
Abstract
Zucchini shoestring virus (ZSSV) has been proposed to be a putative potyvirus in the papaya ringspot virus (PRSV) cluster, based on the sequence similarity of its coat protein to those of related potyviruses. ZSSV has been associated with the outbreak of a damaging disease of baby marrow (Cucurbita pepo L.) that had been observed throughout the province of KwaZulu-Natal, in the Republic of South Africa (RSA). We report the genome sequence of ZSSV, determined by next-generation sequencing of total RNA extracted from an infected baby marrow (Cucurbita pepo L.). The ZSSV genome is 10,295 nucleotides long excluding the poly(A) tail and displays a typical potyvirus organization. Algerian watermelon mosaic virus (AWMV; EU410442.1) was identified as the closest relative of ZSSV, sharing the highest nucleotide sequence identity of 65.68%. The nucleotide and amino acid sequence identity values for each protein support the differentiation of ZSSV as a member of a distinct species in the genus Potyvirus. This taxonomic position was also confirmed using the Pairwise Sequence Comparison online tool from the National Center for Biotechnology Information. Phylogenetic analysis of the polyprotein coding sequence of ZSSV grouped ZSSV together with AWMV and Moroccan watermelon mosaic virus, but in different clusters. ZSSV is the second cucurbit-infecting virus in the PRSV cluster present in RSA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The number of cucurbit-infecting viruses worldwide has increased over the last two decades. Zucchini shoestring virus (ZSSV) has been proposed to be a putative potyvirus infecting cucurbits, based on the nucleotide sequence similarity of its coat protein (CP) to those of related cucurbit-infecting potyviruses [7]. ZSSV was detected in the province of KwaZulu-Natal (KZN) in the Republic of South Africa (RSA) during virus surveys conducted in the cucurbit-growing areas between 2011 and 2013. Symptoms associated with ZSSV include severe leaf filiformy and fruit deformation on baby marrow (Cucurbita pepo L.) [7]. These symptoms were observed throughout the surveys and in all growing areas. Losses up to 100 % were recorded in cases when the infection occurred before fruit formation. The full genome sequence of ZSSV is reported here.
The source of the ZSSV isolate used in this study was a baby marrow leaf displaying filliformy symptoms, randomly selected from the samples collected during virus surveys conducted between 2011 and 2013 in KZN. A NucleoSpin RNA Plant kit (Macherey-Nagel, Germany) was used according to the manufacturer’s instruction to extract total RNA, which was shipped on dry ice to the Agricultural Research Council’s Biotechnology Platform (ARC-BTP) in Pretoria, RSA, for library preparation and next-generation sequencing (NGS) on the Illumina HiSeq platform, using paired-end chemistry 125 × 125-bp reads. FastQC was used to access the quality of the NGS data generated. Removal of the adapters from the reads, trimming, and quality filtering were performed using Trimmomatic version 0.33 [4] with the following settings: {ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:9:1:true LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36}. The paired-end sequences were subsequently used as single reads for de novo assembly, which was performed using SeqMan NGen (software version 12.3.1 build 48; DNASTAR Lasergene) according to the default parameters with the removal of the host data. All generated contigs were used for a BLAST search of the National Center for Biotechnology Information database using the Netsearch function on the program SeqMan pro (software version 12.3.1 build 48 421; DNASTAR Lasergene).
A 10,308-kb contig (median coverage: 162.61) matched the ZSSV CP coding sequence (accession number: KP723639.1) with an E-value of 0.00 and was therefore used as the ZSSV draft genome. Direct sequencing of an overlapping amplicon flanking the draft genome of ZSSV was performed at Inqaba Biotechnical Industries (Pty) Ltd. (Pretoria, RSA) to confirm the integrity of the ZSSV genome sequence. Amplicons were produced by reverse transcription polymerase chain reaction (Table S1). The Open Reading Frame (ORF) Finder was used to predict the ORFs on the ZSSV genome. The cleavage sites were identified using the data provided by Adams et al. [1] and Romay et al. [12]. Information from Chung et al. [5] and from Wen and Hajimorad [15] were useful in identifying the PIPO. Multiple sequence alignments were performed using MUSCLE, implemented in MEGA 6.06 [14]. Nucleotide and amino acid (aa) sequence identity were computed using the SIAS tool (http://imed.med.ucm.es/Tools/sias.html). Any putative recombination junctions were checked using RDP [10], GENECONV [11], MaxChi [13], BootScan [9], and SIScan [6] included in RDP v4.56 package [8]. The full-genome sequences of all potyviruses were used with PAirwise Sequence Comparison (PASC) [3] to confirm the molecular taxonomic position of ZSSV. ZSSV phylogeny was inferred in MEGA 6.06 [14] using the maximum-likelihood method based on the general time-reversible model with a discrete gamma distribution and invariable sites with 500 bootstrap replicates.
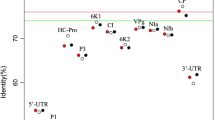
The ZSSV genome consists of 10,295 nucleotides (accession number: KU355553) excluding the poly(A) tail (Fig. 1) and has a genome organization typical of a potyvirus. The ZSSV genome sequence shares the highest nucleotide sequence identity of 65.68 % with Algerian watermelon mosaic virus (AWMV; EU410442.1) (Table S2). Regarding the respective protein-coding sequences, ZSSV shares the highest nucleotide sequence identity with AWMV for eight out of the 11 potyvirus proteins (Table S2). The highest nucleotide sequence identity for the other three potyvirus proteins was shared with Moroccan watermelon mosaic virus (MWMV) isolates (Table S2). The highest amino acid sequence identity (69.22 %) was to AWMV. ZSSV shared the highest aa sequence identity with AWMV for eight potyvirus-encoded proteins (Table S3). Phylogenetic relationships were therefore expected between these isolates (Fig. 2). Two cleavage sites, CI|6K2 and Nia|NIb, were unique to the ZSSV genome. The motifs 624R-I-T-C627 and 882P-T-R884 instead of the highly conserved K-I-T-C and P-T-K, reported in AWMV and MWMV [16, 17], were also identified in the ZSSV genome. No recombination junctions were detected in the ZSSV genome. PASC and the nucleotide sequence identity results qualify ZSSV as a member of a distinct species in the genus Potyvirus according to the demarcation criterion for the genus Potyvirus [2]. ZSSV is the second cucurbit-infecting virus of the PRSV cluster to occur in RSA, and the third one reported in Africa. The shoestring symptom cannot be solely attributed to ZSSV at this stage because a tobamovirus and another potyvirus were also detected in the sample analysed in this study (data not shown).

ZSSV genome organization. The numbers on the diagram indicate the starting nucleotide position predicted for each gene. The cleavage sites are indicated by the symbol “|” between the fourth and the fifth aa

Phylogram of the coding sequences of the ZSSV polyprotein. ZTMV, zucchini tigré mosaic virus
References
Adams MJ, Antoniw JF, Beaudoin F (2005) Overview and analysis of the polyprotein cleavage sites in the family Potyviridae. Mol Plant Pathol 6:471–487
Adams MJ, Zerbini FM, French R, Rabenstein F, Stenger DC, Valkonen JPT (2012) Family Potyviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy: classification and nomenclature of viruses: Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier, San Diego, pp 1069–1089
Bao Y, Chetvernin V, Tatusova T (2014) Improvements to pairwise sequence comparison (PASC): a genome-based web tool for virus classification. Arch Virol 159:3293–3304
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Chung BYW, Miller WA, Atkins JF, Firth AE (2008) An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci USA 105:5897–5902
Gibbs MJ, Armstrong JS, Gibbs AJ (2000) Sister-Scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16(7):573–582. doi:10.1093/bioinformatics/16.7.573
Ibaba JD, Laing MD, Gubba A (2015) First Report of a Novel Potyvirus from the Papaya ringspot virus Cluster Infecting Zucchini (Cucurbita pepo) in KwaZulu-Natal, Republic of South Africa. Plant Dis 99:1289
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1(1):vev003. doi:10.1093/ve/vev003
Martin DP, Posada D, Crandall KA, Williamson C (2005) A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res Hum Retroviruses 21(1):98–102. doi:10.1089/aid.2005.21.98
Martin D, Rybicki E (2000) RDP: detection of recombination amongst aligned sequences. Bioinformatics 16(6):562–563
Padidam M, Sawyer S, Fauquet CM (1999) Possible emergence of new geminiviruses by frequent recombination. Virology 265(2):218–225. doi:10.1006/viro.1999.0056
Romay G, Lecoq H, Desbiez C (2014) Zucchini tigré mosaic virus is a distinct potyvirus in the Papaya ringspot virus cluster: molecular and biological insights. Arch Virol 159:277–289
Smith J (1992) Analyzing the mosaic structure of genes. J Mol Evol 34(2):126–129. doi:10.1007/BF00182389
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
Wen RH, Hajimorad MR (2010) Mutational analysis of the putative pipo of Soybean mosaic virus suggests disruption of PIPO protein impedes movement. Virology 400:1–7
Yakoubi S, Desbiez C, Fakhfakh H, Wipf-Scheibel C, Marrakchi M, Lecoq H (2008) Biological characterization and complete nucleotide sequence of a Tunisian isolate of Moroccan watermelon mosaic virus. Arch Virol 153:117–125
Yakoubi S, Lecoq H, Desbiez C (2008) Algerian watermelon mosaic virus (AWMV): a new potyvirus species in the PRSV cluster. Virus Genes 37(1):103–109
Acknowledgments
We thank Mr. Jonathan Featherston and Ms. Thulile Faith Nhlapo, from the ARC-BTP, for the technical assistance with NGS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ibaba, J.D., Laing, M.D. & Gubba, A. Zucchini shoestring virus: a distinct potyvirus in the papaya ringspot virus cluster. Arch Virol 161, 2321–2323 (2016). https://doi.org/10.1007/s00705-016-2899-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-2899-3