
Abstract
The full-length genome sequences of two novel poleroviruses found infecting cowpea plants, cowpea polerovirus 1 (CPPV1) and cowpea polerovirus 2 (CPPV2), were determined using overlapping RT-PCR and RACE-PCR. Whereas the 5845-nt CPPV1 genome was most similar to chickpea chlorotic stunt virus (73% identity), the 5945-nt CPPV2 genome was most similar to phasey bean mild yellow virus (86% identity). The CPPV1 and CPPV2 genomes both have a typical polerovirus genome organization. Phylogenetic analysis of the inferred P1-P2 and P3 amino acid sequences confirmed that CPPV1 and CPPV2 are indeed poleroviruses. Four apparently unique recombination events were detected within a dataset of 12 full polerovirus genome sequences, including two events in the CPPV2 genome. Based on the current species demarcation criteria for the family Luteoviridae, we tentatively propose that CPPV1 and CPPV2 should be considered members of novel polerovirus species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Members of the family Luteoviridae are distributed worldwide and infect monocotyledonous and dicotyledonous plants [3, 9]. Their 5600- to 5900-nt-long positive-sense single-stranded RNA genomes are encapsidated in icosahedral particles and encode five to seven ORFs [2, 12]. The family includes the genera Luteovirus, Enamovirus and Polerovirus. Whereas members of the genus Polerovirus encode both an ORF0 and an ORF4, members of the genus Luteovirus lack an ORF0, and members of the genus Enamovirus lack an ORF4 [2, 12]. Further, members of the genera Luteovirus and Polerovirus also have a small non-AUG-initiated gene, ORF3a, that is not found in members of the genus Enamovirus [12]. The complete genomes of 28 poleroviruses have been characterized so far (NCBI data). Recently, two novel cowpea-associated poleroviruses provisionally named cowpea polerovirus 1 (CPPV1) and cowpea polerovirus 2 (CPPV2) were identified using a metagenomics-based approach [11]. Specifically, a genomic fragment of 5012 nt was determined for CPPV1 (accession number KX599154) and a fragment of 3164 nt for CPPV2 (accession number KX599164).
The aim of this study was to determine the complete genome sequences of CPPV1 and CPPV2. Cowpea plants BE167 and BE179, from which the two viruses were initially isolated [11], were used to extract total RNA using an RNeasy Plant Mini Kit (QIAGEN, Valencia, CA) as described by the manufacturer. Before determining the 5’ and 3’ ends of the genomes by RACE-PCR, a large internal part of the CPPV2 genome sequence had to be completed by sequencing RT-PCR fragments amplified with a combination of two primer sets: one set designed based on the phasey bean mild yellow virus genome (PBMYV) and another previously used to obtain the partial CPPV2 genome (Supplementary Table 1). To sequence the 5’ region of each genome, cDNAs were produced with primers PoleroNB202R (CPPV1) and Polero2NB1834R (CPPV2) using SuperScript III Reverse Transcriptase (18080-044). These cDNAs were subsequently tailed in parallel with polyA, G or C using a recombinant terminal transferase TdT enzyme mini kit (New England BioLabs, Beverly, MA, USA). The 5’RACE-PCR was performed as described previously [6]. The sequence of the 3’ end was determined for each genome, using a 3’RACE-PCR procedure. Briefly, RNAs were tailed with polyA using a poly(U) polymerase mini kit (New England BioLabs) as described by the manufacturer. Amplicons generated with polyT and virus-specific primers were sequenced directly using automated Sanger sequencing (Beckman Coulter Genomics). Assembly and alignment of the nucleotide sequences and the identification of ORFs was performed as reported previously [11]. Maximum-likelihood phylogenetic trees were produced using PhyML 3.1 [4] implemented in MEGA version 6.06 [14] with the JTT+G amino acid substitution model with branch support tested using 1000 bootstrap replicates. Potential recombination events were detected using the RDP, GENECONV, BOOTSCAN, MAXIMUM CHI SQUARE, CHIMAERA, SISCAN and 3SEQ recombination detection methods, which are implemented in RDP4.79, with default settings [8]. KnotInFrame (http://bibiserv.techfak.uni-bielefeld.de/knotinframe) was used to predict the slippery heptameric -1 ribosomal frameshift [15].
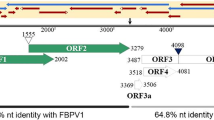
Both the 5845-nt CPPV1 genome (accession number KY364846) and the 5945-nt CPPV2 genome (accession number KY364847) have an organization that is typical of poleroviruses [1, 2, 12], with the presence of the conserved ACAAAAGA motif at the 5’ end and seven ORFs that are characteristic of poleroviruses, including ORF0, ORF4 and ORF3a (Fig. 1). The aa lengths of the putative CPPV1 and CPPV2 proteins are as follows: P0, 246 and 260 amino acids, respectively; P1, 623 and 654 amino acids, respectively; P1-P2, 1024 and 1059 amino acids, respectively; P3a, 45 and 46 amino acids, respectively; P3, 198 and 197 amino acids, respectively; P4, 190 and 189 amino acids, respectively; and P3-P5, 668 and 698 amino acids, respectively. Similar to other known poleroviruses, the -1 ribosomal frameshift for the P1-P2 fusion protein [5] is predicted by the ‘slippery heptamer’ sequence GGGAAAC at positions 1632 to 1638 for CPPV1 and 1684 to 1690 for CPPV2. The putative CPPV1 proteins all share less than 85% identity with their counterparts in other poleroriruses [11]. The putative CPPV2 proteins share 44% (P0), 61% (P1), 76% (P3) and 84% (P1-P2) identity with their homologues in phasey bean mild yellow virus (PBMYV, accession number KT963000), 71% (P3-P5) and 67% (P3a) with their presumed homologues in chickpea chlorotic stunt virus (CpCSV, accession number NC008249) and 56% (P4) with their homologue in suakwa aphid-borne yellows virus (SABYV, accession number KF815677).

Genome organization of CPPV1 (A) and CPPV2 (B). The ORFs that are likely to represent genes expressing characteristic polerovirus proteins (P0-P5) were identified based on comparisons with other members of the genus Polerovirus. RdRp, RNA-dependent RNA polymerase; CP, coat protein; MP, movement protein; RTD, readthrough domain. The predicted -1 slippery ribosomal frameshifting site is indicated by a filled triangle, and the leaky stop codon is indicated by an asterisk. The two unique recombination events (R1 and R2) detected within the CPPV2 sequence are indicated by red bars below the CPPV2 genome diagram
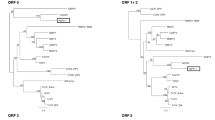
Maximum-likelihood phylogenetic trees based on the P3 protein (Fig. 2A) and the P1-P2 protein (Fig. 2B) confirmed that CPPV1 and CPPV2 cluster with the known poleroviruses. Whereas CPPV1 clusters with groundnut rosette assistor virus (GRAV) with 75% bootstrap support in the P3 tree, in the P1-P2 tree, it does not form a strongly supported cluster with any of the other known poleroviruses. Inversely, CPPV2 clusters with 100% bootstrap support with PBMYV isolates in the P1-P2 tree but forms no strongly supported clusters with any known polerovirus in the P3 tree. It is noteworthy that both CPPV1 and CPPV2 are apparently most closely related to poleroviruses isolated from leguminous plants (phasey bean, groundnut and chickpea).

Maximum-likelihood phylogenetic tree depicting the relationships between CPPV1, CPPV2, and established members of the family Luteoviridae based on the P3 (A) and the P1-P2 (B) proteins. The following members of the genus Polerovirus that exhibited the highest degree of similarity to CPPV1 and CPPV2 were selected for the comparison: chickpea chlorotic stunt virus (CpCSV), groundnut rosette assistor virus (GRAV), phasey bean mild yellow virus (PBMYV), beet western yellow virus (BWYV), beet mild yellowing virus (BMYV-IPP), cucurbit aphid-borne yellows virus, (CABYV), melon aphid-borne yellows virus (MABYV), pepo aphid-borne yellows virus (PABYV-RSA), suakwa aphid-borne yellows virus (SABYV-TW19), cereal yellow dwarf virus (CYDV) and pepper vein yellows virus (PeVYV). One representative of the genus Luteovirus (soybean dwarf virus, SbDV) and one member of the genus Enamovirus (pea enation mosaic virus 1, PEMV-1) were used as outgroups. Branch support values are percentages estimated based on 1000 bootstrap replicates
Using a dataset of 12 full polerovirus genomes, four apparently unique recombination events were detected, including two events in the CPPV2 genomes. The sites of these two events (R1 and R2) are located within the CPPV2 P2-P3a (R1) and P3/P4 (R2) regions (Fig. 2B), and both appear to have involved phasey bean mild yellow virus (accession number NC028793) as the major parent (i.e., the contributor of the larger fraction of the genome) and an undescribed polerovirus as the minor parent (i.e., the contributor of the smaller fraction of the genome; Fig. 2B). The genomic region acquired from the minor parent encompasses the end of the RdRp gene and the beginning of the CP, a genome region that is considered a recombination hot spot in members of the family Luteoviridae [7, 10]. The widespread occurrence of recombination between members of the family Luteoviridae is considered a form of modular evolution that can play a key role in the adaptation of luteoviruses to changing environmental conditions [7, 10, 13].
According to the International Committee of Taxonomy of Viruses, a virus of the family Luteoviridae is considered a member of a new species if it has less than 90% amino acid sequence identity to any previously classified members of the family Luteoviridae, in any of its predicted proteins [2]. Based on this species demarcation criterion, CPPV1 and CPPV2 are likely representatives of two new species in the genus Polerovirus.
References
Chen S, Jiang G, Wu J, Liu Y, Qian Y, Zhou X (2016) Characterization of a novel polerovirus infecting maize in China. Viruses 8(120):1–17
Domier LL, D’Arcy CJ (2010) Luteoviruses. In: Mahy BWJ, Van Regenmortel MHV (eds) Desk encyclopedia of plant and fungal virology. Academic, Oxford, pp 197–204
Domier LL (2012) Family Luteoviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy: ninth report of the International Committee on the Taxonomy of Viruses. Elsevier Academic Press, Amsterdam, pp 1045–1053
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Knierim D, Tsai WS, Deng TC, Green SK, Kenyon L (2013) Full-length genome sequences of four polerovirus isolates infecting cucurbits in Taiwan determined from total RNA extracted from field samples. Plant Pathol 62:633–641
Knierim D, Maiss E, Menzel W, Winter S, Kenyon L (2015) Characterization of the complete genome of a novel Polerovirus infecting Sauropus androgynus in Thailand. J Phytopathol 163:695–702
Lotos L, Maliogka VI, Katis NI (2016) New poleroviruses associated with yellowing symptoms in different vegetable crops in Greece. Arch Virol 161:431–436
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evolution 1(1):1–5
Pagan I, Holmes EC (2010) Long-term evolution of the Luteoviridae: time scale and mode of virus speciation. J Virol 84(12):6177–6187
Pagan I, Holmes EC (2010) Long-term evolution of the Luteoviridae: time scale and mode of virus speciation. J Virol 84:6177–6187
Palanga E, Filloux D, Martin DP, Fernandez E, Gargani D, Ferdinand R, Zabre J, Bouda Z, Neya JB, Sawadogo M, Traore O, Peterschmitt M, Roumagnac P (2016) Metagenomic-based screening and molecular characterization of cowpea-infecting viruses in Burkina Faso. PLoS One 11(10):e0165188
Smirnova E, Firth AE, Miller WA, Scheidecker D, Brault V, Reinbold C, Rakotondrafara AM, Chung BY-W, Ziegler-Graff V (2015) Discovery of a small non-AUG-initiated ORF in poleroviruses and luteoviruses that is required for long-distance movement. PLoS Pathog 11(5):e1004868
Stevens M, Freeman B, Liu HY, Herrbach E, Lemaire O (2005) Beet poleroviruses: close friends or distant relatives? Mol Plant Pathol 6:1–9
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Theis C, Reeder J, Giegerich R (2008) KNOTINFRAME: prediction of −1 ribosomal frameshift events. Nucleic Acids Res 36:6013–6020
Acknowledgements
EP’s PhD fellowship was funded by the French Embassy of Togo (N°: 808087J; 836014F; 864988K) and International Foundation for Science (N°: C/5489-1). PR is supported by EU grant FP7-PEOPLE-2013-IOF (N° PIOF-GA-2013-622571). We acknowledge Ecole Superieur d’Agronomie of the University of Lomé, which accepted to shelter EP’s IFS funds, and the Laboratoire Mixte International Patho-Bios, which supported sample collection.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was funded by the French Embassy of Togo (N°: 808087J; 836014F; 864988K.), International Foundation for Science (N°: C/5489-1) and EU grant FP7-PEOPLE-2013-IOF (N° PIOF-GA-2013-622571).
Conflict of interest
Author EP has received research grants from the French Embassy of Togo (N°: 808087J; 836014F; 864988K) and the International Foundation for Science (N°: C/5489-1). Author PR has received an EU grant FP7-PEOPLE-2013-IOF (N° PIOF-GA-2013-622571). Author EP and PR declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Palanga, E., Martin, D.P., Galzi, S. et al. Complete genome sequences of cowpea polerovirus 1 and cowpea polerovirus 2 infecting cowpea plants in Burkina Faso. Arch Virol 162, 2149–2152 (2017). https://doi.org/10.1007/s00705-017-3327-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3327-z