
Abstract
Avian infectious bronchitis is a contagious viral disease, caused by avian infectious bronchitis virus (IBV), that leads to severe losses in the poultry industry all over the world. Since the 1950s, IBV has circulated in the Middle East and North Africa, and no tangible evidence has shown any effects of measures taken to control its spread or evolution. Furthermore, new IBV variants are continually discovered. Although several genetic studies on IBV have been conducted, many IBV strains from this region have either been misclassified or remain unclassified. The genotype 23 (GI-23) variant emerged and has prevailed in the Middle East by continuously evolving through inter- and/or intra-genotypic recombination. The GI-23 genotype is currently enzootic throughout Europe and Asia. Although many studies of protection against the circulating strains have been conducted, they have not been standardized according to regulatory requirements. In this review, we provide an overview of the evolution and genetic diversity of IBV genotypes and a genetic classification of IBV strains, with a focus on the GI-23 genotype. The high prevalence of IBV GI-23 strains necessitates the adoption of vaccination schemes using GI-23-based vaccines.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Avian infectious bronchitis (IB) is a major viral respiratory disease that occurs in all countries that raise poultry due to its considerable virulence and rapid spread, as well as the existence of several serotypes with poor cross-protection among types. In addition to its severe respiratory effects, IB also produces reproductive and urinary lesions that are associated with increased mortality, depending on the virus strain, age, and immune status of the birds [1].
IB is caused by infectious bronchitis virus (IBV), whose natural hosts include chickens and pheasants [2]. This virus has also been isolated from several types of birds, including peafowl, turkeys, geese, pigeons, quail, ducks, parrots, penguins, Guinea fowl, and other avian species [1, 3, 4]. Chickens of all ages are vulnerable to IBV infection, but younger birds are more susceptible than older ones [5].
IBV is an epitheliotropic virus that attacks organs lined with epithelia, including the respiratory tract (especially the trachea), the alimentary tract, and the urogenital tract [1]. It is believed that the virus replicates primarily in respiratory organs in the ciliated epithelium and mucous-secreting cells of respiratory organs. The virus causes viremia for a short time, followed by systemic dissemination to other organs, where further replication can occur, depending on the virus strain and the immune status of the host [6, 7].
The persistence of the virus and the severity of lesions that form in the trachea depend mainly on the type of IBV strain. [8, 9]. An increased severity of disease occurs mainly due to the activation of secondary infections (such as colibacillosis and mycoplasma infection) following tracheal ciliary stasis [10, 11]. The kidneys are the main replication site for nephropathogenic IBV strains after their initial replication in the respiratory tract. They replicate mainly in the collecting tubules and lower parts of the nephron, leading to the precipitation of urate crystals in the kidneys and ureters [12,13,14]. Some IBV strains disseminate through the bloodstream to the oviduct in young chicks and mature layers [15]. Other affected tissues include the lungs, air sacs, oesophagus, proventriculus, duodenum, jejunum, liver, spleen, bursa of Fabricius, caecal tonsils, liver, ileum, rectum, cloaca, ovaries, and testicles [16,17,18].
A new classification system based on the diversity of the full S1 sequence has been suggested for IBV. In this system, IBV strains are classified into seven genotypes (GI-GVII) with dozens of genetic lineages. Genotype GI has the largest number of genetic lineages (n: 29). The Massachusetts (Mass) type belongs to the GI-1 lineage, while the Egyptian variants I and II and many Middle East IBV strains belong to the GI-23 lineage, which has spread to many countries in Africa, Asia and Europe [19,20,21,22,23]. Although classification based on the full-length S1 is very efficient, it leaves a gap in which a very large number of IBV strains for which only partial S1 sequences are available, remain unclassified.
Here, we provide an overview of the distribution of IBV strains, with a focus on the GI-23 genotype. The validity of IBV genotyping based on both the full S1 nucleotide sequence and that of hypervariable region 3 (HVR3) of S1 are evaluated, and cross-protection is reviewed.
Virus taxonomy and structure
IBV belongs to the subgenus Igacovirus, genus Gammacoronavirus, subfamily Orthocoronavirinae, family Coronaviridae, and order Nidovirales [24]. The virus is a single-stranded, non-segmented, linear positive-sense RNA with a large genome of 27.6 kb that encodes several structural and non-structural proteins (NSP), which are essential for viral replication. IBV has the genome organization 5’UTR-ORF1a/b-S-3a-3b-E-M-4b-4c-5a-5b-N-6b-3’UTR. There is a -1 frameshift at the junction of ORF1a/b (the replicase gene), resulting in translation of the 1a and 1b polyproteins. Post-translational cleavage of the polyproteins gives rise to the individual non-structural proteins that are involved in genome replication and transcription [25].
The viral structural proteins include the spike (S) protein, matrix (M) protein, envelope (E) protein, and nucleocapsid (N) protein [15]. The spike protein is the largest structural glycoprotein in avian IBV. Trimers of the S protein form club-shaped or petal-shaped spikes of 128 kDa that are heavily glycosylated, giving them a total molecular weight of about 200 kDa. The S protein is responsible for viral tropism, attachment of the virus to cells, and fusion of cellular and viral membranes. The S protein is cleaved at the highly basic furin consensus motif RRFRR into the subunits S1 and S2. The S1 subunit is derived from the N-terminal portion of the S protein and is responsible for attachment of the virus to the cellular membrane through its interaction with cellular receptors. It contains three hypervariable regions (HVRs) that are responsible for its variation and escape from the immune defence. HVR1, HVR2 and HVR3 are located at amino acid positions 38-67, 91-141 and 274-387, respectively [26]. The S1 protein is also responsible for eliciting neutralizing antibodies, which interfere with the binding of S1 to its receptor. Its amino acid sequence identity varies between serotypes from 5 to 50%. The S2 subunit consists of a narrow stalk ectodomain, a short transmembrane segment, and a C-terminal domain, and is responsible for fusion of the viral membrane with the cell membrane. [26].
Classification of IBV
IBV classification systems are based on two main types of tests: i) functional or biological tests, and ii) non-functional tests. The functional classification allows viral isolates to be grouped into immunotypes, protectotypes, and antigenic types (serotypes and epitope types). Non-functional tests are based on sequence variations in the S gene that allow them to be assigned to different genotypes. In practical terms, immunotype or protectotype classification is the most important because it provides information about the protective efficacy of vaccine strains [27].
Strains that protect against each other are called protectotypes or immunotypes [27]. The number of protectotypes remains unknown. Protectotyping is usually performed experimentally using in vivo cross-immunization studies [28]. Protectotype experiments have been replaced more recently by the in vitro cross-immunization test (CIT), which uses specific-pathogen-free embryonated chicken eggs (SPF-ECE) or tracheal organ cultures (TOCs) from vaccinated chickens [29]. Both testing procedures may soon be replaced by bioinformatic prediction and structural analysis programs. [30,31,32].
Antigenic types include serotypes and epitope types. Serotype classification is the classical functional classification of IBV based on a strain’s reaction with serotype-specific antibodies raised in chickens. Testing is performed using virus neutralization tests (VNTs) and TOCs or a haemagglutination inhibition (HI) test [29, 33, 34]. Because it is prone to strong and variable cross-reactions, the HI test is considered less trustworthy than VNTs [35]. A VNT is performed using the α or β method, with the α method being more precise and sensitive. However, serotyping shows a lack of standardization [27].
Epitope typing uses monoclonal antibodies (Mabs) to detect the presence of specific epitopes in viral antigens via antigen-capture enzyme-linked immunosorbent assay (ELISA) [36] and immunofluorescent antibody techniques [37]. Mabs are directed specifically against the HVRs of the IBV S1 protein. The main disadvantage of this technique is that false-negative results may be obtained when the Mabs target conserved epitopes; the presence of a mutation in an epitope does not necessarily indicate a change in the serotype [27].
Genotyping is a genetic-based classification technique based on sequencing, detection of nucleic acids (e.g., by RT-PCR), or determining the position of enzyme cleavage sites (e.g., RFLP, RNase T1 fingerprinting) (Table 1) [27]. S1 gene sequencing is the most widely used approach to classifying IBV isolates into genotypes because it is based on differences in the most variable and antigenic region of the viral genome. Recently, phylogenetic analysis based on IBV S1 gene sequencing has revealed the existence of seven genotypes (GI-GVII) consisting of 35 genetic lineages, as well as inter-lineage recombinants (Table 2). Genotype GI includes 29 genetic lineages, while each of the other genotypes has only one lineage. The GI-1 and GI-13 lineages represent the old Massachusetts (Mass) and 793B lineages, respectively. Both lineages are universally distributed in all endemic countries and are most commonly used as vaccine strains, while other lineages are found only in specific parts of the world [19,20,21,22,23]. The GI-23 lineage includes IBV variants circulating in the Middle East. This lineage continues to spread and poses a major challenge not only in the Middle East but also in many countries in Africa, Asia and Europe [19,20,21,22,23].
The relationship between the different classification systems is complicated and has not been fully clarified. However, amino acid changes in the S1 protein may lead to a different serotype designation, depending on the site of the mutation and its effect on cross-immunity. In general, a high degree of amino acid sequence similarity correlates with cross-protection, but some published data have indicated that very small differences can affect the degree of cross-protection [12, 38].
Epidemiological situation in the Middle East and North Africa (MENA)
The epidemiological situation in the Middle East and North Africa (MENA) is still unclear due to a lack of optimized surveillance programs and the absence of adequate full-genome sequence data for the circulating strains. Egypt was the first country to isolate IBV in the 1950s [39], and the virus was then detected in Morocco in the 1980s [40]. During the 1990s, other countries began surveillance programs for the detection and isolation of IBV [25]. However, the scarcity of epidemiological studies in most MENA countries has made precise monitoring of IBV unfeasible.
Egypt
Serotypes related to the Mass, D3128, D274, D-08880 and 4/91 serotypes have been detected in different chicken flocks [41,42,43]; however, none of these isolates have been sequenced. The first IBV sequences for two different genotypes (including D274, an archival sample from 1989, and the new Egyptian genotype Egyptian Var I [Egypt/Beni-Suef/01] represented by two strains from 1998 and 2001) were first published in 2002 [44]. Egyptian Var I (Egypt/Beni-Suef/01) was found to be closely related to the Israel/720/99 strain [44]. From 2006 to 2015, many reports of isolation and identification of IBVs circulating in Egypt were published [38, 45,46,47]. Most of these studies were limited to partial IBV sequences. An IS/1494/06-like strain was detected in Egypt in 2010 [46]. Additionally, a novel IBV genotype was isolated in 2012 and named EGY VAR II. This genotype included both Ck/Eg/BSU-2/2011 and Ck/Eg/BSU-3/2011. The viruses showed 89% amino acid sequence identity in HVR3 to Beni-Suef/01 (EGY VAR I) and IS/885 [45]. The Egyptian VAR II group became the most prevalent group of strains found on chicken farms in Egypt. Egyptian variants I and II were both recently classified as belonging to a wild-type cluster in the GI-23 lineage in the newly established classification system for IBV genotypes [20]. Additionally, a complete genome sequence of the CU/4/2014 strain (EGY VAR II) showed evidence of recombination, with three putative parent strains: the Italian strain 90254/2005 (a QX-like strain) and strains 4/91 and H120 [48].
Lebanon and Syria
National serological surveillance was conducted between 1992 and 1996 in Lebanon for all avian pathogens. The results revealed serological evidence of IBV infection in chicken flocks [49]. No other data about the IBV situation in Lebanon were available until Ganapathy and co-workers identified strains related to Middle East strains (GI-23), 793/B (GI-13) and Mass-like (GI-1) strains that circulated in Lebanon from 2010 to 2012 [50]. The new IBV strain sequences isolated in 2018 from Lebanon were made available in the GenBank database, and our analysis based on these sequences confirmed their clustering with the GI-23 strain. In addition, recently released Syrian IBV strain HVR3 sequences have been determined to belong to the GI-23 genotype. However, no associated publications have been found in either country (Table 3).
Jordan
The first data from Jordan were published in 2007. The presence of the Ark, DE-072, and Mass-like serotypes was demonstrated serologically [51]. Later, IBV strains related to the 4/91 and D274 genotypes were identified in addition to the classical Mass genotype [52]. IBV strains JOA2 and JOA4, which are related to the CK/CH/LDL/97I genotype, were also isolated from chicken flocks in Jordan [53]. Evidence of the circulation of IBV strains related to the GI-23 genotype has been obtained in multiple studies in Jordan since 2009 [50, 54].
The Persian Gulf area
The incidence of IBV in other Gulf countries still remains unclear due to the scarcity of information being published. IBV infection in Saudi Arabia was first detected in 1984 when an IBV isolate was identified using RT-PCR for the N gene without specifying its serotype [55]. IBV infection due to strains related to serotype 793/B (GI-13) was serologically detected in Saudi Arabia in 1997 and 1998 and was further confirmed in 2002 [56]. By 2009 and 2010, another study had characterized two IBV strains (IBV/CHICKEN/KSA/101/2010 and IBV/CHICKEN/KSA/102/2010) related to CH/LDL/011 (GI-16) and IBV/INDIA/TN/92/03 (H120, GI-1), respectively [57]. More recently, evidence of co-circulation of IBV strains related to Mass (GI-1), 4/91 (GI-13), CK/CH/LDL/97I (GI-16), and Middle East IBV (GI-23) has been reported in Saudi Arabia [22, 58].
In the United Arab Emirates, the 793/B (GI-13) strain constitutes the major genetic lineage that infects chicken flocks, while the Mass (GI-1) and D-274 strains are less frequently detected [50]. In Oman, Kuwait and Bahrain, the situation was almost unknown until 2014, but studies published in 2015 detected 793/B (GI-13) as the dominant strains with occasional detection of Mass (G1-1), Middle East (G1-23), and Dutch strains (G1-12) [50, 59].
Iraq
The first isolation of IBV in Iraq occurred from 2008 to 2010 with the detection of two novel genotypes, designated as the Sul/01 and 4/91 IBV strains [60]. Recently, the GI-23 and GI-13 genetic lineages were found to be the most prevalent lineages (IS/1494/06, 46.9%; 793/B, 40.6%). In addition, QX (G1-19) (9.4%) and DY12-2-like (3.1%) strains were also detected among the circulating IBVs [54].
Iran
The close relations of the Persian Gulf countries with Iran may suggest that the IBV epidemiology in Iran could be used as an indicator of the current situation in the Gulf region [61]. The first detection of the Mass type of IBV was in 1994 [62]. Ten years later, 4/91-like strains were reported in addition to the continuously circulating Mass type [24, 63,64,65]. Circulation of diverse genotypes was detected from 2010 to 2014, and partial sequences of the Mass, 793/B, Middle East, Variant 2, QX, IR-I, and IR-II variants were reported [66]. Genotyping of IBV strains between 2015 and 2017 revealed that GI-23 was the dominant genotype in Iran [67, 68].
Arab Maghreb countries
IBV was first isolated in Morocco from 1981 to 1984; the novel IBV strains were related to the Mass serotype and to a unique enterotropic IBV virus designated as Moroccan G [40, 69]. Notably, a potential ancestral relationship between the old North African and the European GI-13 (793/B- and CR88-like strains) was suggested due to the similarity of GI-13 to the Moroccan strains isolated in 1983 [70].
In 2015, two predominant genotypes were detected in Morocco: the Mass type (66%) and the Italy 02 genotype (32%), with only a single detection of the 793B strain [71]. Phylogenetic analysis of all known IBV isolates from 1983 to 2014 in Morocco revealed the circulation of three genetic lineages: GI-1, GI-13 (four genetic clusters), and GI-21 (two genetic clusters). Morocco was the first African country in which the GI-21 lineage was reported (GI-21, cluster 2), and this lineage is genetically related to the gCoV/AvCoV/chicken/Spain/1997 variant [72]. The second incidence of new IBV genotype introductions in Morocco suggested the probability of the introduction of this lineage through the importation of day-old broiler and layer breeders from Europe [72].
No historical data are available about IBV in Algeria. The first data were published in 2015 and indicated the prevalence of GI-1-lineage IBV strains. The Algerian Mass-like nephropathogenic strain showed no genetic relatedness to the North African variant strains and was different from the H120 (GI-1) and 4/91 (GI-13) genotypes, suggesting the possibility of recombination between the nephropathogenic Mass strain and the novel African genotypes [73]. Recently, seroprevalence has indicated a high incidence of IBV antibodies on unvaccinated farms [74] and the circulation of QX (G1-19) and 4/91 (GI-13) IBV variants. [75].
Similarly, in sero-epidemiological studies in Tunisia, new Tunisian variants genetically related to the CR88 and 793/B (GI-13) strains were isolated and characterized [76, 77]. Meanwhile, the Middle East GI-23 genotype strains were first reported in Tunisia in 2018. The first published report from Libya described the isolation of an IBV strain designated as 3382/06 that was closely related to the GI-16 lineage [78]. In 2013, detection of VAR II-like strains (GI-23) was confirmed by the isolation of strains related to the EG/IB1212B and Beni-Suef/02 lineages [79].
Turkey
There were no published data about the situation of IBV in Turkey until 2013. However, in 2011, Kahya and colleagues reported the detection of Middle East–related isolates (GI-23) from broiler and layer flocks [80]. By 2016, the GI-1, GI-12, GI-13 and GI-23 lineages had been detected [81, 82].
Predominant IBV genetic lineages in MENA countries
IBV has been circulating in MENA countries for decades with the continual appearance of escape mutants. Based on the available data, it seems that the GI-23 (especially the Egyptian VAR II genotype) and GI-19 (QX genotype) strains are expanding their geographic distribution [79, 83]. Alongside these prevalent lineages, vaccine strains belonging to GI-1, GI-12, and GI-13 lineages are still circulating at low or undetected levels (Table 3). The co-circulation of both lineages in addition to other detected lineages suggests the continuous evolution of IBV by either mutation and/or recombination. Recently, an analysis of the complete genome sequences of IBV strains from Egypt revealed two new variants: CU/1/2014, an H120-like strain that probably evolved due to accumulated multiple point mutations, and the recombinant strain CU/4/2014, which showed evidence of recombination involving three ancestors belonging to QX-like, 4/91, and H120 IB lineages [48].
More information about the dynamics of the spread of IBV is urgently needed to develop better control strategies. Recently, a Bayesian phylodynamic approach to investigate the spread of the QX-like strains (GI-19) revealed that there was a long period of local circulation in China followed by transmission to European, Asian, and Middle East countries in successive waves beginning in 2011 [84]. In addition, the range of the Q1 (GI-16) viruses seems to extend beyond the MENA countries. Despite reports of sporadic detection, the Q1-like strains are likely to have been introduced through the Asian route (from China to Egypt) according to a phylodynamic analysis constructed for Q1 [85].
These strains were first detected in 2011 in Iraq, Saudi Arabia, and Jordan. In Egypt, two sporadic detections were reported in 2010 and 2017 [86, 87], and the same occurred in Iran in 2019 [88]. However, the absence of enough data about IBV in the MENA region makes it difficult to investigate the current state of this lineage.
Novel genetic lineages in MENA countries
The recent classification of IBV based on full S1 nucleotide sequences conducted by Valastro did not take into consideration the available partial S1 gene sequences in the phylogenetic analysis of MENA IBV strains [20]. Although it would be best to use full S gene sequences for IBV classification, this strict criterion would leave many strains without classification. An in-depth analysis that included Middle Eastern IBV isolates with ≥ 600 bp or full S1 sequences revealed a possible new genetic lineage of IBV in North Africa. These IBV strains were isolated in Algeria in 2013 [73] and in Tunisia in 2016 [89] and found to cluster separately, with ≥ 25% amino acid sequence divergence from the closest genetic lineages in the GI-14 strain (Table 4).
Genetic analysis of MENA IBV strains based on HVR3 sequences was used previously to characterize GI-23 IBV strains in Egypt [45]. Additionally, the HVR3 sequences were employed in a recent evolutionary analysis of Egyptian IBV strains, revealing three subclades of historical IBV strains, including GI-23.1, G23.2 (GI-23.2.1 and GI-23.2.2), and GI-23.3 [90]. However, the study focused only on Egyptian strains. In this review, all available full-length S1 sequences from the Middle East were retrieved from GenBank. Their evolutionary history was inferred using the maximum-likelihood method and the Tamura-Nei model [91]. This analysis involved 200 nucleotide sequences with 1736 total positions in the final dataset and was performed using MEGA X [92]. The analysis revealed that the GI-23 genotype has three sublineages (23.1, 23.2 and 23.3) and that the GI-23.2 sublineage, in turn, has three subclades, GI-23.2.1, GI-23.2.2 and GI-23.2.3 (Supplementary Fig. 1 Fig. 1A and B), with p-distances of 9.8, 13.4, and 15.8%, respectively. The p-distances calculated between the first isolate of sublineage 23.1 and the 23.2 and 23.3 sublineages were 10–14% and 7.7%, respectively (Fig. 1C).

Phylogenetic analysis of the IBV genetic lineages (200 sequences) (A) and GI-23 sequences (B) from the Middle East based on full-length S1 gene sequences using the maximum-likelihood method and the Tamura-Nei model. There were 1736 positions in the final dataset. Distances between the proposed sublineages and subclades (C) were calculated using MEGA X software.
Interestingly, this genetic classification was successfully reproduced using the HVR3 sequences of all IBV genetic lineages (Fig. 2A and B). The only difference was that a few isolates did not cluster with their corresponding genotype as they did with the full sequences. These isolates included the KU238176-D888/2/4/08_IR, DQ386098-Spain/00/336, U29453-Australia/N3/62, and AY296744-AY296746 Japan strains as well as KM660636-GA/10216/2010. Notably, most of these isolates were recombinant IBV strains identified in Iran [93], Spain [94], Australia [95], Japan [96], and the United States [97], respectively. Moreover, the topology of the HVR3 phylogenetic tree was very similar to that of the full S gene except in the case of the GI-22 and GI-29 strains, where their clustering overlapped (Fig. 2A and B). Using the calculated p-distances allowed better discrimination between sublineages (Fig. 2C). GI-29 was recently reported in China [98]. Our analysis showed that this lineage shares 87.5–90.1% nucleotide and 85.6–88.9% amino acid sequence identity with GI-22. This similarity would explain the overlapping clustering of the HVR3 sequences of these two lineages. In addition, the Iranian IS-1494-like strains have multiple deletions and insertions in their HVRs, which led to their separate clustering from the GI-23.1 sublineages (Fig. 2B).

Phylogenetic analysis of the IBV genetic lineages (194 sequences) (A) and GI-23 sequences (B) from the Middle East based on HVR3 sequences using the maximum-likelihood method and the Tamura-Nei model. There were 349 positions in the final dataset. Distances between the proposed sublineages and subclades (C) were calculated using MEGA X software.
Despite the minor limitation of the misclustering of recombinant IBV strains, for the majority of IBV strains, phylogenetic analysis of HVR3 sequences is reliable as a means of IBV classification for rapid diagnostic and broad surveillance purposes.
The spread of IBV GI-23 and its epidemiological implications for Europe, Africa, and Asia.
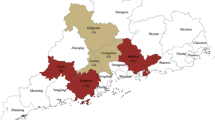
The MENA region is in the centre of the Old World and is situated at the conjunction of Asia, Europe, and Africa (Fig. 3). Historically, some IBV lineages that are endemic in Europe have been isolated in North African countries, and vice versa [70, 72]. Recently, the GI-23 lineage was detected in several European countries, including Ukraine, Lithuania, Poland, Armenia, the Russian Federation, the Republic of Belarus, Tajikistan, Kazakhstan and Germany. Additionally, reports from the Middle Asian countries of Kazakhstan, and Afghanistan have described the isolation of GI-23 strains [99, 100]. GI-23 was isolated in West Africa for the first time in 2013 with the isolation of strains NGA3 (MN082399) and NGA8 (MN082404) in Nigeria. The widespread prevalence of the GI-23 lineage (EGY VAR II-like strains) in the region and recent detections in Asia, Europe, and Africa illustrate the capability of the GI-23 lineage to extend its geographical range and emphasises the need for a comprehensive investigation of the phylodynamic evolution of this lineage, as has been done with other lineages (e.g., GI-19 QX). A recent poster at the XXIst World Veterinary Poultry Association Congress reported the spread of GI-23 in some African and European countries, including Morocco, the Czech Republic, Belgium, Zimbabwe, Zambia, and Uganda; however, there are no published data to confirm the presence of GI-23 in these countries [101]. Therefore, active surveillance is needed to determine the prevalence of the GI-23 IBVs in these nations.

Geographical distribution of genetic lineage 23 of IBV in the Middle East, North Africa, Europe, and Asia. Countries that have reported the detection of GI-23-like strains are indicated in red.
BV is challenging control efforts and will require new approaches
Control of IBV infection is difficult due to the emergence of new variants and the high rate of mutation affecting antigenic on the virion. The subpopulation that remains after replication and any rapid reversion of vaccine strains to pathogenicity also play a role in reducing the ability to control the disease. Additionally, increasing numbers of recombination events further complicate efforts to combat the disease. Therefore, specific and sophisticated control measures need to be established. In addition, strict biosecurity measures must be applied on farms and in their surrounding environments. However, reliable vaccination programs that take into consideration the causative IBV strains, types of birds, and purpose of production should also be implemented.
To satisfy regulatory requirements, different tests have been applied to evaluate the efficacy of IBV vaccines against homologous challenge. In the USA, the Code of Federal regulations (CFR) requires re-isolation of the challenge virus in SPF-ECE after challenge. [102]. However, in Europe, the European Pharmacopeia (Ph. Eur.) requires the performance of a ciliostasis test after a homologous challenge to assess the degree of protection conferred by the tested vaccine [103]. In most MENA countries, the European guidelines have been adopted, including the evaluation of ciliostasis after challenge with endemic viruses, in addition to protection against clinical signs and associated mortalities. The protection studies that have been performed to evaluate available vaccines against the main circulating genetic lineages with respect to the regulatory requirements of the CFR or Ph. Eur. are summarized in Table 5.
While cross-protection studies have indicated the ability of protectotype vaccine strains (especially those belonging to the GI-1 and GI-13 lineages) and combined vaccines to protect against GI-23 viruses, field observations have indicated that variable vaccines have not provided adequate protection; the viruses continue to circulate. Recent studies suggest that the best protection against an IBV challenge can be achieved using a homologous vaccine strain. This has been confirmed with Chinese QX-like IBV strains [104, 105], several Korean nephropathogenic IBV strains [106], and more recently, with GI-23 strains as either live or inactivated vaccines [107,108,109,110].
Few studies have followed the international regulations, and there is a lack of standardized methodology (e.g., virus isolation versus virus molecular detection and/or quantification). For instance, the protection levels in vaccinated birds against shedding of the homologous GI-23 strain, as determined by virus isolation, ranged from 60 to 100% [110]; however, lower levels of protection (40–60%) were found using quantitative real-time reverse transcription polymerase chain reaction [107]. Additionally, while protection studies in commercial birds are thought to reflect the field situation, ciliostasis testing does not take into account possible deleterious effects of live IBV vaccines on tracheal cilia under field conditions or cilostasis due to infections with other respiratory pathogens [11]. Hence, the use of SPF birds in ciliostasis tests remains the gold standard for evaluating live IBV vaccines.
Concluding remarks
Avian IBV remains a challenge for the poultry industry due to its rapid spread and the existence of several serotypes that provide poor cross-protection. Although different classification systems of IBV have been proposed, each system has several limitations that compromise its accuracy. Currently, the genotype classification of IBV based on full-length S1 nucleotide sequences is widely used, but genetic analysis of HVR3 could be a reliable tool for rapid diagnosis of IBV and national surveillance purposes. IBV epidemiology in MENA countries is still unclear due to the lack of optimized programs for surveillance of different genotypes. The wide geographical distribution of GI-23 (EGY VAR II-like strains) in the MENA region and in Asia, Europe, and Africa necessitates the study of its phylodynamic evolution and a re-evaluation of different vaccine efficacy rates against this lineage. Cross-protection studies using protectotype vaccine strains and combined vaccines against GI-23 lineages are not in agreement with the field observations of inadequate protection. A standardized vaccine evaluation protocol (e.g., the use of SPF birds and ciliostasis tests) must be followed to allow studies to be compared. However, as has been seen with the QX-like strain, the use of homologous vaccine strains appears to be necessary for maximal protection.
References
Jackwood MW, de Wit S (2013) Infectious bronchitis. In: Swayne DE, Glisson RJ, McDougald LR, Nolan LK, Suarez, DL, Nair V (eds) Diseases of poultry, 13th edn. Wiley, Hoboken, NJ, USA, pp 139–159
Cavanagh D, Davis PJ, Mockett AP (1988) Amino acids within hypervariable region 1 of avian coronavirus IBV (Massachusetts serotype) spike glycoprotein are associated with neutralization epitopes. Virus Res 11(2):141–150. https://doi.org/10.1016/0168-1702(88)90039-1
Britton P, Evans S, Dove B, Davies M, Casais R, Cavanagh D (2005) Generation of a recombinant avian coronavirus infectious bronchitis virus using transient dominant selection. J Virol Methods 123(2):203–211. https://doi.org/10.1016/j.jviromet.2004.09.017
Bande F, Arshad SS, Omar AR, Hair-Bejo M, Mahmuda A, Nair V (2017) Global distributions and strain diversity of avian infectious bronchitis virus: a review. Anim Health Res Rev 18(1):70–83. https://doi.org/10.1017/S1466252317000044
Crinion RAP, Hofstad MS (2006) Pathogenicity of four serotypes of avian infectious bronchitis virus for the oviduct of young chickens of various ages. Avian Dis 16(2):351. https://doi.org/10.2307/1588800
Naqi SA, Cowen BS, Hattel AL, Wilson RA (1991) Detection of viral antigen following exposure of one-day-old chickens to the holland 52 strain of infectious bronchitis virus. Avian Pathol 20(4):663–673. https://doi.org/10.1080/03079459108418805
Benyeda Z, Szeredi L, Mato T, Suveges T, Balka G, Abonyi-Toth Z et al (2010) Comparative histopathology and immunohistochemistry of QX-like, Massachusetts and 793/B serotypes of infectious bronchitis virus infection in chickens. J Comp Pathol. 143(4):276–283. https://doi.org/10.1016/j.jcpa.2010.04.007
Alvarado IR, Villegas P, Mossos N, Jackwood MW (2005) Molecular characterization of avian infectious bronchitis virus strains isolated in Colombia during 2003. Avian Dis 49(4):494–499. https://doi.org/10.1637/7202-050304R.1
Benyeda Z, Mato T, Suveges T, Szabo E, Kardi V, Abonyi-Toth Z et al (2009) Comparison of the pathogenicity of QX-like, M41 and 793/B infectious bronchitis strains from different pathological conditions. Avian Pathol 38(6):449–456. https://doi.org/10.1080/03079450903349196
Ganapathy K, Bradbury JM (1999) Pathogenicity of Mycoplasma imitans in mixed infection with infectious bronchitis virus in chickens. Avian Pathol 28(3):229–237. https://doi.org/10.1080/03079459994713
Hassan KE, Ali A, Shany SAS, El-Kady MF (2017) Experimental co-infection of infectious bronchitis and low pathogenic avian influenza H9N2 viruses in commercial broiler chickens. Res Vet Sci 115:356–362. https://doi.org/10.1016/j.rvsc.2017.06.024
Cavanagh D, Ellis MM, Cook JKA (1997) Relationship between sequence variation in the S1 spike protein of infectious bronchitis virus and the extent of cross-protection in vivo. Avian Pathol 26(1):63–74. https://doi.org/10.1080/03079459708419194
Abdel-Moneim A, Madbouly H, El-Kady M (2005) In vitro characterization and pathogenesis of Egypt/Beni-Suef/01; a novel genotype of infectious bronchitis virus. Beni-Suef Vet Med J Egypt 15(2):127–133
Reddy VR, Trus I, Desmarets LM, Li Y, Theuns S, Nauwynck HJ (2016) Productive replication of nephropathogenic infectious bronchitis virus in peripheral blood monocytic cells, a strategy for viral dissemination and kidney infection in chickens. Vet Res 47(1):70. https://doi.org/10.1186/s13567-016-0354-9
Cavanagh D (2007) Coronavirus avian infectious bronchitis virus. Vet Res 38(2):281–297. https://doi.org/10.1051/vetres:2006055
Ambali AG, Jones RC (1990) Early pathogenesis in chicks of infection with an enterotropic strain of infectious bronchitis virus. Avian Dis 34(4):809–817
Raj GD, Jones RC (1996) Immunopathogenesis of infection in SPF chicks and commercial broiler chickens of a variant infectious bronchitis virus of economic importance. Avian Pathol 25(3):481–501. https://doi.org/10.1080/03079459608419157
Otsuki K, Huggins MB, Cook JKA (1990) Comparison of the susceptibility to avian infectious bronchitis virus infection of two inbred lines of white leghorn chickens. Avian Pathol 19(3):467–475. https://doi.org/10.1080/03079459008418700
Chen Y, Jiang L, Zhao W, Liu L, Zhao Y, Shao Y et al (2017) Identification and molecular characterization of a novel serotype infectious bronchitis virus (GI-28) in China. Vet Microbiol 198:108–115. https://doi.org/10.1016/j.vetmic.2016.12.017
Valastro V, Holmes EC, Britton P, Fusaro A, Jackwood MW, Cattoli G et al (2016) S1 gene-based phylogeny of infectious bronchitis virus: an attempt to harmonize virus classification. Infect Genet Evol 39:349–364. https://doi.org/10.1016/j.meegid.2016.02.015
Jiang L, Han Z, Chen Y, Zhao W, Sun J, Zhao Y et al (2018) Characterization of the complete genome, antigenicity, pathogenicity, tissue tropism, and shedding of a recombinant avian infectious bronchitis virus with a ck/CH/LJL/140901-like backbone and an S2 fragment from a 4/91-like virus. Virus Res 244:99–109. https://doi.org/10.1016/j.virusres.2017.11.007
Rohaim MA, El Naggar RF, Abdelsabour MA, Mohamed MHA, El-Sabagh IM, Munir M (2020) Evolutionary analysis of infectious bronchitis virus reveals marked genetic diversity and recombination events. Genes (Basel). https://doi.org/10.3390/genes11060605
Ma T, Xu L, Ren M, Shen J, Han Z, Sun J et al (2019) Novel genotype of infectious bronchitis virus isolated in China. Vet Microbiol 230:178–186. https://doi.org/10.1016/j.vetmic.2019.01.020
Cavanagh D (2005) Coronaviruses in poultry and other birds. Avian Pathol 34(6):439–448. https://doi.org/10.1080/03079450500367682
Abdel-Moneim AS (2017) Coronaviridae: infectious bronchitis virus. Emerging and re-emerging infectious diseases of livestock. Springer, Berlin, pp 133–166. https://doi.org/10.1007/978-3-319-47426-7_5
Wickramasinghe IN, van Beurden SJ, Weerts EA, Verheije MH (2014) The avian coronavirus spike protein. Virus Res 194:37–48. https://doi.org/10.1016/j.virusres.2014.10.009
De Wit J (2000) Detection of infectious bronchitis virus. Avian Pathol 29(2):71–93. https://doi.org/10.1080/03079450094108
Cook JK, Orbell SJ, Woods MA, Huggins MB (1999) Breadth of protection of the respiratory tract provided by different live-attenuated infectious bronchitis vaccines against challenge with infectious bronchitis viruses of heterologous serotypes. Avian Pathol 28(5):477–485. https://doi.org/10.1080/03079459994506
Raj GD, Jones RC (1996) Protectotypic differentiation of avian infectious bronchitis viruses using an in vitro challenge model. Vet Microbiol 53(3–4):239–252. https://doi.org/10.1016/s0378-1135(96)01258-8
Ladman BS, Loupos AB, Gelb J Jr (2006) Infectious bronchitis virus S1 gene sequence comparison is a better predictor of challenge of immunity in chickens than serotyping by virus neutralization. Avian Pathol 35(2):127–133. https://doi.org/10.1080/03079450600597865
Kamble NM, Pillai AS, Gaikwad SS, Shukla SK, Khulape SA, Dey S et al (2016) Evolutionary and bioinformatic analysis of the spike glycoprotein gene of H120 vaccine strain protectotype of infectious bronchitis virus from India. Biotechnol Appl Biochem 63(1):106–112. https://doi.org/10.1002/bab.1298
Leyson CLM, Jordan BJ, Jackwood MW (2016) Insights from molecular structure predictions of the infectious bronchitis virus S1 spike glycoprotein. Infect Genet Evol 46:124–129. https://doi.org/10.1016/j.meegid.2016.11.006
Gelb J Jr, Perkins BE, Rosenberger JK, Allen PH (1981) Serologic and cross-protection studies with several infectious bronchitis virus isolates from Delmarva-reared broiler chickens. Avian Dis 25(3):655–666
Wang L, Xu Y, Collisson EW (1997) Experimental confirmation of recombination upstream of the S1 hypervariable region of infectious bronchitis virus. Virus Res 49(2):139–145. https://doi.org/10.1016/s0168-1702(97)01466-4
Cook JKA, Brown AJ, Bracewell CD (1987) Comparison of the haemagglutination inhibition test and the serum neutralisation test in tracheal organ cultures for typing infectious bronchitis virus strains. Avian Pathol 16(3):505–511. https://doi.org/10.1080/03079458708436399
Karaca K, Naqi S, Gelb J Jr (1992) Production and characterization of monoclonal antibodies to three infectious bronchitis virus serotypes. Avian Dis 36(4):903–915
De Wit JJ, Koch G, Kant A, Van Roozelaar DJ (1995) Detection by immunofluorescent assay of serotype-specific and group-specific antigens of infectious bronchitis virus in tracheas of broilers with respiratory problems. Avian Pathol 24(3):465–474. https://doi.org/10.1080/03079459508419086
Abdel-Moneim AS, El-Kady MF, Ladman BS, Gelb J Jr (2006) S1 gene sequence analysis of a nephropathogenic strain of avian infectious bronchitis virus in Egypt. Virol J 3:78. https://doi.org/10.1186/1743-422X-3-78
Ahmed H (1954) Incidence and treatment of some infectious viral respiratory diseases of poultry in Egypt. PhD. Thesis Cairo University, Cairo, Egypt
El-Houadfi M, Jones RC (1985) Isolation of avian infectious bronchitis viruses in Morocco including an enterotropic variant. Vet Rec 116(16):445. https://doi.org/10.1136/vr.116.16.445-a
Sheble A, Sabry M, Davelaar F, Burger A, Khafagy A, Moustafa F et al (1986) Present status of infectious bronchitis in Egypt. J Egypt Vet Med Assoc 4:393–411
El-Kady M (1989) Studies on the epidemiology and means of control of infectious bronchitis disease in chickens in Egypt. PhD. Thesis, Fac. Vet. Med., Cairo Univ., Giza, Egypt
Eid AAM (1998) Infectious bronchitis virus infection in Egypt. In: Kaleta EF, Heffels-Redmann U (eds) Proceedings of the International Symposium on infectious bronchitis and pneumovirus infections in poultry. Rauischholzhausen, Germany, pp 145–156
Abdel-Moneim AS, Madbouly HM, Gelb J Jr, Ladman BS (2002) Isolation and identification of Egypt/Beni-Seuf/01 a novel genotype of infectious bronchitis virus. Vet Med J Giza 50(4):1065–1078
Abdel-Moneim AS, Afifi MA, El-Kady MF (2012) Emergence of a novel genotype of avian infectious bronchitis virus in Egypt. Arch Virol 157(12):2453–2457. https://doi.org/10.1007/s00705-012-1445-1
El-Mahdy SS, El-Hady MM, Soliman YA (2010) Isolation and characterization of nephropathogenic strain of infectious bronchitis virus in Egypt. J Am Sci 6(9):669–675. https://doi.org/10.1006/jtbi.2000.2072
Selim K, Arafa AS, Hussein HA, El-Sanousi AA (2013) Molecular characterization of infectious bronchitis viruses isolated from broiler and layer chicken farms in Egypt during 2012. Int J Vet Sci Med 1(2):102–108. https://doi.org/10.1016/j.ijvsm.2013.10.002
Abozeid HH, Paldurai A, Khattar SK, Afifi MA, El-Kady MF, El-Deeb AH et al (2017) Complete genome sequences of two avian infectious bronchitis viruses isolated in Egypt: evidence for genetic drift and genetic recombination in the circulating viruses. Infect Genet Evol 53:7–14. https://doi.org/10.1016/j.meegid.2017.05.006
Barbour EK, Hamadeh SK, Hilan C, Kallas M, Eid A, Sakr W (1997) National surveillance of poultry diseases in Lebanon. Rev Sci Technol 16(3):770–775. https://doi.org/10.20506/rst.16.3.1070
Ganapathy K, Ball C, Forrester A (2015) Genotypes of infectious bronchitis viruses circulating in the Middle East between 2009 and 2014. Virus Res 210:198–204. https://doi.org/10.1016/j.virusres.2015.07.019
Gharaibeh SM (2007) Infectious bronchitis virus serotypes in poultry flocks in Jordan. Prev Vet Med 78(3–4):317–324. https://doi.org/10.1016/j.prevetmed.2006.11.004
Roussan DA, Totanji WS, Khawaldeh GY (2008) Molecular subtype of infectious bronchitis virus in broiler flocks in Jordan. Poult Sci 87(4):661–664. https://doi.org/10.3382/ps.2007-00509
Ababneh M, Dalab AE, Alsaad S, Al-Zghoul M (2012) Presence of Infectious Bronchitis Virus Strain CK/CH/LDL/97I in the Middle East. ISRN Vet Sci 2012:201721. https://doi.org/10.5402/2012/201721
Seger W, Ghalyanchi-Langeroudi A, Karimi V, Madadgar O, Marandi MV, Hashemzadeh M (2016) Genotyping of infectious bronchitis viruses from broiler farms in Iraq during 2014–2015. Arch Virol 161(5):1229–1237. https://doi.org/10.1007/s00705-016-2790-2
Zwaagstra K, Van der Zeijst B, Kusters J (1992) Rapid detection and identification of avian infectious bronchitis virus. J Clin Microbiol 30(1):79–84. https://doi.org/10.1128/JCM.30.1.79-84.1992
Cavanagh D, Picault JP, Gough R, Hess M, Mawditt K, Britton P (2005) Variation in the spike protein of the 793/B type of infectious bronchitis virus, in the field and during alternate passage in chickens and embryonated eggs. Avian Pathol 34(1):20–25. https://doi.org/10.1080/03079450400025414
Al-Hammad YM, Al-Afaleq AI, Mohamed MHA (2014) Molecular survey and phylogenic analysis of infectious bronchitis virus (IBV) circulating among chicken flocks in Riyadh Province, Saudi Arabia. J Anim Vet Adv 13(16):1002–1008. https://doi.org/10.3923/javaa.2014.1002.1008
Al-Mubarak AIA, Al-Kubati AAG (2020) Cocirculation of four infectious bronchitis virus lineages in broiler chickens in the Eastern Region of Saudi Arabia from 2012 to 2014. Vet Med Int 2020:6037893. https://doi.org/10.1155/2020/6037893
Al-Shekaili T, Baylis M, Ganapathy K (2015) Molecular detection of infectious bronchitis and avian metapneumoviruses in Oman backyard poultry. Res Vet Sci 99:46–52. https://doi.org/10.1016/j.rvsc.2014.12.018
Mahmood ZH, Sleman RR, Uthman AU (2011) Isolation and molecular characterization of Sul/01/09 avian infectious bronchitis virus, indicates the emergence of a new genotype in the Middle East. Vet Microbiol 150(1–2):21–27. https://doi.org/10.1016/j.vetmic.2010.12.015
Shariatmadari F (2000) Poultry production and the industry in Iran. World’s Poult Sci J 56(01):55–65. https://doi.org/10.1079/WPS20000006
Aghakhan SM, Abshar N, Rasoul Nejad Fereidouni S, Marunesi C, Khodashenas M (1994) Studies on avian viral infections in Iran. Arch Razi Inst 44(1):1–10. https://doi.org/10.22092/ARI.1994.109123
Shapouri MRS, Mayahi M, Assasi K, Charkhkar S (2004) A survey of the prevalence of infectious bronchitis virus type 4/91 in Iran. Acta Vet Hung 52(2):163–166. https://doi.org/10.1556/AVet.52.2004.2.4
Shapouri SA, Mayahi MR, Charkhkar S, Assasi K (2002) Serotype Identification of recent Iranian isolates of infectious bronchitis virus by type-specific multiplex RT-PCR. Arch Razi Inst 53(1):79–86
Shoushtari AH, Toroghi R, Momayez R, Pourbakhsh SA (2008) 793/B type, the predominant circulating type of avian infectious bronchitis viruses 1999–2004 in Iran: a retrospective study. Arch Razi Inst 63(1):1–5
Hosseini H, Fard MHB, Charkhkar S, Morshed R (2015) Epidemiology of avian infectious bronchitis virus genotypes in Iran (2010–2014). Avian Dis 59(3):431–435. https://doi.org/10.1637/11091-041515-ResNote.1
Ghalyanchi-Langeroudi A, Karimi V, Jannat A, Hashemzadeh M, Fallah MH, Gholami F, Zabihi MT, Heidarzadeh M (2015) Genotyping of infectious bronchitis viruses in the East of Iran, 2015. Iran J Virol 9(2):31–35
Modiri Hamadan A, Ghalyanchilangeroudi A, Hashemzadeh M, Hosseini H, Karimi V, Yahyaraeyat R et al (2017) Genotyping of avian infectious bronchitis viruses in Iran (2015–2017) reveals domination of IS-1494 like virus. Virus Res 240:101–106. https://doi.org/10.1016/j.virusres.2017.08.002
El-Houadfi M, Jones RC, Cook JK, Ambali AG (1986) The isolation and characterisation of six avian infectious bronchitis viruses isolated in Morocco. Avian Pathol 15(1):93–105. https://doi.org/10.1080/03079458608436269
Jones RC, Savage CE, Naylor CJ, Cook JKA, El-Houadfi M (2004) Possible North African progenitor of the major European infectious bronchitis variant (793B, 4/91, CR88). In Kaleta EF, Heffels-Redmann U (eds) Proceedings of the IV International Symposium on Avian Corona-and Pneumovirus Infections. Rauischholzhausen, Germany, pp 105–111
Fellahi S, Ducatez M, El Harrak M, Guerin JL, Touil N, Sebbar G et al (2015) Prevalence and molecular characterization of avian infectious bronchitis virus in poultry flocks in Morocco from 2010 to 2014 and first detection of Italy 02 in Africa. Avian Pathol 44(4):287–295. https://doi.org/10.1080/03079457.2015.1044422
Fellahi S, El Harrak M, Khayi S, Guerin JL, Kuhn JH, El Houadfi M et al (2017) Phylogenetic analysis of avian infectious bronchitis virus isolates from Morocco: a retrospective study (1983 to 2014). Virol Sin 32(2):155–158. https://doi.org/10.1007/s12250-016-3885-3
Sid H, Benachour K, Rautenschlein S (2015) Co-infection with multiple respiratory pathogens contributes to increased mortality rates in Algerian poultry flocks. Avian Dis 59(3):440–446. https://doi.org/10.1637/11063-031615-Case.1
Barberis A, Alloui N, Boudaoud A, Bennoune O, Ammar A (2018) Seroprevalence of infectious bronchitis virus in broiler farms in Batna, East Algeria. Int J Poult Sci 17(9):418–422. https://doi.org/10.3923/ijps.2018.418.422
Lounas A, Oumouna-Benachour K, Medkour H, Oumouna M (2018) The first evidence of a new genotype of nephropathogenic infectious bronchitis virus circulating in vaccinated and unvaccinated broiler flocks in Algeria. Vet World 11(11):1630–1636. https://doi.org/10.14202/vetworld.2018.1630-1636
Bourogâa H, Miled K, Gribâa L, Behi EI, Ghram A (2009) Characterization of new variants of avian infectious bronchitis virus in Tunisia. Avian Dis 53(3):426–433. https://doi.org/10.1637/8666-022609-Reg.1
Bourogaa H, Miled K, Larbi I, Nsiri J, Gribaa L, El Behi I et al (2009) La Bronchite Infectieuse Aviaire En Tunisie: Seroprevalence, Pathogenicite Et Etude. Arch Inst Pasteur Tunis 86:75–83
Kammon A, Wei TS, Omar AR, Dayhum A, Eldghayes I, Sharif M (2013) Molecular detection and characterization of infectious bronchitis virus from Libya. Int J Anim Vet Sci 7(12):974–976
Awad F, Baylis M, Ganapathy K (2014) Detection of variant infectious bronchitis viruses in broiler flocks in Libya. Int J Vet Sci Med 2(1):78–82. https://doi.org/10.1016/j.ijvsm.2014.01.001
Kahya S, Coven F, Temelli S, Eyigor A, Carli KT (2013) Presence of IS/1494/06 genotype-related infectious bronchitis virus in breeder and broiler flocks in Turkey. Ankara Üniversitesi Veteriner Fakültesi Dergisi 60(1):27–31
Yilmaz H, Altan E, Cizmecigil UY, Gurel A, Ozturk GY, Bamac OE et al (2016) Phylogeny and S1 gene variation of infectious bronchitis virus detected in broilers and layers in Turkey. Avian Dis 60(3):596–602. https://doi.org/10.1637/11346-120915-Reg.1
Yilmaz H, Cizmecigil UY, Gurel A, Faburay B (2017) Variants of infectioous bronchitis virus (IBV) in chickens in Turkey and production of recombinant IBV-N protein by using baculovirus expression system. Bulg J Vet Med 20(1):240–247
Chtioui Z, Bouzouaia M, Belhadj H (eds) (2018) First isolation of IBV variant 2 in Tunisia. VI Mediterranean poultry summit; June 18–20; Turin—Italy
Franzo G, Massi P, Tucciarone CM, Barbieri I, Tosi G, Fiorentini L et al (2017) Think globally, act locally: phylodynamic reconstruction of infectious bronchitis virus (IBV) QX genotype (GI-19 lineage) reveals different population dynamics and spreading patterns when evaluated on different epidemiological scales. PLoS ONE 12(9):e0184401. https://doi.org/10.1371/journal.pone.0184401
Franzo G, Cecchinato M, Tosi G, Fiorentini L, Faccin F, Tucciarone CM et al (2018) GI-16 lineage (624/I or Q1), there and back again: The history of one of the major threats for poultry farming of our era. PLoS ONE 13(12):e0203513. https://doi.org/10.1371/journal.pone.0203513
Mahgoub KM, Khaphagy A, Bassiouni AA, Afify MA, Rabie N (2010) The prevalence of infectious bronchitis (IB) outbreaks in some chicken farms. II. Molecular characterisation of field. J Am Sci 6(9):71–93
Abdel-Sabour MA, Al-Ebshahy EM, Khaliel SA, Abdel-Wanis NA, Yanai T (2017) Isolation and molecular characterization of novel infectious bronchitis virus variants from vaccinated broiler flocks in Egypt. Avian Dis 61(3):307–310. https://doi.org/10.1637/11566-121516-RegR
Ghalyanchilangeroudi A (2020) The emergence of Q1 genotype of Avian Infectious Bronchitis Virus in Iran, 2019: the first report. Iran J Vet Res. https://doi.org/10.22099/IJVR.2020.35858.5252(in press)
Lachheb J, Turki A, Nsiri J, Fathallah I, El Behi I, Larbi I et al (2019) Molecular characterization of a unique variant of avian infectious bronchitis virus in Tunisia. Poult Sci 98(10):4338–4345. https://doi.org/10.3382/ps/pez384
Moharam I, Sultan H, K, Ibrahim M, Shany S, Shehata AA et al (2020) Emerging infectious bronchitis virus (IBV) in Egypt: evidence for an evolutionary advantage of a new S1 variant with a unique gene 3ab constellation. Infect Genet Evol 85:104433. https://doi.org/10.1016/j.meegid.2020.104433
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10(3):512–526. https://doi.org/10.1093/oxfordjournals.molbev.a040023
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549. https://doi.org/10.1093/molbev/msy096
Kiss I, Mato T, Homonnay Z, Tatar-Kis T, Palya V (2016) Successive occurrence of recombinant infectious bronchitis virus strains in restricted area of Middle East. Virus Evol 2(2):vew021. https://doi.org/10.1093/ve/vew021
Dolz R, Pujols J, Ordonez G, Porta R, Majo N (2008) Molecular epidemiology and evolution of avian infectious bronchitis virus in Spain over a fourteen-year period. Virology 374(1):50–59. https://doi.org/10.1016/j.virol.2007.12.020
Sapats SI, Ashton F, Wright PJ, Ignjatovic J (1996) Sequence analysis of the S1 glycoprotein of infectious bronchitis viruses: identification of a novel genotypic group in Australia. J Gen Virol. 77(Pt 3):413–418. https://doi.org/10.1099/0022-1317-77-3-413
Shieh HK, Shien JH, Chou HY, Shimizu Y, Chen JN, Chang PC (2004) Complete nucleotide sequences of S1 and N genes of infectious bronchitis virus isolated in Japan and Taiwan. J Vet Med Sci 66(5):555–558. https://doi.org/10.1292/jvms.66.555
Kulkarni AB (ed) (2016) Characterization of recent infectious bronchitis virus isolates from broilers and backyard flocks of Georgia. In: The 65th western poultry disease conference; 2016; Vancouver, BC, Canada. April 24–27, 2016
Jiang L, Zhao W, Han Z, Chen Y, Zhao Y, Sun J et al (2017) Genome characterization, antigenicity and pathogenicity of a novel infectious bronchitis virus type isolated from south China. Infect Genet Evol 54:437–446. https://doi.org/10.1016/j.meegid.2017.08.006
Scherbakova LO, Kolosov SN, Nikonova ZB, Zinyakov NG, Ovchinnikova YV, Chvala IA (2018) Genetic characterization of Avian infectious bronchitis virus isolates recovered in cis countries in 2015–2017. Vet Sci Today 3:30–39. https://doi.org/10.29326/2304-196x-2018-3-26-30-34
Sadri N, Ghalyanchilangeroudi A, Fallah Mehrabadi MH, Hosseini H, Shayeganmehr A, Sediqian MS et al (2019) Genotyping of avian infectious bronchitis virus in Afghanistan (2016–2017): the first report. Iran J Vet Res 20(1):60–63
Tal E-C, Beny P, Wojciech H, Udi A & Charles C (2019) Epidemiology and Spread of a dominant Infectious Bronchitis Virus (IS/1494/06). The XXIst World Veterinary Poultry Association Congress "WVPAC", September 16–20, 2019 2019 Bangkok; Thailand
CFR “Code of Federal Regulations-9 CFR” (2020) Bronchitis Vaccine. Bronchitis Vaccine. Part 113-Subjgrp - Live Virus Vaccines, Section 113.327. US Government Printing Office, Washington D.C, USA. https://www.gpo.gov/fdsys/granule/CFR-2011-title9-vol1/CFR-2011-title9-vol1-sec113-327
Ph.Eur (2017) Monograph 0442: Avian infectious bronchitis vaccine (live). European pharmacopeia. Strasbourg, France: European Directorate for the Quality of Medicines & Healthcare, pp 1008–1010
Zhao Y, Cheng JL, Liu XY, Zhao J, Hu YX, Zhang GZ (2015) Safety and efficacy of an attenuated Chinese QX-like infectious bronchitis virus strain as a candidate vaccine. Vet Microbiol 180(1–2):49–58. https://doi.org/10.1016/j.vetmic.2015.07.036
Feng K, Xue Y, Wang J, Chen W, Chen F, Bi Y et al (2015) Development and efficacy of a novel live-attenuated QX-like nephropathogenic infectious bronchitis virus vaccine in China. Vaccine 33(9):1113–1120. https://doi.org/10.1016/j.vaccine.2015.01.036
Lee HJ, Youn HN, Kwon JS, Lee YJ, Kim JH, Lee JB et al (2010) Characterization of a novel live attenuated infectious bronchitis virus vaccine candidate derived from a Korean nephropathogenic strain. Vaccine 28(16):2887–2894. https://doi.org/10.1016/j.vaccine.2010.01.062
Sultan HA, Ali A, El Feil WK, Bazid AHI, Zain El-Abideen MA, Kilany WH (2019) Protective efficacy of different live attenuated infectious bronchitis virus vaccination regimes against challenge with IBV variant-2 circulating in the Middle East. Front Vet Sci 6:341. https://doi.org/10.3389/fvets.2019.00341
Erfanmanesh A, Ghalyanchilangeroudi A, Nikaein D, Hosseini H, Mohajerfar T (2020) Evaluation of inactivated vaccine of the variant 2 (IS-1494/GI-23) genotype of avian infectious bronchitis. Comp Immunol Microbiol Infect Dis 71:101497. https://doi.org/10.1016/j.cimid.2020.101497
Elhady MA, Ali A, Kilany WH, Elfeil WK, Ibrahim H, Nabil A et al (2018) Field efficacy of an attenuated infectious bronchitis variant 2 virus vaccine in commercial broiler chickens. Vet Sci. https://doi.org/10.3390/vetsci5020049
Ali A, Kilany WH, Zain El-Abideen MA, Sayed ME, Elkady M (2018) Safety and efficacy of attenuated classic and variant 2 infectious bronchitis virus candidate vaccines. Poult Sci 97(12):4238–4244. https://doi.org/10.3382/ps/pey312
Hofstad MS (1981) Cross-immunity in chickens using seven isolates of avian infectious bronchitis virus. Avian Dis 25(3):650–654. https://doi.org/10.2307/1589995
de Wit JJ, Cook JK (2014) Factors influencing the outcome of infectious bronchitis vaccination and challenge experiments. Avian Pathol 43(6):485–497. https://doi.org/10.1080/03079457.2014.974504
Darbyshire JH (1980) Assessment of cross-immunity dm chickens to strains of avian infectious bronchitis virus using tracheal organ cultures. Avian Pathol 9(2):179–184. https://doi.org/10.1080/03079458008418401
Cowen BS, Hitchner SB (1975) Serotyping of Avian infectious bronchitis viruses by the virus-neutralization test. Avian Dis 19(3):583–595. https://doi.org/10.2307/1589084
Alexander DJ, Chettle NJ (1977) Procedures for the haemagglutination and the haemagglutination inhibition tests for avian infectious bronchitis virus. Avian Pathol 6(1):9–17. https://doi.org/10.1080/03079457708418208
Song C-S, Kim J-H, Lee Y-J, Kim S-J, Izumiya Y, Tohya Y et al (1998) Detection and classification of infectious bronchitis viruses isolated in Korea by dot-immunoblotting assay using monoclonal antibodies. Avian Dis 42(1):92–100. https://doi.org/10.2307/1592580
Naqi SA (1990) A monoclonal antibody-based immunoperoxidase procedure for rapid detection of infectious-bronchitis virus in infected-tissues. Avian Dis 34(4):893–898. https://doi.org/10.2307/1591379
Snyder DB, Marquardt WW (1984) Use of monoclonal antibodies to assess antigenic relationships of avian infectious bronchitis virus serotypes in the United States. Adv Exp Med Biol 173:109–113. https://doi.org/10.1007/978-1-4615-9373-7_9
Montassier MD-FS, Brentano L, Montassier HJ, Richtzenhain LJ (2008) Genetic grouping of avian infectious bronchitis virus isolated in Brazil based on RT-PCR/RFLP analysis of the S1 gene. J Pesquisa Veterinária Brasileira 28:190–194. https://doi.org/10.1590/S0100-736X2008000300011
Chacon RD, Astolfi-Ferreira CS, Chacon JL, Nunez LFN, De la Torre DI, Piantino Ferreira AJ (2019) A seminested RT-PCR for molecular genotyping of the Brazilian BR-I infectious bronchitis virus strain (GI-11). Mol Cell Probes 47:101426. https://doi.org/10.1016/j.mcp.2019.101426
Jones RM, Ellis RJ, Cox WJ, Errington J, Fuller C, Irvine RM et al (2011) Development and validation of RT-PCR tests for the detection and S1 genotyping of infectious bronchitis virus and other closely related gammacoronaviruses within clinical samples. Transbound Emerg Dis 58(5):411–420. https://doi.org/10.1111/j.1865-1682.2011.01222.x
Lee CW, Hilt DA, Jackwood MW (2003) Typing of field isolates of infectious bronchitis virus based on the sequence of the hypervariable region in the S1 gene. J Vet Diagn Investig 15(4):344–348. https://doi.org/10.1177/104063870301500407
Kwon HM, Jackwood MW, Gelb J Jr (1993) Differentiation of infectious bronchitis virus serotypes using polymerase chain reaction and restriction fragment length polymorphism analysis. Avian Dis 37(1):194–202
Clewley JP, Morser J, Avery RJ, Lomniczi B (1981) Oligonucleotide fingerprinting of ribonucleic acids of infectious bronchitis virus strains. Infect Immun 32(3):1227
Kusters JG, Niesters HG, Bleumink-Pluym NM, Davelaar FG, Horzinek MC, Van der Zeijst BA (1987) Molecular epidemiology of infectious bronchitis virus in The Netherlands. J Gen Virol 68(Pt 2):343–352. https://doi.org/10.1099/0022-1317-68-2-343
Parsons LM, Bouwman KM, Azurmendi H, de Vries RP, Cipollo JF, Verheije MH (2019) Glycosylation of the viral attachment protein of avian coronavirus is essential for host cell and receptor binding. J Biol Chem 294(19):7797–7809. https://doi.org/10.1074/jbc.RA119.007532
Zhang X, Deng T, Lu J, Zhao P, Chen L, Qian M et al (2020) Molecular characterization of variant infectious bronchitis virus in China, 2019: Implications for control programmes. Transbound Emerg Dis. 67(3):1349–1355. https://doi.org/10.1111/tbed.13477
Abdel-Sabour MA, Al-Ebshahy EM, Khaliel SA, Abdel-Wanis NA, Yanai T (2017) Isolation and molecular characterization of novel infectious bronchitis virus variants from vaccinated broiler flocks in Egypt. Avian Dis. 61(3):307–310. https://doi.org/10.1637/11566-121516-RegR
Awad F, Forrester A, Baylis M, Lemiere S, Ganapathy K, Hussien HA et al (2015) Protection conferred by live infectious bronchitis vaccine viruses against variant Middle East IS/885/00-like and IS/1494/06-like isolates in commercial broiler chicks. Vet Rec Open 2(2):e000111. https://doi.org/10.1136/vetreco-2014-000111
Bru T, Vila R, Cabana M, Geerligs HJ (2017) Protection of chickens vaccinated with combinations of commercial live infectious bronchitis vaccines containing Massachusetts, Dutch and QX-like serotypes against challenge with virulent infectious bronchitis viruses 793B and IS/1494/06 Israel variant 2. Avian Pathol 46(1):52–58. https://doi.org/10.1080/03079457.2016.1203393
Habibi M, Karimi V, Langeroudi AG, Ghafouri SA, Hashemzadeh M, Farahani RK et al (2017) Combination of H120 and 1/96 avian infectious bronchitis virus vaccine strains protect chickens against challenge with IS/1494/06 (variant 2)-like infectious bronchitis virus. Acta Virol 61(2):150–160. https://doi.org/10.4149/av_2017_02_04
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Handling Editor: Sheela Ramamoorthy.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Houta, M.H., Hassan, K.E., El-Sawah, A.A. et al. The emergence, evolution and spread of infectious bronchitis virus genotype GI-23. Arch Virol 166, 9–26 (2021). https://doi.org/10.1007/s00705-020-04920-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04920-z