
Abstract
We investigated the molecular epidemiology of respiratory syncytial virus (RSV) isolated from children during 28 consecutive seasons (1990-2018) and the genetic variability of the duplication region of RSV genotypes ON1 and BA in South Korea. RSV was identified using culture-based methods in Hep-2 cells and was grouped as RSV-A or RSV-B by an immunofluorescence assay. The second hypervariable region of the G gene was sequenced for genotyping. The nucleotide and deduced amino acid sequences of the duplication region of RSV ON1 and BA were analyzed. A total of 670 RSV-A and 233 RSV-B isolates were obtained. For RSV-A, the NA1 genotype predominated during the 2004/2005-2011/2012 seasons. The ON1 genotype was first detected in 2011 and has since replaced all other genotypes. For RSV-B, the GB3 genotype predominated during the 1999/2000-2005/2006 seasons, but the BA genotype also replaced all other genotypes of RSV-B after the first season in which it was isolated (2005/2006). In ON1 and BA genotype RSV strains, novel sequence types of the duplication region of the G gene were identified in 50-95% and 33-80% of the isolates, respectively, in each season. The ON1 and BA9 genotypes are responsible for the current epidemics of RSV infection in South Korea. The sequences in the duplication region of the G gene have evolved continuously and might be sufficient for the identification of specific strains of the RSV-A ON1 and RSV-B BA genotypes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Respiratory syncytial virus (RSV), a major cause of bronchiolitis and pneumonia in infants, is associated with substantial morbidity in children [1]. Additionally, in South Korea, RSV is the most common lower respiratory tract infection (LRTI)-related pathogen in hospitalized children younger than 5 years of age [2]. RSV usually circulates from October to March, with peak incidence in December in South Korea [2, 3]. This virus can infect young children in the presence of maternal antibodies, and recurrent infections occur throughout life because natural infection results in only partial protection [4, 5].
RSV is an enveloped single-stranded negative-sense RNA virus belonging to the species Human orthopneumovirus, genus Orthopneumovirus, family Pneumoviridae. The virions are pleomorphic and include spheres and long, fragile filaments [5, 6]. Two subgroups, namely, RSV-A and RSV-B, have been described on the basis of reactions with monoclonal antibodies. Extensive analyses of clinical isolates worldwide have demonstrated substantial antigenic and genetic variation both between and within these subgroups [7, 8].
Surface glycoprotein G is mainly involved in virus attachment to host cell receptors. The G protein is also a potent immunogen and a candidate vaccine target for RSV. Due to immunological pressure exerted on the G protein, its nucleotide and amino acid sequences have been shown to be the most divergent in the RSV genome [9, 10]. Therefore, RSV epidemics can be investigated by sequencing the G gene. Tracking the emergence and spread of new RSV genotypes is valuable for obtaining new insight into how RSV persistently causes recurrent infections and for providing future directions for the treatment and prevention of RSV infection [4, 5, 11].
However, previous Korean studies on the molecular epidemiology of RSV have reported sequences of isolates from a relatively short period and from a limited number of isolates [3, 12, 13]. Thus, in this study, we evaluated genotype changes in a large number of RSV-A and RSV-B isolates from children from 28 consecutive seasons in South Korea and investigated the genetic variability of the ON1 and BA genotypes, which are the dominant strains worldwide.
Materials and methods
Viral isolates
The viral isolates in this study consisted of all RSV isolates obtained from nasopharyngeal aspirates (NPA) of children < 18 years of age who were hospitalized or visited the emergency room (ER) with a diagnosis of LRTI in Seoul National University Children’s Hospital (SNUCH) between November 1990 and July 2018. All RSVs were identified using culture-based methods in Hep-2 cells and were subgrouped using an immunofluorescence assay with monoclonal antibodies as described previously [14]. Isolated RSV was kept frozen at -80 °C until use. The RSV data collected from 1990-1999 were retrieved from a previous study [15]. The handling and propagation method for RSV was identical throughout the study period, including 1990-1999. This study was approved by the Institutional Review Board of SNUCH (IRB registration number 1102-084-353). The Ethics Committee waived informed consent because this study included only information about the virus and the age of the patients from whom the viruses were obtained.
Amplification of the G gene
RSV-A isolates collected since the 1999/2000 season were selected for G gene analysis based on the following criteria: For seasons in which more than 15 viruses were isolated, all viruses from months in which no more than five viruses were isolated were included, but only half of the viruses were included from months in which more than five viruses were isolated. For seasons with no more than 15 isolates, all of the isolates were included in the analysis. RSV-B isolates were selected in the same manner as RSV-A isolates.
The frozen stocks of the selected RSV-A and RSV-B isolates were propagated in Hep-2 cells, and viral RNA was extracted when an extensive cytopathic effect was evident, using a QIAamp Viral RNA Mini Kit according to the manufacturer’s recommendations (QIAGEN, Valencia, USA). The extracted viral RNA was converted to cDNA, and PCR was performed with Taq DNA polymerase (Takara Bio Inc., Shiga, Japan). The amplification was performed for 35 cycles in a C1000 Touch Thermal Cycler (Bio-Rad, Foster City, USA). The complete G gene was amplified for both RSV-A and RSV-B using the same primers: A1F (5’-GGCAAATGCAAACATGTCCAAAA-3’) and F164 (5’-GTTATGACACTGGTATACCAACC-3’).
Nucleotide sequencing
A Wizard SV Gel and PCR Cleanup System was used for PCR product purification (Promega, Madison, USA). Cycle sequencing was performed using a BigDye Terminator 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, USA). The PCR primers (A1F and F164) were also used for sequencing the G gene for both RSV-A and RSV-B, and two additional primers (A482F [5’-ATGATTTTCACTTTGAAGTGTT-3’] and A650R [5’-TGGTTGTCTTGATGGTTGGTT-3’]) were used for RSV-A. Nucleotide sequencing was carried out on both strands, and primary editing was performed using Sequencher version 4.6 (Gene Codes Corporation, Ann Arbor, USA). When a double peak was observed without baseline noise, the cDNA was reamplified and resequenced. Then, the base that gave a higher peak on the chromatogram was finally called. If there were more than two mixed sequences with considerable baseline noise even after resequencing, the sequencing quality was regarded as low, and the ends, including this location, were trimmed off. No sequencing errors were present within the final sequenced region of the G gene for more than 10 RSV strains, which were checked with the original and resequenced cDNAs.
Further editing, alignment, and analysis were performed using CLC Main Workbench ver. 6.6.5 software (CLC bio, Aarhus, Denmark). Complete or partial sequences of the G gene were deposited in the GenBank database under accession numbers MK633970 to MK634300 for RSV-A and MK947219 to MK947359 for RSV-B.
Genotyping and phylogenetic analysis
The 264 nucleotides at the 3’ end (the second hypervariable region [HVR]) of the G gene of both RSV-A and RSV-B, except the ON1 (336 nucleotides) and BA (324 nucleotides) genotypes, are commonly used to determine the genotype and perform phylogenetic analysis of RSV [16]. The nucleotide and deduced amino acid sequences of the second HVR of all RSV-A and RSV-B strains in this study were compared to those of the prototype strains A2 (accession number JX198138) and CH18537 (accession number M17213), respectively. In addition, the sequences of the second HVR of RSV-A and RSV-B with genotype information were retrieved from GenBank and compared with the sequences of RSVs in this study for genotyping (Supplementary Table 1 and Supplementary Figs. 1 and 2). Furthermore, a comprehensive collection of the sequences of the second HVR of RSV-A ON1 and RSV-B BA strains identified worldwide during the 2009/2010-2012/2013 (Supplementary Table 2) and 2005/2006-2008/2009 (Supplementary Table 3) seasons, respectively, was obtained from GenBank to establish relationships between early Korean and international isolates. Phylogenetic analysis using the maximum-likelihood method and determination of the statistical significance of the tree topology by bootstrapping (1,000 replicates) were performed using CLC Main Workbench ver. 6.6.5 software.
Genotypes were assigned as described previously [17]. First, RSV-A with a 72-nt duplication and RSV-B with a 60-nt duplication in the second HVR were assigned to the well-known genotypes ON1 and BA, respectively. Sequences were then arbitrarily assigned to a genotype if they clustered together with bootstrap values > 70%. The criteria were further refined to include strains with a pairwise nucleotide distance (p-distance) < 0.07 to all other members in the same phylogenetic cluster. The p-distance is the number of pairwise nucleotide differences divided by the total number of nucleotides in the sequenced segment, and it was calculated using CLC Main Workbench ver. 6.6.5 software.
Sequence analysis of the ON1 and BA genotypes
To investigate the genetic variability of the ON1 and BA genotypes, the nucleotide and amino acid sequences of 144-nt and 120-nt duplication regions, respectively, were further analyzed. The original and duplicated regions were compared with each other within the individual strain, and the amino acid sequences in the entire duplication region of all ON1 and BA strains in this study were aligned and compared by season.
Results
Frequency of isolation of RSV groups and genotypes during the 1990/1991-2017/2018 seasons
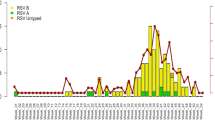
Of the 903 RSV isolates obtained from 28 consecutive seasons, 670 (74.2%) were RSV-A and 233 (25.8%) were RSV-B. As shown in Fig. 1, RSV-A was more frequently isolated than RSV-B, except in seven (25.0%) seasons. In the seasons when RSV-A was dominant, the median proportion of RSV-A isolates was 89.8% (range, 61.8-100%). When RSV-B was dominant, the median proportion of RSV-B isolates was 66.7% (range, 59.1-100%). Among all the RSV isolates, 355 (53.0%) RSV-A and 153 (65.7%) RSV-B isolates were sequenced for genotyping. Six genotypes of RSV-A were detected: GA2, GA5, GA7, NA1, NA2, and ON1 (Fig. 2a and Supplementary Table 4). Until the 2003/2004 season, GA2 or GA7 predominated, but during the 2004/2005-2011/2012 seasons, NA1 predominated. The ON1 genotype after the first detected in November 2011, replacing all genotypes of RSV-A since 2012/2013 season.

Number of respiratory syncytial virus (RSV)-A and RSV-B strains isolated during the study period

Proportions of genotypes of respiratory syncytial virus (RSV)-A (a) and RSV-B (b) in the study period. Among all the RSV isolates, 355 (53.0%) RSV-A and 153 (65.7%) RSV-B isolates were sequenced for genotyping
In total, 12 genotypes of RSV-B were detected: GB1, GB2, GB3, GB4, SAB2, SAB3, SAB4, URU1, URU2, BA7, BA9, and BA10 (Fig. 2b and Supplementary Table 5). Although a small number of RSV-B isolates were typed during the 1990/1991-1998/1999 seasons, three genotypes, namely, GB2 (n = 1), GB4 (n = 5), and SAB2 (n = 5), were identified. Between the 1999/2000 and 2005/2006 seasons, various genotypes were detected, but GB3 predominated, representing 44.2% of the total (n = 19). Similar to the ON1 genotype of RSV-A, the BA genotype replaced all other genotypes of RSV-B after the first season in which it was isolated (2005/2006). In particular, BA9 was exclusively identified in high numbers during the last three seasons (2015/2016-2017/2018).
Characterization of South Korean RSV-A ON1 genotype strains
Among the 355 RSV-A isolates sequenced, full-length G gene sequences (969 bp in ON1 and 897 bp in others) were obtained for 162 (45.6%) isolates. In the other 193 RSV-As, partial sequences (30-400 bp) of the 5’ region of the G gene were trimmed due to low sequencing quality. For the ON1 genotype, the G gene was sequenced fully in 38.8% (n = 40) and partially (759-919 bp) in the remainder (n = 63, 61.2%). We compared the partial sequences (870 bp, 89.8% of the whole sequence) of the G gene of most (n = 94, 91.3%) RSV-A ON1 strains in this study, which showed high 95.6-100% sequence identity at the nucleotide level and 91.0-100% at the amino acid level.
In the phylogenetic tree constructed for the second HVR with international RSV-A ON1 strains from the 2009/2010 to 2012/2013 seasons (Fig. 3a), most (n = 12, 83.3%) Korean RSV-A ON1 strains from 2011/2012 (n = 3) and 2012/2013 (n = 12) seasons are clustered together and separated from the earliest international strains (09/10 MEX and 10//11 CAD).

Phylogenetic tree of our respiratory syncytial virus (RSV)-A ON1 isolates during the 2009-2013 season (a) and RSV-B BA isolates during the 2005-2009 season (b) with international isolates from the same seasons. The dotted red circles indicate the grouping of most Korean RSV-A ON1 and RSV-B BA genotypes in this study. The tree was generated using the second hypervariable region of the G gene. Strains isolated in the same month of the year from the same country were not included. A total of 38 sequences from international RSV-A ON1 strains and 15 sequences from our strains were included (a); 28 sequences from international RSV-B BA strains and 19 sequences from our strains were included (b). Blue asterisks indicate our RSV-A ON1 and RSV-B BA strains from the first season in which they were detected. Isolates are labeled as ‘Isolated season_isolated country_genotype’ for international strains obtained from GenBank and as ‘KOR_strain name_genotype’ for our strains in this study. Abbreviations: RSV, respiratory syncytial virus; MEX, Mexico; CND, Canada; PAN, Panama; KOR, South Korea; JPN, Japan; TLD, Thailand; GMN, Germany; ITL, Italy; SAF, South Africa
We compared two duplicated 72-nt sequences and deduced 23-aa sequences within all individual RSV-A ON1 strains. None of the RSV-A ON1 strains in this study had an identical nucleotide sequence between the original and duplicated regions (data not shown). However, the original and duplicated sequences were identical in the first Korean RSV-A ON1 at amino acid level. This repeating unit of 23 aa, QEETLHSTTSEGYLSPSQVYTTS, exactly matched those of the earliest ON1 strains in 2009 from Mexico. This sequence type of the duplication region, arbitrarily named here as “11/12 (ON1) A01”, was identified in all subsequent seasons except 2015/2016, exhibiting particularly high incidence in the 2013/2014 (45.5%) and 2014/2015 (28.9%) seasons. In the 2012/2013 season and thereafter, amino acid substitutions occurred in the duplication region of the G gene, and the amino acid substitutions were asymmetric between the two duplicates. As a result, a total of 40 sequence types were identified in the duplication region of the G gene of the ON1 genotype from the 2011/2012 to 2017 2018 seasons. Among the sequence types identified in each season, 50-95% were novel types.
Characterization of South Korean RSV-B BA genotype strains
Among the 152 RSV-B isolates sequenced, 108 (71.1%) were of the BA genotype. A full G gene sequence (954 bp) was retrieved from 34 (31.5%) BA isolates, and partial sequences (875-915 bp) were retrieved from the remainder of the BA isolates. We compared the partial sequence of the G gene (870 bp, 97.0% of the whole sequence) of all BA genotype RSV-B strains in this study. The overall nucleotide and amino acid identity levels were over 93.5% and 88.3%, respectively.
The BA genotype was first detected in our study subjects in November 2005 as the BA9 genotype. In the phylogenetic tree constructed for the second HVR with international RSV-B BA strains from the 2005/2006 to 2008/2009 seasons (Fig. 3b), our isolates from the first detection season (2005/2006) were found to be closely related to those from Japan and Spain in the same season. Most Korean RSV-B BA strains in the following seasons clustered with our BA strains in the previous season.
The 60-nt (20-aa) duplication was not an exact replicate of the preceding original region in both nucleotide and amino acid sequences in all RSV-B BA strains in this study (Fig. 4b). There were 52 different amino acid sequences in the duplication region of the BA genotypes. None of these sequences were the same as that of the first identified BA strain from Argentina (BA/1370/99). The mutations also occurred asymmetrically between the two duplicates, as observed in the ON1 genotype.

Comparison of duplication region amino acid sequences in respiratory syncytial virus (RSV)-A genotype ON1 (a) and RSV-B genotype BA (b). The reference sequences at the top of the alignment of RSV-A and RSV-B were extracted from the prototype strains A2 (accession number JX198138) and BA/1370/99 (accession number DQ227364), respectively. Nucleotides that are identical to those in the corresponding position of the reference sequence are shown as a dot. Each line indicates the sequence types in the duplication regions. The sequence types are labeled as ‘First isolated season_genotype_arbitrarily given number_(number of strain).’ Other sequences are all nonidentical sequences detected during the corresponding season
In the 2008/2009 season and thereafter, the novel sequence types in the duplication region were identified in each season at a frequency of 33.3-100.0%. Sequence type 05/06 (BA10) 01 was detected during the longest time period from the 2005/2006 to 2014/2015 seasons, and the prevalent sequence type in the most recent years was sequence type 13/14 (BA09) 04: 2014/2015 (n = 1, 12.5%), 2015/2016 (n = 1, 6.7%), 2016/2017 (n = 6, 42.9%), and 2017/2018 (n = 18, 51.4%).
Discussion
In this study, we investigated genotypic variation in the second HVR of RSV-A and RSV-B over the past 28 seasons. Although different genotypes have cocirculated within most individual seasons, the RSV-A ON1 and RSV-B BA genotypes have completely replaced the previous types since 2013/2014 and 2006/2007, respectively. Korean RSV-A ON1 and RSV-B BA strains appear to have evolved within the domestic niche, and novel sequence types of the duplication region of the G gene of these strains were detected in the following season.
The G protein consists of three regions, including the cytoplasmic tail, transmembrane domain, and ectodomain. RSV-A and RSV-B are further characterized into several genotypes based on the antigenic and genetic variability of the second variable region in the C-terminal ectodomain of the G protein [7, 9]. To date, at least 14 genotypes of RSV-A have been identified, including GA1-7, SAA1-2, NA1-4, and ON1. For RSV-B, at least 24 genotypes have been identified, including GB1-4, SAB1-4, URU1-2, and BA1-14 [18, 19]. In this study, six RSV-A and eight RSV-B genotypes were detected.
Throughout the past 28 consecutive epidemic seasons, RSV A was dominant in three times as many seasons as RSV-B. The dominance of either RSV-A or RSV-B and the relative size of the epidemic of RSV infection in a particular season were not the same as those observed in other countries [18,19,20,21,22,23] but matched those reported for previous Korean studies [3, 13]. For RSV-A, a relatively large number of isolates (n > 50) was obtained in the 2001/2002, 2004/2005, 2011/2012, and 2014/2015 seasons. GA7, NA1, and ON1 were dominant genotypes in the 2001/2002, 2004/2005 and 2011/2012, and 2014/2015 seasons, respectively. Although the NA1 genotype was dominant from the 2004/2005 to 2011/2012 seasons, it mostly cocirculated with the GA5 genotype based on the data obtained in this study. Since the 2013/2014 season, the ON1 genotype was exclusively detected.
The RSV-A ON1 genotype was first detected in Canada in December 2010 and has since extended worldwide [24]. However, two additional early RSV-A ON1 viruses detected in Panama in November 2010 and in Mexico in November 2009 were reported later [21, 25]. It has been speculated that the ON1 genotype separated from the GA2/NA1 genotype between 2005 and 2009 [25]. In this study, we detected the first RSV-A ON1 virus in November 2011. This isolate is the second-earliest RSV-A ON1 strain detected in South Korea; the first strain was isolated in August 2011 by others [26]. Previous Korean studies based on nationwide surveillance of outpatients of all ages with acute respiratory illness showed that the ON1 genotype rapidly replaced previously circulating strains and became the predominant strain in the season following the one in which it was first isolated [12, 13]. The same replacement pattern was also shown for children who were hospitalized or visited an ER in this study: the proportion of RSV-A genotype ON1 increased from 11.8% in 2011/2012 to 91.7% in the next season. A similar pattern has been observed in other countries as well [21, 22].
The number of RSV-B isolates was relatively high in the 2005/2006 (n = 26) and 2017/2018 (n = 59) seasons in this study. In the 2005/2006 season, six different genotypes cocirculated, and the BA genotype first appeared in this season. In 1999, the RSV-B BA genotype was first detected in Buenos Aires, Argentina [27]. Since then, BA has become the predominant RSV-B genotype around the world [28]. In this study, BA was the exclusive genotype of RSV-B since the 2006/2007 season, which was the season immediately following that in which the BA genotype first appeared in South Korea. This phenomenon is the same as that for the change that occurred in South Africa: all previously identified RSV-B genotypes have been replaced by the BA genotype since 2006, when the first BA strain was recovered [23]. Together with these findings, the results show that the duplication in the G gene might confer survival and circulation advantages upon RSV strains, at least for the current populations. Other researchers have also suggested that genetic and antigenic variability in the G protein might enhance the fitness of RSV [20, 29, 30]. However, it is not certain whether the RSV strains with a duplication have higher virulence than the others [11, 31].
Analysis of the nucleotide sequences of the duplication regions in the G gene of the earliest ON1 strains from Korea and those from other countries showed that Korean RSV-A ON1 strains from the 2012/2013 season clustered together with Korean strains from the 2011/2012 season in the phylogenetic tree rather than with those from other countries. This suggests that Korean RSV-A ON1 strains have evolved in the domestic niche independently after their first introduction. Because the 72-nt duplication was not an exact replicate of the preceding 72 nucleotides in the first RSV-A ON1 strain in South Korea, the Korean ON1 strain might have been introduced from other countries rather than having developed domestically.
Additionally, Korean RSV-B BA strains from the 2006/2007-2008/2009 seasons clustered together with the previous Korean strains rather than with those from other countries in the phylogenetic tree, implying an independent domestic evolution of Korean RSV-B BA as well.
In this study, we focused on the duplication region to characterize the genetic variability of the RSV-A ON1 and RSV-B BA genotypes. Because these two genotypes arose by duplication, the two duplicates might have been identical in the first ON1 and BA strains. Over time, substitutions must have started to occur and accumulated in this region. Because the ON1 and BA genotypes appeared recently, the sequence variation in the duplication region might be less complex than that in other parts of the HVR of the G gene. Therefore, we assumed that this recently created short region is as good a surrogate for characterizing specific strains of the ON1 and BA genotypes as the second HVR or even the entire G gene. We observed high genetic variability in the duplication region of the G gene in both the RSV-A ON1 and RSV-B BA genotypes, and the novel amino acid sequences of the duplication region subsequently developed in the ensuing seasons.
This study has several limitations. First, because the viruses studied were obtained from children who were hospitalized or visited the ER at a single center, the epidemiology of the RSV genotypes might not be exactly representative of Korean strains, and these strains might be genetically different from and more virulent than RSV isolates that circulated in the community. The isolation and collection rate of viruses might have fluctuated during the long study period, even though we maintained almost the same procedure and personnel. The numbers of isolates in each season were also possibly influenced by more frequent hospitalization/ER visits and the lower threshold for viral study using rapid molecular diagnosis for children with LRTI in recent years than in the early study period. In addition, the RSVs studied were collected only by tissue culture. Therefore, the number of isolated viruses may not exactly represent the magnitude of the RSV epidemics in that season. Last, there is a validity issue regarding the sequencing of cultured RSV rather than RSV in clinical specimens directly, which may result in spontaneous mutation in the G gene. However, RSV seems to have a mutational rate of 10−5-10−6 per nucleotide per infected cell [32], and we used RSV strains from no later than passage 2. Therefore, the probability of amino acid substitutions or even nucleotide substitutions in the short region of interest (duplication region) in this study is very low.
In conclusion, the results of this study suggest that the RSV-A ON1 and RSV-B BA genotypes, including the 72- and 60-nt duplications, respectively, led to the current epidemics of RSV infection in children in South Korea. The duplication region of the G gene of the ON1 and BA genotypes evolved continuously, and this region alone might be sufficient for identification of specific strains of these genotypes. Understanding the actual effect of G gene duplication on fitness, virulence and transmissibility might allow changes in viral phenotype and immunogenicity to be predicted. It will also provide insight into the vaccine potential of the G protein.
Data availability
The datasets generated and/or analyzed in the current study are available from the corresponding author on reasonable request.
References
Hall CB, Weinberg GA, Iwane MK et al (2009) The burden of respiratory syncytial virus infection in young children. N Engl J Med 360:588–598. https://doi.org/10.1056/NEJMoa0804877
Choi EH, Lee HJ, Kim SJ et al (2006) The association of newly identified respiratory viruses with lower respiratory tract infections in Korean children, 2000–2005. Clin Infect Dis 43:585–592. https://doi.org/10.1086/506350
Baek YH, Choi EH, Song MS et al (2012) Prevalence and genetic characterization of respiratory syncytial virus (RSV) in hospitalized children in Korea. Arch Virol 157:1039–1050. https://doi.org/10.1007/s00705-012-1267-1
Tregoning JS, Schwarze J (2010) Respiratory viral infections in infants: causes, clinical symptoms, virology, and immunology. Clin Microbiol Rev 23:74–98. https://doi.org/10.1128/CMR.00032-09
Collins PL, Graham BS (2008) Viral and host factors in human respiratory syncytial virus pathogenesis. J Virol 82:2040–2055. https://doi.org/10.1128/JVI.01625-07
Rima B, Collins P, Easton A et al (2017) ICTV virus taxonomy profile: Pneumoviridae. J Gen Virol 98:2912–2913. https://doi.org/10.1099/jgv.0.000959
Sullender WM (2000) Respiratory syncytial virus genetic and antigenic diversity. Clin Microbiol Rev 13:1–15
Wertz GW, Moudy RM (2004) Antigenic and genetic variation in human respiratory syncytial virus. Pediatr Infect Dis J 23:S19–S24. https://doi.org/10.1097/01.inf.0000108189.87181.7c
Johnson PR, Spriggs MK, Olmsted RA, Collins PL (1987) The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: extensive sequence divergence between antigenically related proteins. Proc Natl Acad Sci USA 84:5625–5629
Cane PA, Pringle CR (1995) Evolution of subgroup A respiratory syncytial virus: evidence for progressive accumulation of amino acid changes in the attachment protein. J Virol 69:2918–2925
Anderson LJ, Peret TC, Piedra PA (2019) RSV strains and disease severity. J Infect Dis 219:514–516. https://doi.org/10.1093/infdis/jiy498
Kim YJ, Kim DW, Lee WJ et al (2014) Rapid replacement of human respiratory syncytial virus A with the ON1 genotype having 72 nucleotide duplication in G gene. Infection, genetics and evolution. Infect Genet Evol 26:103–112. https://doi.org/10.1016/j.meegid.2014.05.007
Park E, Park PH, Huh JW et al (2017) Molecular and clinical characterization of human respiratory syncytial virus in South Korea between 2009 and 2014. Epidemiol Infect 145:3226–3242. https://doi.org/10.1017/S0950268817002230
Park JY, Lee HJ (1999) Development of monoclonal antibodies to respiratory syncytial virus (RSV) and application for study of antigenic variation of RSV. Korean J Infect Dis 31:111–121
Choi EH, Lee HJ (2000) Genetic diversity and molecular epidemiology of the G protein of subgroups A and B of respiratory syncytial viruses isolated over 9 consecutive epidemics in Korea. J Infect Dis 181:1547–1556. https://doi.org/10.1086/315468
van Niekerk S, Venter M (2011) Replacement of previously circulating respiratory syncytial virus subtype B strains with the BA genotype in South Africa. J Virol 85:8789–8797. https://doi.org/10.1128/JVI.02623-10
Venter M, Madhi SA, Tiemessen CT, Schoub BD (2001) Genetic diversity and molecular epidemiology of respiratory syncytial virus over four consecutive seasons in South Africa: identification of new subgroup A and B genotypes. J Gen Virol 82:2117–2124. https://doi.org/10.1099/0022-1317-82-9-2117
Hu P, Zheng T, Chen J et al (2017) Alternate circulation and genetic variation of human respiratory syncytial virus genotypes in Chengdu, West China, 2009–2014. J Med Virol 89:32–40. https://doi.org/10.1002/jmv.24603
Gimferrer L, Vila J, Pinana M et al (2019) Virological surveillance of human respiratory syncytial virus A and B at a tertiary hospital in Catalonia (Spain) during five consecutive seasons (2013–2018). Future Microbiol 14:373–381. https://doi.org/10.2217/fmb-2018-0261
Okamoto M, Dapat CP, Sandagon AMD et al (2018) Molecular characterization of respiratory syncytial virus in children with repeated infections with subgroup B in the philippines. J Infect Dis 218:1045–1053. https://doi.org/10.1093/infdis/jiy256
Comas-Garcia A, Noyola DE, Cadena-Mota S et al (2018) Respiratory syncytial virus-A ON1 genotype emergence in Central Mexico in 2009 and evidence of multiple duplication events. J Infect Dis 217:1089–1098. https://doi.org/10.1093/infdis/jiy025
Luo HJ, Huang XB, Zhong HL et al (2020) Epidemiological characteristics and phylogenic analysis of human respiratory syncytial virus in patients with respiratory infections during 2011–2016 in southern China. Int J Infect Dis 90:5–17. https://doi.org/10.1016/j.ijid.2019.10.009
Pretorius MA, van Niekerk S, Tempia S et al (2013) Replacement and positive evolution of subtype A and B respiratory syncytial virus G-protein genotypes from 1997–2012 in South Africa. J Infect Dis 208:S227–S237. https://doi.org/10.1093/infdis/jit477
Eshaghi A, Duvvuri VR, Lai R et al (2012) Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS One 7:e32807. https://doi.org/10.1371/journal.pone.0032807
Duvvuri VR, Granados A, Rosenfeld P et al (2015) Genetic diversity and evolutionary insights of respiratory syncytial virus A ON1 genotype: global and local transmission dynamics. Sci Rep 5:14268. https://doi.org/10.1038/srep14268
Lee WJ, Kim YJ, Kim DW et al (2012) Complete genome sequence of human respiratory syncytial virus genotype A with a 72-nucleotide duplication in the attachment protein G gene. J Virol 86:13810–13811. https://doi.org/10.1128/JVI.02571-12
Trento A, Galiano M, Videla C et al (2003) Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J Gen Virol 84:3115–3120. https://doi.org/10.1099/vir.0.19357-0
Trento A, Viegas M, Galiano M et al (2006) Natural history of human respiratory syncytial virus inferred from phylogenetic analysis of the attachment (G) glycoprotein with a 60-nucleotide duplication. J Virol 80:975–984. https://doi.org/10.1128/JVI.80.2.975-984.2006
Hirano E, Kobayashi M, Tsukagoshi H et al (2014) Molecular evolution of human respiratory syncytial virus attachment glycoprotein (G) gene of new genotype ON1 and ancestor NA1. Infect Genet Evol 28:183–191. https://doi.org/10.1016/j.meegid.2014.09.030
Hotard AL, Laikhter E, Brooks K et al (2015) Functional analysis of the 60-nucleotide duplication in the respiratory syncytial virus Buenos Aires strain attachment glycoprotein. J Virol 89:8258–8266. https://doi.org/10.1128/JVI.01045-15
Streng A, Goettler D, Haerlein M et al (2019) Spread and clinical severity of respiratory syncytial virus A genotype ON1 in Germany, 2011–2017. BMC Infect Dis 19:613. https://doi.org/10.1186/s12879-019-4266-y
Sanjuan R, Nebot MR, Chirico N et al (2010) Viral mutation rates. J Virol 84:9733–9748. https://doi.org/10.1128/JVI.00694-10
Acknowledgements
We thank Sun Joong Kim and Sung Yeon Lee for their technical support. This work was supported by grant number 0420170430 (2017-1169) from the SNUH Research Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by grant number 0420170430 (2017-1169) from the SNUH Research Fund.
Conflict of interest
None of the authors have any competing interests to declare.
Additional information
Handling Editor: Bert K. Rima.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yun, K.W., Choi, E.H. & Lee, H.J. Molecular epidemiology of respiratory syncytial virus for 28 consecutive seasons (1990-2018) and genetic variability of the duplication region in the G gene of genotypes ON1 and BA in South Korea. Arch Virol 165, 1069–1077 (2020). https://doi.org/10.1007/s00705-020-04580-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04580-z