
Abstract
Human respiratory syncytial virus (RSV) is recognized as a leading pathogen responsible for severe respiratory infections in the pediatric population, particularly in infants and young children. A previous study by the same study group revealed that the RSV BA strain was prevalent in southwestern China in epidemic seasons from 2008 to 2009. The aim of this study was to investigate the epidemiology of RSV in the following two years, the genetic variability of the G gene, and mutations at the 276th amino acid in the fusion (F) protein of RSV strains. Nine BA substrains were found in 16 subgroup B viruses by phylogenetic analysis. The G gene of genotype BA was predicted to encode proteins with five different lengths. The findings indicate that subgroup A and B RSVs alternately circulate in southwestern China and that genotype BA strains appear to be the long-term circulating ones. These epidemiological data may help in future vaccine design and further investigation of G protein function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human respiratory syncytial virus (RSV), a member of the genus Pneumovirus in the family Paramyxoviridae, is recognized as a leading pathogen responsible for severe respiratory infections in the pediatric population, particularly in infants and young children [1]. RSV is estimated to infect 64 million people and cause 160,000 deaths every year. This virus causes regular seasonal epidemics, usually in the winter in temperate countries or during the rainy season in tropical areas [2]. A peculiar aspect of RSV is that the RSV-induced immune response does not confer long-lasting protection, which is why reinfection is common throughout life [3].
RSV strains are separated into two major subgroups (A and B) based on antigenic differences. The main differences between RSV groups A and B are found in the attachment glycoprotein G, and the RSV G protein shows the highest degree of divergence, both between and within the two groups. The G protein is a type II glycoprotein that consists of 289–299 amino acids. It has two hypervariable regions in the extracellular domain, leading to much higher variability than other proteins, both between and within the major antigenic groups of RSV. Diversity occurs mainly in the G ectodomain, which has only 44 % amino acid sequence identity between the two subgroups, as compared to 83 % for the transmembrane and cytoplasmic domains [4]. It has been suggested that the antigenic differences within this protein are the main cause of repeated RSV infections [4, 5]. In addition, positive selection for amino acid changes has been observed in the two hypervariable regions of the G protein ectodomain [6, 7]. One of the hypervariable regions is located in the C-terminal region and contains multiple epitopes recognized by monoclonal antibodies, suggesting that immune selection of new variants by antibodies may contribute to the generation of RSV diversity. This is also seen in the diversity of the F protein. Since the start of the clinical use of a monoclonal antibody against F protein, palivizumab (PZ), a panel of variants has been described with or without resistance to PZ [8, 9]. An alteration at amino acid 276, which is very close to the epitope recognized by PZ, is suspected to be involved in the generation of PZ resistance. The distribution of this genetic mutation in circulating viruses and its clinical significance remain unknown.
Recent studies have demonstrated a worldwide prevalence of RSV of very distinct genotypes with temporal rather than geographical clustering. However, less information is available on the distribution pattern of epidemic RSV strains in China. In our previous study, during three consecutive epidemic periods (2006–2009), we showed the genetic diversity of the G gene and identified the predominance of a distinct strain, genotype BA RSV, in southwestern China [10]. In the current study, we attempt to describe the epidemiologic features of RSV in 2009–2011 and the genetic variability of the G gene in circulating virus strains.
Materials and methods
Collection of specimens
Nasopharyngeal aspirates (NPAs) were obtained from children with symptoms of acute respiratory infections (ARIs) on three fixed days during the week after admission to the Children’s Hospital of Chongqing Medical University from April 2009 to March 2011. ARIs were diagnosed according to the criteria developed by the World Health Organization. The specimens were immediately added to tubes on ice containing 3 ml of cold viral transport medium (phosphate-buffered saline [PBS], 100 U/μl penicillin, 100 μg/ml streptomycin) and transported to the virology lab within 4–6 h. Upon arrival, the specimens were vortexed and centrifuged at 1,500×g for 10 min at 4 °C. The supernatants of the specimens were collected and stored at −70 °C, followed by RNA extraction. The whole protocol was approved by the Ethics and Research Council of Children’s Hospital of Chongqing Medical University, and informed consent was obtained from the patient’s parents or guardians.
RNA extraction and cDNA synthesis
Total RNA was extracted from 140 μl of the supernatants of the clinical specimens using an RNeasy Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Total RNA was reverse transcribed into cDNA using a PrimeScript RT kit (TaKaRa, Japan). The synthesis of cDNA was carried out for 15 min at 37 °C, followed by 5 s at 80 °C, and the cDNA was stored at −20 °C and subsequently used to amplify the G and F genes and determine their genotypes.
PCR amplification and sequencing of the F gene
The F gene was amplified using the primers F622For (5’-GTTACCTATTGTGAACAAGC-3’) and F1225Rev (5’-GCTGCTTACATCTGTTTTTG-3’) [11], which spanned the F gene from nucleotides 622 to 1225 and yielded a 603-bp fragment after amplification at 50 °C for 45 min, 95 °C for 15 min, 40 cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 1 min followed by a final step at 72 °C for 5 min. The PCR products were purified by agarose gel electrophoresis using a QIAquick Gel Extraction Kit (QIAGEN, Hilden, Germany) and then sequenced on an ABI 3100 sequencer (Applied Biosystems, Foster City, CA, USA).
Subgroup identification by PCR
Subgroup-specific products were amplified with 5’ primer P1, subgroup-A-specific 3’ primer P2, and subgroup-B-specific 3’ primer P3 [12]. Three μl of cDNA was added to a 23-μl PCR master mix containing 1.5 mM MgCl2, 200 μM deoxynucleoside triphosphates, 1.5 U of Taq DNA polymerase, and 50 pM each primer. Amplification was carried out at 94 °C for 2 min, followed by 35 cycles of 94 °C for 1 min, 55 °C for 1 min and 72 °C for 1 min, with a final extension for 5 min at 72 °C. The expected amplicon size for subgroup A and B was 250 and 341 bp, respectively. The PCR products were subjected to 1.5 % agarose gel electrophoresis, stained with GoldView nucleic acid stain (SBS Genetech), and visualized under UV light. The viruses detected were designated by city/group/year of isolation/isolate number.
PCR amplification of the C-terminal half of the G gene
A number of subgroup A and B RSV-positive samples were selected for amplification of the G gene by PCR. The primer GA480 (5’-ACAAACCACCAAACAAACCC-3’, corresponding to bases 480 to 499 of the G gene of strain A2) or GB496 (5’-GATGATTACCATTTTGAAGTGTTCA-3’, corresponding to bases 496 to 520 of the G gene of strain CH18537) and primer F164 (5’-GTTATGACACTGGTATACCAACC-3’, corresponding to bases 164 to 186 of the F gene of strain 18537, with one mismatch in the G gene of strain A2) were used. 3 μl of cDNA was added to a 22-μl PCR master mix containing 1.5 mM MgCl2, 200 μM deoxynucleoside triphosphates, 1.5 U of Taq DNA polymerase, and 50 pM each primer. Amplification was carried out at 94 °C for 3 min, followed by 35 cycles of 94 °C for 1 min, 57 °C for 1 min, and 72 °C for 1 min, with a final extension for 10 min at 72 °C. The PCR products were subjected to agarose gel electrophoresis and purified using a QIAquick Gel Extraction Kit (QIAGEN) according to the manufacturer’s instructions. The purified PCR products were sequenced directly.
Phylogenetic analysis
The nucleotide and amino acid sequences of subgroup A and B viruses were edited manually independently using the BioEdit program (version 7.0.1) and compared with the available human RSV sequences in the GenBank database using the ClustalX program (version 1.81) [13]. Phylogenetic trees were constructed by the neighbor-joining method using the MEGA (version 3.1) software [14]. The statistical significance of the tree topology was tested by bootstrapping (1,000 replicates).
Phylogenetic trees were constructed using the nine subgroup A viruses based on 270 nucleotides of the C-terminal variable region of the G gene, and these sequences were compared to the sequences of G gene of 25 reference strains representing all known subgroup A genotypes (eight genotypes) available in GenBank. These sequences included strain A2 and isolates from South Africa (SA), the United States (TX, MO, AL, NY), Kenya (Ken), Canada (CN), and elsewhere [4, 5, 15–19].
For subgroup B viruses, phylogenetic trees were constructed using the sequence of 16 subgroup A viruses in this study along with reference sequences of 31 strains representing all known genotypes by using 252 nucleotides from the C-terminal variable region of the G gene. The sequences were selected so that the representatives of each genotype of subgroup B viruses (nine genotypes) included strain CH18537 and isolates from South Africa; the United States (MO, NY, Rochester, CH, AL); Buenos Aires, Argentina (BA); Niigata, Japan (NG); Quebec, Canada (QUE); Belgium (BE); Montevideo, Uruguay (MON); Beijing; South Africa (SA); and elsewhere [4, 5, 15, 17, 18, 20, 21].
Results
Specimen collection and epidemiology of RSV
From April 2009 to March 2011, 921 specimens were collected from patients with ARIs, among which 210 (22.8 %) were found to be positive for RSV by RT-PCR. Strains of subgroups A and B were identified in 75 (35.7 %) and 134 (63.8 %) specimens, respectively, and viruses of both subgroups were found in one specimen (0.5 %).
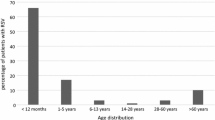
The majority of RSV-positive individuals were younger than 6 months of age and accounted for 50.0 % of RSV-positive samples. RSV-positive individuals aged 6–12 months, 1–2 years and 2–5 years accounted for 24.3 %, 12.9 % and 8 % of positive samples, respectively. The rates of RSV infection in the two consecutive epidemic seasons were 19.8 % and 28.0 %, respectively. The RSV outbreak activity in Chongqing was high in November, December, and January and reached a peak in December. The detection rate of RSV infection declined in April, and RSV infection was not found in May and July 2009. The predominant strain of RSV in the epidemic season 2009–2010 belonged to subgroup B, and the rate of subgroup B RSV infection was as high as 78.0 % (92/118) at this time. The rates for subgroup A and B RSV infection in the epidemic season 2010–2011 were 54.3 % and 45.7 %, respectively (Fig. 1).

A Seasonal distribution of subgroup A and B RSV in infants and children with acute lower respiratory tract infection in Chongqing, China, between April 2009 and March 2011. B Monthly distribution of genotypes of subgroup A and B RSV in Chongqing, China, between April 2009 and March 2011
Clinical symptoms were observed in all 210 patients with RSV infection. The major clinical symptoms included fever (n=67; 31.9 %), cough (n=198; 94.3 %), wheezing (n=150; 71.4 %), and cyanosis (n=156; 74.3 %). The RSV-infected patients were diagnosed with bronchiolitis (n=85; 40.5 %), bronchopneumonia (n=63; 30.0 %), interstitial pneumonia (n=47; 22.4 %), bronchialasthma exacerbation (n=24; 11.4 %), asthmatic bronchitis (n=5; 2.4 %) and bronchitis (n=3; 1.4 %).
Analysis of the F gene
A total of 45 subgroup A (n=30) and B (n=15) viruses were selected for amplification and sequencing of a fragment of the F gene. In a large proportion of subgroup A viruses (56.7 %, 17/30), asparagine (N) was changed to serine (S) at amino acid 276 of the F protein. However, this was not found in 15 subgroup B viruses. Of the patients infected with the suspected mutated virus, nine were males (52.9 %, 9/17). The majority (64.7 %) of these patients were younger than 6 months of age. Notably, none of these patients had a history of PZ treatment, indicating that the genetic alteration was not related to the use of PZ.
Phylogenetic analysis and genotyping
Sequences of the second variable region of the G genes of 9 subgroup A and 16 subgroup B strains obtained in this study were used for phylogenetic analysis. The results revealed that all nine subgroup A viruses were clustered into genotype GA2 (Fig. 2).

Phylogenetic trees of nucleotide sequences from the second variable region of the G gene of Chinese subgroup A (A) and subgroup B (B) RSV. Bootstrap values greater than 50 % are shown at the branch nodes. The value represents the bootstrap probability. The genotypes are indicated on the right by brackets. Reference sequences describing each genotype (genotypes GA1 to GA7, SAA1, GB1 to GB4, SAB1 and SAB3) were obtained from the GenBank database and included in the tree. Additional sequences from around the world were included in the comparison by selecting representatives of distinct clusters found in previous studies and selecting isolate sequences from GenBank that gave the best hits in BLAST searches with each of the Chinese clusters. Prototype strains (strain A2 for subgroup A and strain CH18537 for subgroup B) were used as the outgroup sequences
Between the prototype strain A2 and the Chinese subgroup A viruses, the rates of divergence were 10.6 % to 14.7 % at the nucleotide level and 17.5 to 23.6 % at the amino acid level. Differences at the nucleotide level and at the amino acid level were observed among the Chinese subgroup A viruses, which were 8.8 % and 5.7 %, respectively.
Two genotypes were identified among the 16 subgroup B strains: seven with genotype GB2 and nine with genotype BA (Fig. 2). Eight genotype BA strains were the same as the originally identified genotype, which has a 60-nucleotide duplication in the second variable region of the G gene [17, 22, 23]. Interestingly, the BA-like strain (Chongqing/B/10/11) had a 42-nucleotide duplication in the second variable region and an 18-nucleotide deletion in the middle of the 60-nucleotide duplication region. In addition, a six-nucleotide deletion after residue 489 was seen in five BA-like strains, resulting in a two-amino-acid deletion, which had also been identified in our previous study. The rates of divergence between the Chinese B strains and the prototypic genotype B strain CH18537 were 7.1–11.7 % at the nucleotide level and 12.5–20.3 % at the amino acid level. Up to 8.7 % nucleotide differences and up to 23.0 % amino acid differences were observed in the subgroup B strains. There were 2.4–5.7 % divergence at the nucleotide level and 4.7–14.9 % divergence at the amino acid level between the BA strains and prototypic genotype BA strain BA/4128/99B. The prototypic genotype B2 strain CH93-9B and the genotype B2 strains showed 2.7–3.7 % divergence at the nucleotide level and 8.9–9.2 % divergence at the amino acid level. The rate of divergence at the nucleotide level and at the amino acid level was 1.8 % and 5.9 %, respectively, between the genotype B2 strains and H5598 and H5601, which were the first GB2 strains detected in mainland China in December 2004.
Amino acid analysis
The predicted amino acid sequences of the subgroup A and subgroup B strains were compared with those of the prototype strains A2 and CH18537, respectively (Fig. 3). Eight out of nine Chinese subgroup A viruses had the same amino acids as the prototypic strain A2. A two-amino acid insertion was observed in the strain Chongqing/A/11/24, which led to the strain having 300 amino acids.

Deduced amino acid sequence alignments of the second variable region of the G gene of Chinese subgroup A (A) and B (B) RSV. Alignments relative to the sequences of prototype strain A2 and prototype B strain CH18537 are shown. The relative lengths (number of amino acids [aa]) of these regions are indicated in parentheses on the right
There were significant differences in the lengths of the deduced G protein amino acid sequences among the subgroup B viruses. The predicted complete G protein sequences of the Chinese subgroup B viruses had six different lengths (292, 294, 295, 306, 312 and 315 amino acids) (Fig. 3). Differences in G protein length were attributed to alternative termination codons and the presence of in-frame duplication, deletion, and insertion. Among the 16 subgroup B viruses, five were found to possess a G protein with 295 amino acids, five were found to have a G protein with 312 amino acids, and two were found to have a G protein with 294 and 315 amino acids, respectively. Only three viruses were found to have a G protein with 306 amino acids. Three different sizes of G protein (306, 312 and 315 amino acids) were detected in the nine genotype BA strains. With the exception of the Chongqing/B/10/11 strain, which had a G protein with 306 amino acids, the genotype BA strains had G proteins with 312 (n=5) or 315 (n=3) amino acids. In addition, a six-nucleotide deletion was found after residue 489 in five genotype BA strains, resulting a in two-amino acid deletion. The same deletion was observed in all seven genotype GB2 strains.
Nucleotide sequence accession numbers
The GenBank accession numbers of the nucleotide sequences obtained in the present study are JX885106 to JX885130.
Discussion
RSV is the most common pathogen causing lower respiratory tract infections in infants and young children [5]. The present study is an epidemiological description of RSV infections in the past two years. A total of 921 samples were collected from April 2009 to March 2011. In this study, 210 (22.8 %) were positive for RSV, which is consistent with the previous report from April 2006 to March 2009, in which 439 (31.7 %) were positive for RSV in 1,387 specimens from April 2006 to March 2009. In this study, most infected individuals were infants aged 0–6 months, accounting for 50.0 % of all samples, and individuals aged 6–12 months accounted for 24.3 %. There was no significant difference in age distribution, clinical manifestations, or seasonality of RSV infections in southwestern China compared to developed countries. The molecular epidemiology of RSV has largely been performed in developed countries [21, 24–27]; therefore, the present study, together with the previous one, shows the circulating patterns of RSV strains over the last five consecutive epidemic seasons, from April 2006 to March 2011, which may improve the understanding of RSV epidemiology in the countries with a large population.
A genetic alteration, asparagine (N) to serine (S) at position 276 in the F protein, has been observed recently. This study detected a lineage harboring an N276S mutation in 56.7 % of subgroup A RSV sequences from isolates collected from April 2009 to March 2011. A similar proportion of the same mutation was seen in subgroup A viruses in Canada, where the rates of the N276S mutation were 44.4 % during 2008–2009 and 100 % during 2009–2010 [28]. In the present study, of 30 subgroup A viruses detected, 17 were found to have N276S in the F protein, but the alteration was not completely the same in 15 subgroup B viruses. Adams et al. [29] have proposed that this mutation from asparagine (N) to serine (S) adjacent to the A epitope at position 276 in the F protein for subgroup A RSV could also confer partial resistance to PZ. In a subsequent study [8], it was reported that the variant N276S was detected in both subgroup A and B RSV. Moreover, variant N276S in subgroup A and B RSV did not confer resistance to PZ, and thus was a possible polymorphism [8]. At the same mutation site, more than half of subgroup A viruses showed the variant N276S. Interestingly, none of the infected individuals had a history of PZ administration. These findings suggest that this mutation is a spontaneous one and unlikely to be associated with PZ resistance.
Most of studies on the pattern of circulation of RSV have shown that subgroup A and B viruses co-circulate in the same area during epidemic periods and have various patterns of predominance [10, 15, 17, 19]. In a previous study in Chongqing, subgroup A viruses were predominant during two epidemic seasons (2006–2008), while subgroup B viruses prevailed during the subsequent 2008–2009 epidemic season. In the present study, subgroup B viruses predominated during the epidemic season 2009–2010. However, in the epidemic season 2010–2011, the rates for subgroup A and B viruses were 54.3 % and 45.7 %, respectively. The same pattern of RSV circulation was noted in a past epidemic season (2005–2006) in Beijing, China. In a community, a shift in the prevalence of an RSV group can occur at different time intervals, and different patterns of prevalence of subgroup A and B viruses have been described. Similar patterns of circulation of RSV groups have been reported in Niigata, Japan, where the subgroup B RSV was predominant in the 2002–2003 epidemic season, but there was a shift to subgroup A viruses in 2003–2007 [27]. However, in Kenya, Uruguay, and the United States, subgroup B RSV became predominant after two consecutive seasons, with a high prevalence of subgroup A infections [24, 29, 30]. The shift of RSV groups correlated in part with the variability of the G gene.
In the present study, genotype GA2 strains remained the dominant RSV genotype in two consecutive epidemic seasons, which is similar to what was observed in a previous study (2006–2009) in Chongqing. Thus, we propose that genotype GA2 strains were predominant in Chongqing in the last five consecutive epidemic seasons (2006–2011), which was consistent with the results in other geographic areas, including Beijing, China [17, 31–33]. These findings show that genotype GA2 strains were the most common genotype in subgroup A RSV.
In terms of subgroup B viruses, seven strains with genotype GB2 and nine strains with genotype BA were identified from April 2009 to March 2011. The high frequency of the BA strain in the subgroup B viruses was similar to findings in the previous study from April 2006 to March 2009 in Chongqing, in which 20 strains with genotype BA, 7 with genotype GB1 and 3 with genotype GB3 were identified in 30 subgroup B RSV strains.
The genotype BA strain, with a 60-nucleotide duplication in the G gene, was first identified in Argentina in 1999 [18]. In recent years, Shobugawa et al. [27] and Agrawal et al. [22] also identified 143 and 22 genotype BA strains in Japan and eastern India, respectively. In mainland China, the BA strain was first identified in Beijing in 2004. We identified genotype BA RSV in December 2007 in Chongqing, China [10]. Genotype BA of RSV rapidly became the dominant one in the 2008–2009 epidemic season. No studies were found that tracked this new subgroup B genotype in mainland China since then. In the present study, genotype BA RSV continued to account for the majority of subgroup B RSV, which was consistent with other studies conducted in other countries such as Argentina and India [17, 23], which showed that genotype BA RSV has been prevalent for at least 10 years since 1999. The predominance of genotype BA RSV at least in subgroup B may suggest a selective advantage of genotype BA RSV over RSV with other genotypes, but the mechanism is unknown. An antigenic change in the G protein and avoidance of the host immune response may be associated with the selective advantage [23]. It is possible that the additional residues significantly modify the antigenic characteristics of the G protein, allowing escape from neutralization by antibodies in humans as a result of previous infections. An additional possibility is that the duplication affects G-protein-mediated attachment of the viruses to host cells, thereby enhancing the fitness of the virus.
The predicted G proteins of the subgroup B RSV had six different lengths (292, 294, 295, 306, 312 and 315 amino acids). The mechanisms responsible for this variability include amino acid substitution, insertion, deletion, duplication, and change in stop codon usage. Six nucleotides were deleted after residue 489 in five out of nine strains with genotype BA and in all seven strains with genotype GB2, which resulted in a two-amino-acid deletion. Agrawal et al. [22] reported that six nucleotides were deleted after residue 489 for all 22 strains with genotype BA in the 2005–2006 epidemic season in eastern India. Zhang et al. [10] found the same nucleotide deletion in 15 out of 20 strains with genotype BA. The findings reinforce the notion of the transfer of viruses within close communities and reveal that this deletion exists all the time.
In conclusion, different groups of RSVs co-circulated in southwestern China from 2006 to 2011, probably in an alternating pattern. Genotype BA of RSV with a 60-nucleotide insertion and a few more variations in the duplication is the dominant genotype in the subgroup B RSV. These findings may improve our understanding of the molecular epidemiology of RSV in mainland China and aid in the further investigation of G protein function and vaccine development in the future.
References
Glezen WP, Taber LH, Frank AL, Kasel JA (1986) Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child 140:543–546
Cane PA (2007) Molecular epidemiology and evolution of RSV. In: Cane PA (ed) Perspectives in medical virology. Respiratorysyncytial virus. Elsevier, Salisbury, pp 89–113
Glezen P, Denny FW (1973) Epidemiology of acute lower respiratorydisease in children. N. Engl. J. Med 288:498–505
Johnson PR, Spriggs MK, Olmsted RA, Collins PL (1987) The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: extensive sequence divergence between antigenically related proteins. Proc. Natl. Acad. Sci. USA 84:5625–5629
Sullende WM (2000) Respiratory syncytial virus genetic and antigenic diversity. J. Clin. Microbiol Rev. 13:1–15
Botosso VF, Zanotto PM, Ueda M, Arruda E, Gilio AE, Vieira SE, Stewien KE, Peret TC, Jamal LF, Pardini MI, Pinho JR, Massad E, Sant’anna OA, Holmes EC, Durigon EL (2009) Positive selectionresults in frequent reversible amino acid replacements in the G protein geneof human respiratory syncytial virus. PLoS. Pathog 5:e1000254. doi:10.1371/journal.ppat.1000254
Zlateva KT, Lemey P, Vandamme AM, Ranst MV (2004) Molecular evolution and circulation patterns of human respiratory syncytial virus subgroup A: positively selected sites in the attachment G glycoprotein. J. Virol 78:4675–4683
Zhu Q, Patel NK, McAuliffe JM, Zhu W, Wachter L, McCarthy MP, Suzich JA (2012) Natural Polymorphisms and resistance-associated mutations in the fusion protein of respiratory syncytial virus (RSV): Effects on RSV Susceptibility to Palivizumab. J. Infect. Dis. 205:635–638
Arbiza J, Delfraro A, Frabasile S (2005) Molecular epidemiology of human respiratory syncytial virus in Uruguay: 1985–2001-a review. Mem. Inst. Oswaldo Cruz 100:221–230
Zhang Z, Du L, Chen X, Zhao Y, Liu E, Yang X, Zhao X (2010) Genetic variability of respiratory syncytial viruses (RSV) prevalent in Southwestern China from 2006 to 2009: emergence of subgroup B and A RSV as dominant strains. J Clin Microbiol 48:1201–1207
Zhao X, Sullender WM (2005) In vivo selection of respiratory syncytial viruses resistant to palivizumab. J. Virol 79:3962–3968
Zhu RN, Geng XH, Wang ZL (1998) Identification of subgroups of respiratory syncytial virus by reverse transcription polymerase chain reaction. Chin. J. Pediatr 36:538–540
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform 5:150–163
Kuroiwa Y, Nagai K, Okita L, Yui I, Kase T, Nakayama T, Tsutsumi H (2005) A phylogenetic study of human respiratory syncytial viruses group A and B strains isolated in two cities in Japan from 1980 to 2002. J. Med. Virol 76:241–247
Mufson MA, Orvell C, Rafnar B, Norrby E (1985) Two distinct subtypes of human respiratory syncytial virus. J. Gen. Virol 66:2111–2124
Parveen S, Sullender WM, Fowler K, Lefkowitz EJ, Kapoor SK, Broor S (2006) Genetic variability in the G protein gene of group A and B respiratory syncytial viruses from India. J. Clin. Microbiol 44:3055–3064
Trento A, Galiano M, Videla C, Carballal G, Garcia-Barreno B, Melero JA, Palomo C (2003) Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J. Gen. Virol 84:3115–3120
Venter M, Madhi SA, Tiemessen CT, Schoub BD (2001) Genetic diversity and molecular epidemiology of respiratory syncytial virus over four consecutive seasons in South Africa: identification of new subgroup A and B genotypes. J. Gen. Virol 82:2117–2124
Peret TC, Hall CB, Schnabel KC, Golub JA, Anderson LJ (1998) Circulation patterns of genetically distinct group A and B strains of human respiratory syncytial virus in a community. J. Gen. Virol 79:2221–2229
Zlateva KT, Lemey P, Moes E, Vandamme AM, Van Ranst M (2005) Genetic variability and molecular evolution of the human respiratory syncytial virus subgroup B attachment G protein. J. Virol 79:9157–9167
Agrawal AS, Sarkar M, Ghosh S, Chawla-Sarkar M, Chakraborty N, Basak M, Naik TN (2009) Prevalence of respiratory syncytial virus group B genotype BA-IV strains among children with acute respiratory tract infection in Kolkata, Eastern India. J. Clin. Virol 45:358–361
Trento A, Viegas M, Galiano M, Videla C, Carballal G, Mistchenko AS, Melero JA (2006) Natural history of human respiratory syncytial virus inferred from phylogenetic analysis of the attachment (G) glycoprotein with a 60-nucleotide duplication. J. Virol 80:975–984
Coggins WB, Lefkowitz EJ, Sullender WM (1998) Genetic variability among group A and group B respiratory syncytial viruses in a children’s hospital. J. Clin. Microbiol 36:3552–3557
Frabasile S, Delfraro A, Facal L, Videla C, Galiano M, Sierra MJ, Ruchansk D, Vitureira N, Berois M, Carballal G, Russi J, Arbiza J (2003) Antigenic and genetic variability of human respiratory syncytial viruses (group A) isolated in Uruguay and Argentina: 1993–2001. J. Med. Virol 71:305–312
Muelenaer PM, Henderson FW, Hemming VG, Walsh EE, Anderson LJ, Prince GA, Murphy BR (1991) Group-specific serumantibody responses in children with primary and recurrent respiratory syncytial virus infections. J. Infect. Dis 164:15–21
Shobugawa Y, Saito R, Sano Y, Zaraket H, Suzuki Y, Kumaki A, Dapat I, Oguma T, Yamaguchi M, Suzuki H (2009) Emerging genotypes of human respiratory syncytial virus subgroup A among patients in Japan. J. Clin. Microbiol 47:2475–2482
Papenburg J, Carbonneau J, Hamelin MÈ, Isabel S, Bouhy X, Ohoumanne N, Déry P, Paes BA, Corbeil J, Bergeron MG, Serres GD, Boivin G (2012) Molecular evolution of respiratory syncytial virus fusion gene, Canada, 2006–2010. Emerg. Infect. Dis. 18(1):120–124
Adams O, Bonzel L, Kovacevic A, Mayatepek E, Hoehn T, Vogel M (2010) Palivizumab-resistant human respiratory syncytial virus infection in infancy. Clin. Infect. Dis. 51:185–188
Scott PD, Ochola R, Ngama M, Okiro EA, Nokes DJ, Medley GF, Cane PA (2004) Molecular epidemiology of respiratory syncytial virus in Kilifi District, Kenya. J. Med. Virol 74:344–354
Ostlund MR, Lindell AT, Stenler S, Riedel HM, Wirgart BZ, Grillner L (2008) Molecular epidemiology and genetic variability of respiratory syncytial virus (RSV) in Stockholm, 2002–2003. J Med Virol 80:159–167
Rebuffo-Scheer C, Bose M, He J, Khaja S, Ulatowski M, Beck ET, Fan J, Kumar S, Nelson MI, Henrickson KJ (2011) Whole genome sequencing and evolutionary analysis of human respiratory syncytial virus A and B from Milwaukee, WI 1998–2010. PLoS One 6:e25468. doi:10.1371/journal.pone.0025468
Viegas M, Mistchenko AS (2005) Molecular epidemiology of human respiratory syncytial virus subgroup A over a six-year period (1999–2004) in Argentina. J Med Virol 77:302–310
Acknowledgments
We thank all of the families whose members enrolled in this study. We appreciate Qiang-lin Duan from Tongji Hospital for critical reading of the manuscript.
This work was supported by the National Natural Science Foundation of China (30730098).
Author information
Authors and Affiliations
Corresponding author
Additional information
X. Qin and C. Zhang contribute equally to the article
This work was supported by National Natural Science Foundation of China (30730098)
Rights and permissions
About this article
Cite this article
Qin, X., Zhang, C., Zhao, Y. et al. Genetic variability of subgroup A and B respiratory syncytial virus strains circulating in southwestern China from 2009 to 2011. Arch Virol 158, 1487–1495 (2013). https://doi.org/10.1007/s00705-012-1552-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-012-1552-z