited was removed from the filter surface and transferred into a 3-mm EPR tube. The tube was pumped out to 10−3 torr and sealed.
2.2 EPR and PELDOR Measurements
An X-band Bruker ELEXSYS E580 EPR spectrometer was used. PELDOR experiments were carried out using a split-ring Bruker ER 4118 X-MS-3 resonator and an Oxford Instruments CF-935 cryostat. In the used three-pulse PELDOR setup, the length of pumping pulse was 28 ns; the lengths of the π/2 and π pulses of the echo-forming detection pulse sequence were 16 and 32 ns, respectively. The time delay between two detection pulses was 650 ns. The pumping pulse was scanned starting at a time delay d 0 = 200 ns preceding the first detection pulse, with a step of 8 ns. The starting delay T for the PELDOR time trace analysis (T = 0) was determined as described in Ref. [24]. The pumping pulse in all cases was applied at the frequency ν B corresponding to the maximum of the echo-detected EPR spectrum, and the difference ν A − ν B between the detection and pumping frequencies was set to 70 MHz. The turning angle of the pumping pulse was set to π in measurements in which ν A was set equal to ν B, the pumping pulse positioned at time delay d 0, and its amplitude varied to adjust the inverted echo signal to its minimum. The in-phase part of the echo was integrated with a time gate of 80 ns. The changes in the PELDOR signal V(T) upon passage of the pumping pulse through the detecting pulses were corrected by the method described in Ref. [20].
The resonator was cooled with gaseous nitrogen. The sample temperature was kept near 77 K.
2.3 Theoretical Background for the PELDOR Data Analysis
For doubly spin-labeled molecules, the PELDOR time trace depends on two contributions: the intramolecular one, arising from interactions between two labels in the molecule, and the intermolecular one, arising from interactions between labels in different molecules. These two contributions can be assumed to be independent so that the PELDOR time trace is presented as a product:
The theory [17, 25] predicts that
where the factor p B is determined by the parameters of the pumping pulse (it characterizes the portion of spins excited), and
where n(r) is the distance distribution function between the two spin labels in the molecule. This function is assumed to be normalized, \(\int_{0}^{{^{\infty}}} {n\left(r \right){\text{d}}r = 1}\) (so making f(0) = 1).
Note that for a rectangular pumping pulse the factor p B can be calculated by formula [25]
where τ p is the pumping pulse length, g(ν) reflects a normalized EPR line shape (\(\int_{0}^{\infty} {g\left(\nu \right){\text{d}}\nu = 1}\)), and ω 1 is the pulse amplitude in the angular frequency units.
The Fourier transform of the f(T) function,
is a frequency-domain PELDOR lineshape, which can be called as a Pake resonance pattern [26]. Note that from Eq. (3) it follows that F(ν) is also normalized, \(\int_{- \infty}^{\infty} {F\left(\nu \right){\text{d}}\nu = 1}\).
V INTER(T) often obeys a simple exponential dependence,
where α ≤ 1 is a parameter reflecting the dimensionality of the space [17]: α = 1 for spins in three-dimensional space and α = 2/3 for spins on the two-dimensional surface (for other cases it is called sometimes fractal dimension). Note that the PELDOR measurements of mono-labeled molecules adsorbed on surfaces [19] indicate that the α value is somewhat larger than 2/3, which is probably related to some excursions of spins from the two-dimensional plane.
The distance distribution function n(r) can be obtained by solving the integral Eq. (3) [26]. To avoid instability of solution because of the ill-posed nature of the problem, the methods based on Tikhonov regularization are employed—see, e.g., [27]. Here, we use a simple Monte-Carlo approach to get the solution. This approach consists of the following steps:
(1) From the experimentally obtained F(ν) Pake resonance pattern the interval of distances r was assessed in which n(r) has significant values; (2) this interval was divided into N − 1 small subintervals with a set of boundaries r i , i = 1\(\ldots\) N; (3) for each r i value, a random value p i = ξ i was ascribed, where 0 < ξ i < 1; (4) the trial distance distribution function was obtained as a set of values n trial(r i ) = p i /S, where \(S = 0.5\left\{{p_{1} \left({r_{2} - r_{1}} \right) + \sum\nolimits_{i = 2}^{i = N - 1} {p_{i} \left({r_{i + 1} - r_{i - 1}} \right) + p_{N} \left({r_{N} - r_{N - 1}} \right)}} \right\}\) is the integral sum for normalization of this function; (5) for this trial distribution the F trial(ν) function was calculated as an integral sum in line with Eqs. (3) and (5); (6) the mean-squared deviation between F trial(ν) and the experimental F(ν) function was calculated; (7) the set of n trial(r i ) values providing the smallest mean-squared deviation was selected as a solution.
Calculations could be repeated for different r i sets. If calculations reveal a smooth behavior of n(r i ), then, for better convergence, a correlation may be established between the consequent adjacent points, in the form of p i+1 = p i (1 − λ) + λξ i+1. If λ = 0, the solution becomes a constant (full correlation), if λ = 1, the adjacent points are uncorrelated.
Normally, the N value between 8 and 16 [depending on smoothness of the calculated n trial(r i ) set] and the number of n trial(r i ) sets of 107 were enough to obtain a reproducible solution. The calculations took about 1 h with an ordinary PC. For this approach, the proper choice of N is crucial: if N is too small, a good agreement with experiment cannot be achieved; if N is too large, the solution oscillates because of the ill-posed nature of the problem. The precise mathematical treatment of convergence of the suggested Monte-Carlo approach will be presented elsewhere.
3 Results and Discussion
3.1 Transmission Electron Microscopy
Results of TEM investigation of silica powder before and after peptide (Tri 1,8) adsorption are presented in Fig. 1. Data show that powder consists of monodisperse spheres of ~20 nm diameter. One can notice some tendency for particle aggregation after adsorption (the property which allows particle sedimentation on a 0.1-μm centrifugal filter—see the Sect. 2).

TEM snapshots for silica powder before (left) and after (right) peptide (Tri 1,8) adsorption
3.2 Spin-Labeled Trichogin GA IV
Figure 2 presents CW EPR spectra of Tri 1,8 on the silica surface immediately after adsorption from a methanol solution and after washing out of the surface by excess of the solvent. The broad line for the initially deposited Tri 1,8 peptide unambiguously highlights that the peptide molecules are assembled in closely-packed clusters in which spatial separation is small enough to essentially induce EPR line broadening due to dipole–dipole and exchange interactions between electron spins. Washing out of the surface reduces remarkably the peptide concentration on the surface so that the close assemblies disappear. This phenomenon results in a substantial narrowing of the EPR lines, with acquisition of a shape typical for dispersed, non-interacting nitroxides.

CW EPR spectra of Tri 1,8 on the silica surface and in the methanol solutions at 77 K: in solution before adsorption (1), at the surface after adsorption (2), at the surface after washing by use of an excess of solvent (3), and in the supernatant methanol solution after washing out of the surface (4)
Results of PELDOR measurements for the doubly spin-labeled Tri 1,8 are given in a semi-logarithmic plot in Fig. 3 for three different conditions: in methanol solution, after adsorption from this solution on the silica surface (with the solution removed), and in the supernatant methanol solution after washing out of the surface.

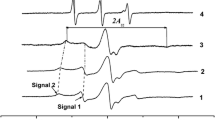
Semi-logarithmic plot of PELDOR V(T) time traces (solid lines) for Tri 1,8 in methanol solution (1), in supernatant methanol solution after washing out of the surface (2), and on the silica surface (3). Curves 2 and 3 are shifted downwards by 0.2 and 0.4, respectively. Dashed lines present asymptotics which linearly depend on T for curves 1 and 2 and linearly depend on T 0.7 for curve 3. Inset results of subtraction of these asymptotics, shifted to the zero initial value
As stated earlier in the text, the experimental PELDOR time trace V(T) may be considered as a product of two contributions, see Eq. (1). The intermolecular contribution to the PELDOR signal, V INTER(T), can be obtained from the asymptotical behavior of the PELDOR time traces. For spin-labeled peptides in solutions, these experiments are known to provide a simple exponential decay, \(V(T) /V(0) = \exp (- {\text{const}}{\kern 1pt} {\kern 1pt} {\kern 1pt} T)\) [17]. This behavior indicates a linear T-dependence of the asymptotics in the semi-logariphmic scale in Fig. 3, which is indeed observed for curves (1) and (2). For Tri 1,8 adsorbed on the silica surface (curve 3) data can be fitted according to Eq. (6) with α = 0.70 which is close to the expected values of 2/3 for the two-dimensional surface.
By subtracting these asymptotical T-dependences in the semi-logarithmic plot reported in Fig. 3, the ln(V INTRA(T)) contribution can be refined (see inset to Fig. 3). As \(f(T)\mathop{\longrightarrow}\limits{}0\) for large T, it follows from Eq. (2) that p B can be obtained from experiment as
From time traces presented in Fig. 3, one can obtain that p B = 0.31 ± 0.05 in methanol solution, p B = 0.29 ± 0.05 in supernatant methanol solution after washing out of the surface, and p B = 0.24 ± 0.05 on the silica surface. Note that our computer simulations employing Eq. (4) and the experimentally obtained g(ν) EPR line shape (data not given) for Tri 1,8 in methanol solution resulted in p B = 0.27 while for Tri 1,8 on silica surface we calculated p B = 0.23 which is in a good agreement with experiment.
In principle, in the adsorption/desorption process a partial modification of the molecule can take place. The modification with the onset of mono spin-labeled residues, for which p B = 0, would immediately be detected by decrease of p B. The closeness of the p B values obtained for all the cases described above allows one to conclude that the biradical structure of the Tri 1,8 molecule during the adsorption/desorption process onto the silica surface could be destroyed only slightly, by ~6 %, which is close to experimental uncertainty.
The closeness of experimental and calculated pumping factor p B evidences the absence of peptide clusters that allows directly analyze peptide conformation. To compare the experimentally obtained V INTRA(T) with the theoretical formula for f(T) in Eq. (3), the V INTRA(T) time dependence must be transformed into its normalized form:
Note [19] that the normalized form (8) allows one to avoid the effects induced by overlapping of excitation and detection in the PELDOR experiments [28].
The cosine Fourier transforms of the V N (T) time traces are given in Fig. 4, along with simulations employing Eqs. (3) and (4), which are based on the Monte-Carlo approach described above. The inset shows the obtained n(r) distribution functions.

Cosine Fourier transforms of the experimental time dependences V N (T) for Tri 1,8 obtained from the data in Fig. 3 (solid lines), and their Monte-Carlo simulations (dashed lines), for Tri 1,8 in methanol solution (a), and on the silica surface (b) (shifted downwards by 0.05 MHz−1). Inset shows the best-fitted distance distribution functions n(r)
From Fig. 4 one can see that distances are distributed around a value close to 2.03 nm in solution and to ~1.9 nm on the surface and that on the surface the distribution is noticeably wider. The result in solution is in agreement with that reported for Tri 1,8 in frozen alcohols [29] where the distance of 1.97 nm in methanol solution was found. This result was ascribed in [30] to a 2.27–/310-helix mixture of conformations of the Tri molecule.
3.3 Spin-Labeled Ampullosporin A
Amp 3,13 was adsorbed on the silica surface optionally either from methanol or from CH2Cl2 solution (in both cases the surface was later washed out by methanol). The PELDOR time traces are shown in Fig. 5.

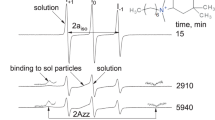
Semi-logarithmic plot of PELDOR V(T) time traces (solid lines) for doubly spin-labeled Amp 3,13 in methanol solution (1), in supernatant methanol solution after washing out of the surface with peptide adsorbed from methanol (2a), in supernatant methanol solution after washing out of the surface with peptide adsorbed from CH2Cl2 (2b), and on the silica surface (3). Curve 3 is shifted downwards by 0.3. Dashed lines present asymptotics which linearly depend on T for curves 1, 2a, 2b and linearly depend on T 0.7 for curve 3. Inset on the right side results of subtraction of these asymptotics, shifted to the zero initial value (data extracted from curve 3 are additionally shifted upwards by 0.05). Inset on the left side CW EPR lineshape for the peptide in methanol solvent (1) and adsorbed on the surface (3)
Using Eq. (7), from data in Fig. 6 one can obtain that p B = 0.32 in methanol solution, p B = 0.31 in supernatant methanol solution after washing out of the surface with peptide adsorbed from methanol, p B = 0.24 in supernatant methanol solution after washing out of the surface with peptide adsorbed from CH2Cl2, p B = 0.23 on the silica surface. Note that our computer simulations employing Eq. (4) and experimentally obtained g(ν) EPR line shape for Amp 3,13 in methanol solution and on the silica surface (data not given) resulted in p B values of 0.29 and 0.23, respectively, which are in a good agreement with experimental values and mean that peptides are non-aggregated. As was mentioned above, the p B decrease can be unambiguously interpreted as a result of the loss of the peptide biradical character during the adsorption/desorption process. Therefore, we conclude from comparison of experimental values that the portion of modified molecules is ~6 % in the supernatant after washing out of the surface with peptide adsorbed from methanol (which is close to the experimental uncertainty), and ~25 % in the supernatant after washing out of the surface with peptide adsorbed from CH2Cl2.

Cosine Fourier transforms of the experimental time dependences V N (T) (solid lines), and their Monte-Carlo simulations (dashed lines), for Amp 3,13 in methanol solution (a) and on the silica surface (b) (shifted downwards by 0.05 MHz−1). Inset shows the best-fitted distance distribution functions n(r)
Loss of the biradical character upon adsorption of the Amp 3,13 molecules may imply either that one of the spin label is lost or that the original whole peptide structure is severely modified during this process. In our opinion, the latter possibility is more reasonable for this compound because this phenomenon strongly depends on which solvent is used for adsorption—methanol or CH2Cl2. We suggest that binding to the surface would be mediated by solvent molecules and that this mediation may be responsible for the loss of the biradical character.
The cosine Fourier transforms of normalized PELDOR time traces V N (T), obtained from data in Fig. 5 in the same way as described above, are given in Fig. 6 for Amp 3,13 in initial methanol solution and on the silica surface after adsorption, along with simulations based on Monte-Carlo fitting of n(r) (see above).
Note that for the data in solution, the found distribution function n(r) is in good agreement with that obtained for the same system by standard approach of solving integral Eq. (3) employing the Tikhonov regularization method [19]. Indeed, data [19] (see Fig. 5 there) give for n(r) the Gaussian line centering at 1.80 nm and attaining the maximal intensity of 10.5 nm−1, while data in inset to Fig. 6 give the line centered also at 1.80 nm and attaining a maximal intensity of about ~9 nm−1.
3.4 Comparison with Distances Calculated from Molecular-Dynamics Simulations
The experimental distances found here can be compared with the distances between label sites calculated using molecular dynamics simulations for different peptide conformations [30, 31]. The conformational space of peptides is restricted to a set of backbone torsion angles defining the α-, 310-, 2.27- (multiple γ-turns) helices and β-sheet conformations [32–34]. Aib-rich peptides are known to prefer the 310-helix. The length of the different conformations for a given peptide increases in the order α- < 310- < 2.27- < β-sheet. Distances between the Cα atoms of the terminal residues of peptides calculated for standard α-, 310-, 2.27-helix, and β-sheet conformations vary between 1.7 and 3.4 nm for a 10-mer (trichogin) and between 2.4 and 4.9 for a 14-mer (ampullosporin). One can see that the disordered conformations found in this work for peptides on the surface give distances between these boundaries.
4 Conclusions
In this work, spin-label CW EPR and PELDOR techniques were employed to investigate adsorption of the short-sequence trichogin GA IV and the medium-length sequence ampullosporin A peptaibiotics on the inorganic SiO2 surface. CW EPR data show that adsorption of trichogin occurs via formation of close-packed peptide clusters. After washing of the surface using excess of solvent, these clusters are replaced by non-associated peptide molecules which can be studied employing the PELDOR technique. PELDOR data were analyzed using a Monte-Carlo approach for deriving the spatial distribution function. The results indicate that both peptides adsorbed on the silica surface adopt a more disordered conformation (exist in a multi-conformational state) than that in solution. For ampullosporin, it was found that disordering is much more marked than that for trichogin. After desorption, the peptides restore their original conformations. PELDOR data for the supernatant solution support the view that up to ~25 % of ampullosporin molecules can be modified in their biradical properties upon adsorption.
References
C. Toniolo, H. Brückner (eds.), Peptaibiotics: Fungal Peptides Containing α-Dialkyl α-Amino Acids (Verlag Helvetica Chimica Acta, Zurich and Wiley-VCH, Weinheim, 2009)
H. Duclohier, Curr. Pharm. Design 16, 3212–3223 (2010)
G. Bocchinfuso, A. Palleschi, B. Orioni, G. Grande, F. Formaggio, C. Toniolo, Y. Park, K.-S. Hahm, L. Stella, J. Pept. Sci. 15, 550–558 (2009)
N.K.N. Neumann, N. Stoppacher, S. Zeilinger, T. Degenkolb, H. Brückner, R. Schuhmacher, Chem. Biodivers. 12, 743–751 (2015)
K.S. Soppimath, T.M. Aminabhavi, A.R. Kulkarni, W.E. Rudzinski, J. Control. Release 70, 1–20 (2001)
C.P. Reis, R.J. Neufeld, A.J. Ribeiro, F. Veiga, Nanomedicine 2, 53–65 (2006)
S.C. Yadav, A. Kumari, R. Yadav, Peptides 32, 173–187 (2011)
Z. Mao, L. Wan, L. Hu, L. Ma, C. Gao, Coll. Surf. B Biointerfaces 75, 432–440 (2010)
R. Coppage, J.M. Slocik, M. Sethi, D.B. Pacardo, R.R. Naik, M.R. Knecht, Angew. Chem. Int. Ed. 49, 3767–3770 (2010)
J.M. Slocik, A.O. Govorov, R.R. Naik, Nano Lett. 11, 701–705 (2011)
L. Lenoci, P.J. Camp, J. Am. Chem. Soc. 128, 10111–10117 (2006)
L.B. Wright, T.R. Walsh, Phys. Chem. Chem. Phys. 15, 4715–4726 (2013)
P. Rigler, W.-P. Ulrich, P. Hoffmann, M. Mayer, H. Vogel, ChemPhysChem 4, 268–275 (2003)
K.T. Nguyen, J.T. King, Z. Chen, J. Phys. Chem. B 114, 8291–8300 (2010)
G. Goobes, P.S. Stayton, G.P. Drobny, Prog. Nucl. Magn. Reson. Spectrosc. 50, 71–85 (2007)
P.A. Mirau, R.R. Naik, P. Gehring, J. Am. Chem. Soc. 133, 18243–18248 (2011)
A.D. Milov, A.G. Maryasov, Yu.D. Tsvetkov, Appl. Magn. Reson. 15, 107–143 (1998)
M. Pannier, S. Veit, A. Godt, G. Jeschke, H.W. Spiess, J. Magn. Reson. 142, 331–340 (2000)
A.D. Milov, R.I. Samoilova, Yu.D. Tsvetkov, C. Peggion, F. Formaggio, C. Toniolo, J. Phys. Chem. B 118, 7085–7090 (2014)
A.D. Milov, Yu.A. Grishin, S.A. Dzuba, Yu.D. Tsvetkov, Appl. Magn. Reson. 41, 59–67 (2011)
J.E. Lovett, B.W. Lovett, J. Harmer, J. Magn. Reson. 223, 98–106 (2012)
V. Monaco, F. Formaggio, M. Crisma, C. Toniolo, P. Hanson, G. Millhauser, C. George, J.R. Deschamps, J.L. Flippen-Anderson, Bioorg. Med. Chem. 7, 119–131 (1991)
A.D. Milov, Y.D. Tsvetkov, M. Bortolus, A.L. Maniero, M. Gobbo, C. Toniolo, F. Formaggio, Biopolymers (Pept. Sci.) 102, 40–48 (2014)
N.A. Kuznetsov, A.D. Milov, V.V. Koval, R.I. Samoilova, Yu.A. Grishin, D.G. Knorre, Yu.D. Tsvetkov, O.S. Fedorova, S.A. Dzuba, Phys. Chem. Chem. Phys. 11, 6826–6832 (2009)
A.D. Milov, K.M. Salikhov, M.D. Schirov, Fiz. Tverd. Tela (in Russian) 23, 975–982 (1981)
G. Jeschke, A. Koch, U. Jonas, A. Godt, J. Magn. Reson. 155, 72–82 (2002)
N.A. Kuznetsov, A.D. Milov, N.P. Isaev, Yu.N. Vorobjev, V.V. Koval, S.A. Dzuba, O.S. Fedorova, Yu.D. Tsvetkov, Mol. BioSyst. 7, 2670–2680 (2011)
K.M. Salikhov, I.T. Khairuzhdinov, R.B. Zaripov, Appl. Magn. Reson. 45, 573–619 (2014)
A.D. Milov, A.G. Maryasov, R.I. Samoilova, Yu.D. Tsvetkov, J. Raap, V. Monaco, F. Formaggio, M. Crisma, C. Toniolo, Dokl. Akad. Nauk 370, 265–268 (2000)
A.D. Milov, Y.D. Tsvetkov, E.Y. Gorbunova, L.J. Mustaeva, T.V. Ovchinnikova, J.W. Handgraaf, J. Raap, Chem. Biodivers. 4, 1243–1255 (2007)
A.D. Milov, Y.D. Tsvetkov, J. Raap, M. De Zotti, F. Formaggio, C. Toniolo, Biopolymers (Pept. Sci.) (2015) (in press)
C. Toniolo, E. Benedetti, Trends Biochem. Sci. 16, 350–353 (1991)
M. Crisma, F. Formaggio, A. Moretto, C. Toniolo, Biopolymers (Pept. Sci.) 84, 3–12 (2006)
M. Crisma, M. De Zotti, A. Moretto, C. Peggion, B. Drouillat, K. Wright, F. Couty, C. Toniolo, F. Formaggio, New J. Chem. 39, 3208–3216 (2015)
Acknowledgments
We thank Dr. A.D. Milov for helpful discussion and assistance in some of the measurements. This work was supported by the Russian Science Foundation, project no. 15-15-00021.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Syryamina, V.N., Samoilova, R.I., Tsvetkov, Y.D. et al. Peptides on the Surface: Spin-Label EPR and PELDOR Study of Adsorption of the Antimicrobial Peptides Trichogin GA IV and Ampullosporin A on the Silica Nanoparticles. Appl Magn Reson 47, 309–320 (2016). https://doi.org/10.1007/s00723-015-0745-5
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-015-0745-5