
Abstract
Coronaviruses can have a devastating impact on the health of humans and animals. Porcine epidemic diarrhea virus (PEDV) causes extremely high fatality rates in neonatal piglets, whereas severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the current COVID-19 pandemic in humans. As a critical component of the host antiviral innate immune response, type I interferon production and signaling play a very important role, especially in the initial phase of the antiviral responses. Coronaviruses have evolved multiple ways to counteract type I interferon responses. Although the primary functions of the nucleocapsid protein are to facilitate viral RNA replication and package viral genomic RNA into virions, recent studies have shown that the nucleocapsid protein is also involved in virus-host interactions. The aim of this review is to summarize our current understanding of how the nucleocapsid proteins of PEDV and SARS-CoV-2 modulate type I interferon responses. This knowledge will be useful for developing strategies to combat coronavirus infections.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coronaviruses are important RNA viruses, and members of the subfamily Orthocoronavirinae are classified into four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus [1]. Coronaviruses have a very significant impact on both humans and animals such as pigs. For example, porcine epidemic diarrhea virus (PEDV), an enteric alphacoronavirus, was first reported in 1978 in Belgium [2]. Since then, PEDV has spread across Europe and Asia, causing sporadic outbreaks. Notably, highly pathogenic PEDV strains were identified in 2010 when severe porcine epidemic diarrhea (PED) outbreaks were reported in China and in other countries. PEDV first entered North America in 2013, spreading rapidly [3, 4]. PED is characterized by vomiting, diarrhea, dehydration, and anorexia, causing up to 100% mortality in suckling piglets and impaired growth in finishing pigs [4].
The recently identified severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a respiratory betacoronavirus that is causing the ongoing global COVID-19 pandemic [5]. Major symptoms of COVID-19 include shortness of breath, fever, and muscular discomfort [6]. There are major economic implications of both PEDV and SARS-CoV-2. Specifically, PEDV causes devastating economic losses in the swine industry [7], whereas SARS-CoV-2 has led to negative growth rates in the economy, with closing of many businesses and schools and travel restrictions as a result of public health concerns [6]. While the origins of PEDV and SARS-CoV-2 have not been clearly established, the identification of closely related bat coronaviruses suggests a zoonotic origin [8,9,10,11].
Coronaviruses are enveloped, single-stranded, positive-sense RNA viruses [1]. Their genome is a single linear RNA molecule with a 5′ cap structure and a polyadenylated tail at the 3′ end. The PEDV genome is approximately 28 kb in length, whereas the genome of SARS-CoV-2 is about 30 kb long [12, 13]. The open reading frames are flanked by untranslated regions at both the 5′ and 3′ ends. The N-terminal two-thirds of the RNA genome contains the large open reading frames ORF1a and ORF1ab, which, due to a -1 frameshift, are translated into two polyproteins, pp1a and pp1ab. The polyproteins of both PEDV and SARS-CoV-2 undergo proteolytic cleavage to generate up to 16 mature nonstructural proteins [13, 14]. The remaining portion of the genome codes for four structural proteins – spike (S), envelope (E), membrane (M), and nucleocapsid (N) – as well as a number of accessory proteins. PEDV encodes only one accessory protein (the ORF3 protein), whereas SARS-CoV-2 encodes up to nine accessory proteins [13, 14]. While the wide-ranging effects of the accessory proteins on the virus life cycle and virus-host interactions are still under investigation, the roles of structural proteins are better understood [13, 14]. One of these, the N protein, has been shown to play a very important role in coronavirus biology, virus-host interaction, and pathogenesis, and modulating host innate antiviral responses such as the type I interferon (IFN) response is one of its critical functions. Therefore, in this article, we discuss the mechanisms by which N proteins of alpha- and betacoronaviruses regulate type I IFN responses. We chose PEDV and SARS-CoV-2 because they possess distinct pathogenic features (enteric vs. respiratory) and target different species (swine vs. human). We begin with an overview of type I IFN responses to coronavirus infections, continue with a discussion on our current understanding of how the N proteins of PEDV and SARS-CoV-2 regulate this response, and end with our thoughts on future directions.
Host type I interferon responses to coronavirus infections
Innate immunity is responsible for the immediate response of the host to coronavirus infections. This response begins with the recognition of danger signals by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), NOD-like receptors (NLRs), C-type lectin receptors (CLRs), absent in melanoma 2 (AIM2)-like receptors, and RIG-I-like receptors (RLRs) [15,16,17]. Nod-, LRR- and pyrin-domain-containing protein 3 (NLRP3) is one of the well-characterized NLRs [18]. NLRP3 responds to cell stress and virus infections by assembling an NLRP3 inflammasome. The inflammasome is a multiprotein complex that induces the expression of pro-inflammatory mature IL-1β and IL-18, as well as pyroptotic cell death [18]. While inflammation is protective against virus infections, uncontrolled inflammation has been documented to contribute to viral pathogenesis, such as severe COVID-19 disease after SARS-CoV-2 infection [19]. In this regard, SARS-CoV-2 N protein has been shown to induce NLRP3 inflammasome assembly through direct binding to NLRP3, resulting in hyperinflammation [20]. The SARS-CoV-2 N protein has also been shown to promote hyperactivation of NF-κB and, consequently, excessive inflammation [21].
Cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS) is also an important molecule in the host innate immune system [22]. cGAS functions through stimulator of interferon genes (STING), which triggers the production of interferons and inflammatory cytokines [22]. It has been shown that PEDV antagonizes STING activation [23], whereas STING contributes to immunopathology after SARS-CoV-2 infection [24].
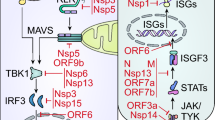
As a vital component of the host innate immune response, type I IFNs are a group of signaling cytokines produced by the host for antiviral defense [25]. Upon coronavirus infection, the activation of the type I IFN response occurs in three stages: PRRs recognize pathogen-associated molecular patterns (PAMPs), type I IFNs are released through paracrine and autocrine pathways, and many antiviral IFN-stimulated genes (ISGs) are expressed, causing the host to enter an antiviral state [17]. RIG-I, MDA5, and Laboratory of Genetics and Physiology 2 (LGP2) are the major RLRs that can sense viral RNA [26]. RIG-I and MDA5 contain two N-terminal caspase activation and recruitment domains (CARDs), a helicase domain, and a C-terminal domain. Upon binding to viral RNA by the helicase and C-terminal domains, the CARD undergoes a conformational change and is activated by polyubiquitination by several ubiquitin ligases, such as TRIM25. The activated RIG-I/MDA5 binds to MAVS/IPS-1, which is a mitochondrial adaptor protein, to form a complex called the MAVS signalosome [15]. This complex activates TNF receptor-associated factor 3 (TRAF3), TANK-binding kinase 1 (TBK1), and IκB1 kinase (IKK), leading to the phosphorylation of interferon regulatory factor 3 (IRF3) [27]. Phosphorylated IRF3 translocates into the nucleus, where it promotes type I IFN gene transcription [27]. LGP2 lacks a CARD domain and therefore does not mediate signaling. However, LGP2 can modulate the functions of RIG-I and MDA5 both positively and negatively [26]. Secreted extracellular type I IFNs bind to the IFNα receptor (IFNAR), a heterodimeric complex consisting of two subunits, IFNAR-1 and IFNAR-2, on the cell surface. This in turn activates tyrosine kinase 2 (TYK2)/Janus kinase 1 (JAK1), leading to the formation of IFN-stimulated gene factor 3 (ISGF3), composed of STAT1-STAT2 and IRF9 [25, 28]. The cytoplasmic ISGF3 then moves into the nucleus to turn on the expression of IFN-stimulated genes.
Coronaviruses, including PEDV and SARS-CoV-2, have evolved multiple means to modulate host type I IFN responses [17, 28,29,30,31,32]. In this review, we focus on the nucleocapsid proteins of PEDV and SARS-CoV-2.
Domain structure and biological functions of the nucleocapsid protein
The N proteins of coronaviruses, including PEDV and SARS-CoV-2, share a similar domain structure, consisting of an N-arm, an N-terminal domain (NTD), a Ser/Arg-rich linker (LKR), a C-terminal domain (CTD), and a C-tail [33,34,35,36]. NTD and CTD are structured domains, whereas the N-arm, LKR, and C-tail are intrinsically disordered regions (IDRs). The primary and conserved function of the nucleocapsid is to package viral genomic RNA into the virion. As such, it is not surprising that both NTD and CTD are involved in RNA binding, with the three IDRs playing a modulating role [35]. CTD also mediates homodimerization of the N protein [36]. In addition to RNA encapsidation, other functions have been attributed to the N protein. It plays multiple roles in the virus life cycle and virus-host interactions, including enhancing viral RNA transcription and replication [37, 38], and modulating host antiviral responses (see below).
N proteins can be post-translationally modified by phosphorylation, sumoylation, and ADP-ribosylation [39]. These modifications play important roles in regulating the functions of the N protein. It has been shown that phosphorylation of the LKR domain regulates N protein phase separation, resulting in different forms of condensates with diverse functions in terms of RNA-protein and protein-protein interactions [40, 41]. A very recent study identified Ser79 as a new phosphorylation site outside the LKR domain of the SARS-CoV-2 N protein [42]. This phosphorylation event enhances the binding of the N protein to the host prolyl-isomerase Pin1. Knocking down Pin1 expression reduces viral RNA levels, suggesting a functional role of the N-Pin1 interaction [42].
Sumoylation has been documented for the N protein when it is ectopically expressed [43]. Furthermore, it has been shown that N protein sumoylation enhances its homo-oligomerization and nucleolar localization [43]. N proteins of a few coronaviruses, including PEDV and SARS-CoV, are also modified by ADP-ribosylation [44]. Interestingly, N protein ADP-ribosylation only occurs in the context of viral infections, and ADP-ribosylated N proteins are detected in virions [44]. Whether N protein ADP-ribosylation increases its incorporation into the virions and whether any other functions are associated with this modification remain to be investigated.
PEDV N protein
There are a few studies documenting the effects of PEDV N protein on type I IFN responses as well as how the PEDV N protein is targeted by several ISG proteins. It has been shown that PEDV N antagonizes IFN-β production after Sendai virus infection [45, 46]. Furthermore, the PEDV N protein can inhibit IFN-β promoter activation by TBK1 and its upstream molecules RIG-I, MDA5, MAVS, and TRAF3 [45, 46]. Interestingly, however, the PEDV N protein does not directly impede IFN-β promoter activation by IRF3 [45, 46]. Instead, it interferes with the interactions between TBK1 and IRF3, resulting in a lack of IRF3 activation and subsequent inhibition of IFN-β expression.
Given the antagonizing role of PEDV N protein in the type I IFN response, it is not unexpected that this protein becomes a target of the host innate immune system. One study showed that viperin, an ISG induced by IRF1 and IRF3, interacts with the PEDV N protein and inhibits PEDV proliferation [47]. IRAV (IFN-regulated antiviral), BST-2 (bone marrow stromal cell antigen-2), and TRIM21 are three additional ISG proteins that have been shown to interact with the PEDV N protein [48,49,50]. These protein-protein interactions lead to N protein degradation and inhibition of viral replication [48,49,50].
SARS-CoV-2 N protein
In comparison to PEDV, we have a better understanding of the complex interactions between the SARS-CoV-2 N protein and the host type I IFN system. It has been shown that the SARS-CoV-2 N protein inhibits IFN-β activation by RIG-I or Sendai virus infection [51,52,53,54,55]. Mechanistically, the SARS-CoV-2 N protein interacts directly with RIG-I [51, 52]. Furthermore, it has been shown that the interaction of SARS-CoV-2 N with RIG-I interferes with the binding of TRIM25 to RIG-I as well as the interaction between TBK1 and IRF3 [52, 54]. These interactions abrogate the activation of downstream effectors of RIG-I signaling such as IRF3 phosphorylation and nuclear translocation, which in turn dampens IFN-β expression [51, 52]. Along the same line, it has been shown that SARS-CoV-2 N, via the dimerization domain, is enriched in the MAVS signalosome and interferes with its signaling capacity [55]. Interestingly however, two studies have shown that the SARS-CoV-2 N protein had no effect on IFN-β promoter activity when RIG-I was overexpressed [56, 57]. The reason for the discrepancy is not clear, but it is most likely due to differences in the N protein sequences and cell lines used in different laboratories.
Formation of stress granules (SGs) is another mechanism by which the host mounts an antiviral response [58], and SARS-CoV-2 N has been shown to interfere with SG formation [57, 59]. Mechanistically, it was found that SARS-CoV-2 N interacts with protein kinase R (PKR) to inhibit PKR autophosphorylation and activation, which is required for SG formation [59]. Furthermore, Ras-GTPase-activating protein-binding protein 1 (G3BP1), a key nucleating component of SGs, is also targeted by the SARS-CoV-2 N through direct protein-protein interaction [59]. A recent crystal structure analysis demonstrated that the N-terminal 25 amino acid residues of N contribute to binding to the nuclear transport factor 2 (NTF2)-like domain of G3BP1 [60]. One of the functions of SGs is type I interferon production [58]. Zheng et al. showed that the SARS-CoV-2 N protein antagonizes IFN-β activation by coexpressing RIG-I and G3BP1, as well as RIG-I and the PKR-activating protein PACT [57]. Consistent with the antiviral function of SG, it has been shown that knocking down PKR or G3BP1 expression increases SARS-CoV-2 replication [59].
SARS-CoV-2 N has also been shown to inhibit interferon signaling by reducing STAT1 and STAT2 phosphorylation and nuclear translocation upon interacting with these two proteins [54, 61]. More importantly, it has been demonstrated that overexpression of the N protein enhances the replication of SARS-CoV-2 by antagonizing type I IFN signaling in infected human HepG2 cells [61].
All of these studies except for one [61] were performed after ectopic expression of the N protein, and the relevance of these findings in the context of SARS-CoV-2 infection should be investigated. This is critically important, as one study showed various effects of SARS-CoV-2 N protein on IFN-β expression as well as IFN-β signaling, depending on the levels of the N protein [62]. Specifically, at low levels, the SARS-CoV-2 N protein inhibits IFN-β production by disturbing the interaction of TRIM25 and RIG-I and inhibiting the phosphorylation and nuclear translocation of IRF3, STAT1, and STAT2, whereas at high levels, the N protein promotes IFN-β expression by enhancing the phosphorylation and nuclear translocation of STAT1 and STAT2 [62].
Summary and future directions
The type I IFN response is a critical component of the host innate immune response and plays a very important role in counteracting coronavirus infections. Coronaviruses, on the other hand, have evolved multiple mechanisms to suppress the type I IFN response mounted by the host. Among them, the role of the N protein in modulating the IFN response has been largely underappreciated. In our efforts to promote more studies on this important area, we have summarized our current knowledge on how the N proteins of PEDV and SARS-CoV-2 interact with the type I IFN regulatory system. Although the N proteins of both PEDV and SARS-CoV-2 have been demonstrated to suppress IFN-β activation by RIG-I, detailed mechanistic studies are lacking, especially for the PEDV N protein. Meanwhile, we have a better understanding of how the PEDV N protein is targeted by ISGs, whereas this has not been studied for the SARS-CoV-2 N protein. In addition, it is noteworthy that almost all published investigations have relied on ectopic overexpression of the N protein and the components of the type I IFN system of interest. While data obtained in this manner are valuable, studies in the context of viral infection and the endogenous type I IFN system should also be performed.
It is worth mentioning that type I IFN has been evaluated in clinical trials to treat COVID-19 patients [63]. The results are mixed in terms of disease outcome and timing of IFN therapy. This calls for a better understanding of the interactions between the type I IFN regulatory system and coronaviral components, including the N protein. On the other hand, the N protein has been increasingly explored as a drug target to control viral replication and as a vaccine antigen to induce an effective immune response [36, 48, 49, 64,65,66,67]. Zoonotic transmission of coronaviruses is another dimension that needs attention when it comes to developing countermeasures against coronavirus infections. Since the N proteins of members of the family Coronaviridae share relatively high sequence similarity [46, 66], targeting the N protein might be a viable option for zoonosis management. In summary, we believe that a thorough understanding of how the N proteins modulate type I IFN responses will provide additional insights to guide our efforts toward eliminating the detrimental impact of coronaviruses on human and animal health.
References
V’Kovski P, Kratzel A, Steiner S, Stalder H, Thiel V (2021) Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol 19:155–170
Pensaert MB, de Bouck P (1978) A new coronavirus-like particle associated with diarrhea in swine. Arch Virol 58:243–247
Gerdts V, Zakhartchouk A (2017) Vaccines for porcine epidemic diarrhea virus and other swine coronaviruses. Vet Microbiol 206:45–51
Jung K, Saif LJ (2015) Porcine epidemic diarrhea virus infection: etiology, epidemiology, pathogenesis and immunoprophylaxis. Vet J 204:134–143
Chams N, Chams S, Badran R, Shams A, Araji A, Raad M, Mukhopadhyay S, Stroberg E, Duval EJ, Barton LM, Hajj HI (2020) COVID-19: a multidisciplinary review. Front Public Health 8:383
Khan M, Adil SF, Alkhathlan HZ, Tahir MN, Saif S, Khan M, Khan ST (2020) COVID-19: a global challenge with old history, epidemiology and progress so far. Molecules 26:39
Turlewicz-Podbielska H, Pomorska-Mol M (2021) Porcine coronaviruses: overview of the state of the art. Virol Sin 36:833–851
Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, Li G, Dong BQ, Liu W, Cheung CL, Xu KM, Song WJ, Vijaykrishna D, Poon LL, Peiris JS, Smith GJ, Chen H, Guan Y (2006) Prevalence and genetic diversity of coronaviruses in bats from China. J Virol 80:7481–7490
Han Y, Du J, Su H, Zhang J, Zhu G, Zhang S, Wu Z, Jin Q (2019) Identification of diverse bat alphacoronaviruses and betacoronaviruses in China provides new insights into the evolution and origin of coronavirus-related diseases. Front Microbiol 10:1900
Delaune D, Hul V, Karlsson EA, Hassanin A, Ou TP, Baidaliuk A, Gambaro F, Prot M, Tu VT, Chea S, Keatts L, Mazet J, Johnson CK, Buchy P, Dussart P, Goldstein T, Simon-Loriere E, Duong V (2021) A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat Commun 12:6563
Zhou H, Ji J, Chen X, Bi Y, Li J, Wang Q, Hu T, Song H, Zhao R, Chen Y, Cui M, Zhang Y, Hughes AC, Holmes EC, Shi W (2021) Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 184:4380-4391 e14
Bar-On YM, Flamholz A, Phillips R, Milo R (2020) SARS-CoV-2 (COVID-19) by the numbers. Elife 9:e57309
Jantraphakorn Y, Viriyakitkosol R, Jongkaewwattana A, Kaewborisuth C (2021) Interaction between PEDV and its hosts: a closer look at the ORF3 accessory protein. Front Vet Sci 8:744276
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Huttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C et al (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583:459–468
Diamond MS, Kanneganti TD (2022) Innate immunity: the first line of defense against SARS-CoV-2. Nat Immunol 23:165–176
Manes NP, Nita-Lazar A (2021) Molecular mechanisms of the toll-like receptor, STING, MAVS, inflammasome, and interferon pathways. mSystems. https://doi.org/10.1128/mSystems.00336-21:e0033621
Li S, Yang J, Zhu Z, Zheng H (2020) Porcine epidemic diarrhea virus and the host innate immune response. Pathogens 9:E367
Swanson KV, Deng M, Ting JP (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19:477–489
Hsu RJ, Yu WC, Peng GR, Ye CH, Hu S, Chong PCT, Yap KY, Lee JYC, Lin WC, Yu SH (2022) The role of cytokines and chemokines in severe acute respiratory syndrome coronavirus 2 infections. Front Immunol 13:832394
Pan P, Shen M, Yu Z, Ge W, Chen K, Tian M, Xiao F, Wang Z, Wang J, Jia Y, Wang W, Wan P, Zhang J, Chen W, Lei Z, Chen X, Luo Z, Zhang Q, Xu M, Li G, Li Y, Wu J (2021) SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun 12:4664
Wu Y, Ma L, Cai S, Zhuang Z, Zhao Z, Jin S, Xie W, Zhou L, Zhang L, Zhao J, Cui J (2021) RNA-induced liquid phase separation of SARS-CoV-2 nucleocapsid protein facilitates NF-kappaB hyper-activation and inflammation. Signal Transduct Target Ther 6:167
Ablasser A, Chen ZJ (2019) cGAS in action: Expanding roles in immunity and inflammation. Science 363:eaat8657
Xing Y, Chen J, Tu J, Zhang B, Chen X, Shi H, Baker SC, Feng L, Chen Z (2013) The papain-like protease of porcine epidemic diarrhea virus negatively regulates type I interferon pathway by acting as a viral deubiquitinase. J Gen Virol 94:1554–1567
Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, Nass T, Guenova E, Schaller M, Conrad C, Goepfert C, de Leval L, Garnier CV, Berezowska S, Dubois A, Gilliet M, Ablasser A (2022) The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 603:145–151
Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14:36–49
Carty M, Guy C, Bowie AG (2021) Detection of viral infections by innate immunity. Biochem Pharmacol 183:114316
Al Hamrashdi M, Brady G (2022) Regulation of IRF3 activation in human antiviral signaling pathways. Biochem Pharmacol 200:115026
Lee S, Channappanavar R, Kanneganti TD (2020) Coronaviruses: innate immunity, inflammasome activation, inflammatory cell death, and cytokines. Trends Immunol 41:1083–1099
Jouvenet N, Goujon C, Banerjee A (2021) Clash of the titans: interferons and SARS-CoV-2. Trends Immunol 42:1069–1072
Schultze JL, Aschenbrenner AC (2021) COVID-19 and the human innate immune system. Cell 184:1671–1692
Zanoni I (2021) Interfering with SARS-CoV-2: are interferons friends or foes in COVID-19? Curr Opin Virol 50:119–127
Zhang Q, Yoo D (2016) Immune evasion of porcine enteric coronaviruses and viral modulation of antiviral innate signaling. Virus Res 226:128–141
Bai Z, Cao Y, Liu W, Li J (2021) The SARS-CoV-2 nucleocapsid protein and its role in viral structure, biological functions, and a potential target for drug or vaccine mitigation. Viruses 13:1115
Chang CK, Hou MH, Chang CF, Hsiao CD, Huang TH (2014) The SARS coronavirus nucleocapsid protein–forms and functions. Antiviral Res 103:39–50
McBride R, van Zyl M, Fielding BC (2014) The coronavirus nucleocapsid is a multifunctional protein. Viruses 6:2991–3018
Tseng YY, Liao GR, Lien A, Hsu WL (2021) Current concepts in the development of therapeutics against human and animal coronavirus diseases by targeting NP. Comput Struct Biotechnol J 19:1072–1080
Liwnaree B, Narkpuk J, Sungsuwan S, Jongkaewwattana A, Jaru-Ampornpan P (2019) Growth enhancement of porcine epidemic diarrhea virus (PEDV) in Vero E6 cells expressing PEDV nucleocapsid protein. PLoS One 14:e0212632
Nguyen HT, Falzarano D, Gerdts V, Liu Q (2021) Construction of a noninfectious SARS-CoV-2 replicon for antiviral-drug testing and gene function studies. J Virol 95:e0068721
Fung TS, Liu DX (2018) Post-translational modifications of coronavirus proteins: roles and function. Future Virol 13:405–430
Carlson CR, Asfaha JB, Ghent CM, Howard CJ, Hartooni N, Safari M, Frankel AD, Morgan DO (2020) Phosphoregulation of phase separation by the SARS-CoV-2 N protein suggests a biophysical basis for its dual functions. Mol Cell 80:1092-1103 e4
Lu S, Ye Q, Singh D, Cao Y, Diedrich JK, Yates JR 3rd, Villa E, Cleveland DW, Corbett KD (2021) The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat Commun 12:502
Ino Y, Nishi M, Yamaoka Y, Miyakawa K, Jeremiah SS, Osada M, Kimura Y, Ryo A (2022) Phosphopeptide enrichment using Phos-tag technology reveals functional phosphorylation of the nucleocapsid protein of SARS-CoV-2. J Proteomics 255:104501
Li FQ, Xiao H, Tam JP, Liu DX (2005) Sumoylation of the nucleocapsid protein of severe acute respiratory syndrome coronavirus. FEBS Lett 579:2387–2396
Grunewald ME, Fehr AR, Athmer J, Perlman S (2018) The coronavirus nucleocapsid protein is ADP-ribosylated. Virology 517:62–68
Ding Z, Fang L, Jing H, Zeng S, Wang D, Liu L, Zhang H, Luo R, Chen H, Xiao S (2014) Porcine epidemic diarrhea virus nucleocapsid protein antagonizes beta interferon production by sequestering the interaction between IRF3 and TBK1. J Virol 88:8936–8945
Liu Y, Liang QZ, Lu W, Yang YL, Chen R, Huang YW, Wang B (2021) A comparative analysis of coronavirus nucleocapsid (N) proteins reveals the SADS-CoV N protein antagonizes IFN-beta production by inducing ubiquitination of RIG-I. Front Immunol 12:688758
Wu J, Chi H, Fu Y, Cao A, Shi J, Zhu M, Zhang L, Hua D, Huang J (2020) The antiviral protein viperin interacts with the viral N protein to inhibit proliferation of porcine epidemic diarrhea virus. Arch Virol 165:2279–2289
Kong N, Shan T, Wang H, Jiao Y, Zuo Y, Li L, Tong W, Yu L, Jiang Y, Zhou Y, Li G, Gao F, Yu H, Zheng H, Tong G (2020) BST2 suppresses porcine epidemic diarrhea virus replication by targeting and degrading virus nucleocapsid protein with selective autophagy. Autophagy 16:1737–1752
Wang H, Kong N, Jiao Y, Dong S, Sun D, Chen X, Zheng H, Tong W, Yu H, Yu L, Zhang W, Tong G, Shan T (2021) EGR1 suppresses porcine epidemic diarrhea virus replication by regulating IRAV To degrade viral nucleocapsid protein. J Virol 95:e0064521
Wang H, Chen X, Kong N, Jiao Y, Sun D, Dong S, Qin W, Zhai H, Yu L, Zheng H, Tong W, Yu H, Tong G, Shan T (2021) TRIM21 inhibits porcine epidemic diarrhea virus proliferation by proteasomal degradation of the nucleocapsid protein. Arch Virol 166:1903–1911
Chen K, Xiao F, Hu D, Ge W, Tian M, Wang W, Pan P, Wu K, Wu J (2020) SARS-CoV-2 nucleocapsid protein interacts with RIG-I and represses RIG-mediated IFN-beta production. Viruses 13:47
Gori Savellini G, Anichini G, Gandolfo C, Cusi MG (2021) SARS-CoV-2 N protein targets TRIM25-mediated RIG-I activation to suppress innate immunity. Viruses 13:1439
Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L, Guo F, Zhao Z, Zhou Z, Xiang Z, Wang J (2020) Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun 11:3810
Oh SJ, Shin OS (2021) SARS-CoV-2 nucleocapsid protein targets RIG-I-like receptor pathways to inhibit the induction of interferon response. Cells 10:530
Wang S, Dai T, Qin Z, Pan T, Chu F, Lou L, Zhang L, Yang B, Huang H, Lu H, Zhou F (2021) Targeting liquid-liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat Cell Biol 23:718–732
Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, Menachery VD, Rajsbaum R, Shi PY (2020) Evasion of type I interferon by SARS-CoV-2. Cell Rep 33:108234
Zheng Y, Deng J, Han L, Zhuang MW, Xu Y, Zhang J, Nan ML, Xiao Y, Zhan P, Liu X, Gao C, Wang PH (2022) SARS-CoV-2 NSP5 and N protein counteract the RIG-I signaling pathway by suppressing the formation of stress granules. Signal Transduct Target Ther 7:22
Eiermann N, Haneke K, Sun Z, Stoecklin G, Ruggieri A (2020) Dance with the devil: stress granules and signaling in antiviral responses. Viruses 12:984
Zheng ZQ, Wang SY, Xu ZS, Fu YZ, Wang YY (2021) SARS-CoV-2 nucleocapsid protein impairs stress granule formation to promote viral replication. Cell Discov 7:38
Biswal M, Lu J, Song J (2022) SARS-CoV-2 nucleocapsid protein targets a conserved surface groove of the NTF2-like domain of G3BP1. J Mol Biol 434:167516
Mu J, Fang Y, Yang Q, Shu T, Wang A, Huang M, Jin L, Deng F, Qiu Y, Zhou X (2020) SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov 6:65
Zhao Y, Sui L, Wu P, Wang W, Wang Z, Yu Y, Hou Z, Tan G, Liu Q, Wang G (2021) A dual-role of SARS-CoV-2 nucleocapsid protein in regulating innate immune response. Signal Transduct Target Ther 6:331
Jhuti D, Rawat A, Guo CM, Wilson LA, Mills EJ, Forrest JI (2022) Interferon treatments for SARS-CoV-2: challenges and opportunities. Infect Dis Ther. https://doi.org/10.1007/s40121-022-00633-9
Jiao Y, Kong N, Wang H, Sun D, Dong S, Chen X, Zheng H, Tong W, Yu H, Yu L, Huang Y, Wang H, Sui B, Zhao L, Liao Y, Zhang W, Tong G, Shan T (2021) PABPC4 broadly inhibits coronavirus replication by degrading nucleocapsid protein through selective autophagy. Microbiol Spectr 9:e0090821
Peng Y, Du N, Lei Y, Dorje S, Qi J, Luo T, Gao GF, Song H (2020) Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. EMBO J 39:e105938
Thura M, Sng JXE, Ang KH, Li J, Gupta A, Hong JM, Hong CW, Zeng Q (2021) Targeting intra-viral conserved nucleocapsid (N) proteins as novel vaccines against SARS-CoVs. Biosci Rep 41:BSR20211491
Wang L, Sola I, Enjuanes L, Zuniga S (2021) MOV10 helicase interacts with coronavirus nucleocapsid protein and has antiviral activity. MBio 12:e0131621
Acknowledgements
Research in our laboratory is supported by the Natural Sciences and Engineering Research Council of Canada (RGPIN-2018-04138) and the Saskatchewan Agriculture Development Fund (20180101). PY is supported by a Mitac Globalink Research Internship. LH is partially supported by the Vaccinology and Immunotherapeutics graduate scholarship. VIDO receives operational funding from the Government of Saskatchewan through Innovation Saskatchewan and the Ministry of Agriculture, and from the Canada Foundation for Innovation through the Major Science Initiatives for its CL3 facility. This article is published with the permission of the Director of VIDO, journal series no. 979.
Funding
This study was partially funded by the Natural Sciences and Engineering Research Council of Canada (RGPIN-2018-04138) and a Mitac Globalink Research Internship. The funding sources had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: QL. Investigation: PY, HL, QL. Writing – original draft: PY, HL, QL. Writing – review and editing: QL.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants and animals performed by any of the authors.
Additional information
Handling Editor: T. K. Frey.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yelemali, P., Hao, L. & Liu, Q. Mechanisms of host type I interferon response modulation by the nucleocapsid proteins of alpha- and betacoronaviruses. Arch Virol 167, 1925–1930 (2022). https://doi.org/10.1007/s00705-022-05513-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05513-8