
Abstract
Bovine leukaemia virus (BLV) is an oncogenic member of the genus Deltaretrovirus of the family Retroviridae. Recent studies revealed that BLV strains can be classified into six different genotypes and raised the possibility that another genotype may exist. In order to gain insight into the degree of genetic variability of BLV strains circulating in the South American region, a phylogenetic analysis was performed using gp51 env gene sequences. The results of these studies revealed the presence of seven BLV genotypes in this geographic region and the suitability of partial gp51 env gene sequences for phylogenetic inference. A significant number of amino acid substitutions found in BLV strains isolated in South America map to the second neutralization domain of gp51. A 3D molecular model of BLV gp51 revealed that these substitutions are located on the surface of the molecule. This may provide a selective advantage to overcome immune host neutralization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
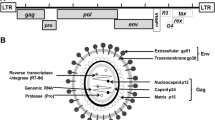
Bovine leukaemia virus (BLV) is an oncogenic member of the genus Deltaretrovirus of the family Retroviridae. BLV is the causative agent of enzootic bovine leukaemia [18] and infects cattle worldwide, imposing a severe economic impact on the dairy cattle industry. Most of the infected animals (60%) never show haematological signs of infection and become asymptomatic aleukemic (AL) carriers of the virus. Roughly one-third of the infected cattle show persistent lymphocytosis (PL) characterized by a non-malignant polyclonal expansion of CD5+ B-cells. Only 5–10% of the infected animals develop malignant monoclonal B-cell lymphosarcoma (LS) [5, 19, 23]. As in all members of the family Retroviridae, BLV envelope (env) glycoproteins play a crucial role in determining viral infectivity and syncytium formation [8, 28], since they probably contain the receptor-binding domain (RBD), which interacts with the cell-surface receptor required for virus entry [16]. The BLV envelope gene codes for a polyprotein precursor (gp72), which is cleaved into signal peptide and gp51 surface (SU) and gp30 transmembrane (TM) glycoproteins [24].
The localization of env glycoproteins at the surface of viral particles determines that they are the natural target of specific neutralizing antibodies. After BLV infection, the typical immunological response involves a strong and permanent production of antibodies directed against env glycoprotein. However, it has been reported in several cases that cattle were persistently infected with BLV without displaying a detectable antibody response [11].
Early studies on the genetic variability of the BLV env gene had shown very little variation among isolates [9, 28]. This low genetic diversity might be due to the relative low substitution rate of the virus [33]. However, studies based on restriction fragment length polymorphism analysis (RFLP) allowed the classification of BLV strains in up to seven different genotypes, suggesting that BLV is more diverse than previously anticipated [2, 11, 21]. The analysis of BLV gp51 env gene sequences from different geographic regions revealed the presence of different genetic groups that correlated with the geographic origin of the isolates [4, 6, 7, 12, 24–26, 34]. Based on analysis of the full-length BLV env gene, a new classification of BLV strains into seven genotypes has been proposed recently [29].
There are few studies on the genetic variability and heterogeneity of BLV in the South American region. In a recent study carried out in Chile by using partial BLV env sequences, the presence of a specific genetic group composed by strains isolated in that country was proposed [12]. Evolutionary studies done in Brazil suggested that Brazilian BLV strains belong to at least two different phylogenetic clusters [7]. Similar studies done in Argentina revealed the presence of two types of BLV strains in dairy herds, denoted Australian and Argentine [26]. Very recent studies done in Argentina revealed that at least four genotypes circulate in that country [29].
New gp51 env gene sequence data have been obtained recently from BLV field isolates from Argentina, Brazil, Chile and Uruguay. In order to gain insight into the degree of genetic variability of BLV in the South American region, phylogenetic analysis was performed. The results of these studies revealed the presence of seven BLV genotypes in this region of the world and the suitability of partial gp51 env gene sequences for phylogenetic inference.
Materials and methods
Sampling and BLV antibody detection
Milk and blood samples were collected from farms located in the northern and southern regions of Uruguay. Somatic cells, sera, and peripheral blood mononuclear cells (PBMCs) were obtained from these samples. All serum samples (n = 456) were analyzed by using an enzyme-linked immunosorbent assay (ELISA) test (VMRD Inc., Pullman, WA, USA) designed to detect bovine antibodies against viral gp51 env glycoprotein.
DNA extraction
Genomic DNA was extracted from PBMC and milk somatic cells from BLV-infected dairy cattle by using a QIAmp DNA Blood Mini kit from QIAGEN.
BLV env gp51 PCR amplification
PCR amplification of the full-length BLV gp51 env gene (903 bp) was performed by using the following primers: forward, 5′-ATG CCY AAA GAA CGA CGG-3′; and reverse, 5′-CGA CGG GAC TAG GTC TGA CCC-3′. The final reaction mixture contained 20 mM Tris HCl, pH 8.4, 50 mM KCl, 1.5 mM MgCl2, 200 μM dNTPs, 200 nM of each primer, and 1 U Taq polymerase (Invitrogen, USA). Conditions for PCR amplification were as follows: 95°C for 4 min, 30 cycles of denaturation at 95°C for 60 s, annealing at 55°C for 60 s, and extension at 72°C for 60 s, followed by a final extension at 72°C for 10 min. Partial BLV gp51 env gene fragments (413 and 311 bp) were amplified according to the nested PCR developed by Ballagi-Pordány et al. [3].
BLV DNA cloning and sequencing
Amplicons were resolved by 2% agarose gel electrophoresis, purified using a QIAquick PCR Purification Kit from QIAGEN, and cloned into pGEM-T Easy Vector (Promega, USA). Escherichia coli XL1-Blue cells were transformed by electroporation (BTX Electroporator, Genetronics Biomedical Ltd). Positive colonies were expanded, and small-scale plasmid purification was performed by using a GFX DNA purification kit (GE Healthcare, Piscataway, NJ, USA). Both strands of the purified plasmids were sequenced using universal T7 or SP6 primers and a Big Dye DNA sequencing kit (Perkin-Elmer) on a 373 DNA Sequencer (Perkin-Elmer).
Phylogenetic analysis
Sequences were aligned using the CLUSTAL W program [32]. Once aligned, the program Model Generator [17] was used to identify the optimal evolutionary model that described our sequence dataset. Akaike information criteria and hierarchical likelihood ratio test indicated that the HKY + Γ model best fit the sequence data.
Maximum-likelihood phylogenetic trees were constructed under the HKY + Γ model using software from the PhyML program [15]. As a measure of the robustness of each node, we used an approximate Likelihood Ratio Test (aLRT), which demonstrates that the branch studied provides a significant likelihood against the null hypothesis that involves collapsing that branch of the phylogenetic tree but leaving the rest of the tree topology identical [1]. aLRT was calculated using three different approaches: (a) minimum of Chi square-based calculations; (b) a Shimodaira-Hasegawa-like procedure (SH-like) [30, 31], which is non-parametric, and (c) a combination of both (SH-like and the minimum Chi square-based calculations), which is the most conservative option for these calculations. In addition, the bootstrap method was also used.
3D structural homology modeling of BLV gp51
A 3D structural homology model of the BLVgp51 RBD was produced using MODELLER version 9v6 [27]. As a template, we used the closest crystallographic structure (PDB ID: 1AOL_A, identity 12.3%), corresponding to the RBD of the env glycoprotein of Friend murine leukemia virus (F-MuLV-RBD) [10]. The sequence containing the putative BLVgp51-RBD (amino acid positions 1–152) was first automatically aligned with F-MuLV-RBD by using Clustal W [32] and then corrected manually to remove potential alignment errors by comparison with the sequence alignment of related proteins (ENV polyprotein domain) found in the Pfam database [13]. Homology modeling was performed by running an automated comparative modeling routine, and the best model generated was chosen on the basis of the lowest discrete optimized protein energy (DOPE potential). To improve accuracy, a second optimization round was performed using the appropriate MODELLER scripts by manual inspection/correction of secondary structure elements on the basis of observed structure-sequence relationships between Fr-MLV and BLV RBDs. Five models were obtained, and the one with the lowest DOPE score was selected. Visualization, manual correction and figure preparation were done with PyMol (http://www.pymol.org).
Nucleotide sequence accession numbers
The sequences reported in this work have been submitted to the EMBL Database. For names, accession numbers and geographic location of all sequences involved in this study, see Supplementary Material Table 1.
Results
Phylogenetic analysis of BLV strains
In order to gain insight into the degree of genetic variability of BLV strains isolated in the South American region, we first obtained full-length gp51 env gene sequences from 8 BLV field strains from Uruguay. These sequences were aligned with 31 corresponding sequences of strains isolated in Argentina and Brazil, as well as 37 sequences from BLV strains isolated elsewhere, representing all BLV genotypes.
Once aligned, maximum-likelihood phylogenetic trees were constructed under the HKY + Γ model. The results of these studies are shown in Fig. 1.

Maximum-likelihood phylogenetic tree analysis of BLV strains using full-length gp51 env gene sequences. Strains in the tree are shown by accession numbers, and the country of isolation is shown in parentheses. Genotypes are indicated by numbers on the right of the figure according to Rodriguez et al. [29]. BLV strains isolated in South America are shown in italics. Numbers at the branches show aLRT values using an SH-like calculation according to Anisimova and Gascuel [1], as implemented in the PhyML program [15]. For results found for aLRT using Chi square-based, a combination of minimum of SH-like and Chi square-based or bootstrap calculations, see Supplementary Material Fig. 1. The bar at the bottom of the figure denotes distance
All strains in the tree were assigned to seven clusters, representing the seven genotypes described recently [29]. All clusters were supported by very high aLRT values (see Fig. 1). All Uruguayan isolates were clustered together into genotype 1 (see Fig. 1, middle). Strains isolated in Argentina were assigned to genotypes 1, 2, 4 and 6. Interestingly, genotype 2 is only one represented by strains isolated in that country (see Fig. 1, bottom). Strains isolated in Brazil were assigned to genotypes 1 and 6. This former genotype is composed only of strains isolated in Argentina and Brazil (see Fig. 1, bottom). The results of these studies suggest the diversification of BLV strains in four different genotypes.
A significant number of partial gp51 env gene sequences have been obtained recently from BLV field strains from Argentina, Brazil and Chile [7, 12, 26]. In this work, we have characterized 42 partial gp51 env gene sequences from BLV field isolates from Uruguay. In order to gain insight into the degree of genetic variability of BLV in that region, as well as to establish the suitability of partial gp51 env gene sequences for establishing phylogenetic relations among BLV strains, the same studies were repeated using 123 sequences from strains isolated in South America (for 33 of them, their genotype had been established previously using full-length gp51 env gene sequences) and 24 sequences from BLV strains isolated elsewhere, representing all BLV genotypes, for which their genotype had also been established previously [29]. The results of these studies are shown in Fig. 2.

Maximum-likelihood phylogenetic tree analysis of BLV strains using partial gp51 env sequences. Strains in the tree are shown by accession numbers for strains reported previously and for whom their genotype was previously established. Uruguayan strains are shown by name. For accession numbers, see Supplementary Material Table 1. Numbers at the branches show aLRT values using an SH-like calculation according to Anisimova and Gascuel [1], as implemented in the PhyML program [15]. For results found for aLRT using Chi square-based or a combination of minimum SH-like and Chi square-based calculations, see Supplementary Material Fig. 2. The rest is the same as in Fig. 1
Again, all strains included in these studies were assigned to seven genetic lineages, corresponding to the seven genotypes established recently for BLV [29]. All of these clusters were supported by very high aLRT values (see Fig. 2). All BLV strains isolated in Uruguay were assigned to genotype 1, in agreement with previous results using full-length gp51 env sequences (compare Figs. 1, 2).
A different situation was found in Argentina, where at least three different genotypes co-circulate (1, 2 and 4). All genotype 2 strains included in these studies were found to be BLV strains isolated in Argentina (see Fig. 2, middle). This is also in agreement with the results found using full-length gp51 env sequences (compare Figs. 1, 2).
Interestingly, strain S83530, isolated in Italy, the only strain defining a proposed BLV genotype 7, shares a well-supported cluster with BLV strains isolated in Chile and Brazil (see Fig. 2, bottom). This finding suggests that this genotype indeed exists and is present in the South American region.
At least four different genotypes (1, 5, 6 and 7) circulate in Brazil (see Fig. 2). This result is in agreement with previous studies on BLV genetic variability carried out in Brazil [6, 7].
In addition, two different genotypes were found in Chile (4 and 7). Genotype 4 Chilean strains seem to have a close genetic relationship among themselves and a more comparatively distant relationship with other genotype 4 strains. This may relate to the observed specific genetic group composed of strains isolated in Chile that was proposed by Felmer et al. [12].
Taking all these results together, we can detect the presence of all seven BLV genotypes in the South American region.
Amino acid substitutions in gp51 env protein of BLV strains isolated in South America
To get insight into the amino acid changes observed in BLV strains isolated in South America, partial gp51 env sequences from strains isolated in that region were aligned using the Clustal W program [32] and translated to amino acids sequences using the MEGA 4 program [20]. The results of these studies are shown in Fig. 3.

Alignment of BLV gp51 env amino acid sequences from strains isolated in South America. Strains are indicated by accession number at the left side of the figure, followed by country of isolation. Amino acid sequences are indicated by the one-letter code. Identity to reference strain K02120, isolated in Japan, is indicated by a dash. Positions of CD4+ and CD8+ T cell epitopes, first and second neutralization domains (ND), E linear epitope, G conformational epitope, and a Zn-binding peptide, according to Zhao and Buehring [34], are indicated at the top of the sequence alignment. Underlined residues correspond to D 134, N 141 and F 146
Interestingly, amino acid substitutions in the gp51 protein of BLV strains isolated in South America are not scattered throughout the protein but mostly located in the second neutralization domain of the gp51 protein (see Fig. 3). One significant substitution can be observed in some BLV strains isolated in Uruguay and Brazil at position 134 of the gp51 protein, where aspartic acid (D) is substituted by asparagine (N) (see Fig. 3). These strains also share a substitution of phenylalanine (F) by serine (S) at position 146. Strains isolated in Argentina and some of the BLV strains isolated in Brazil share a substitution of N by D at position 141 of the gp51 protein (see Fig. 3).
Tridimensional model of gp51 env RBD
The amino acid changes in the gp51 protein of BLV strains isolated in South America, along with their remarkable clustering within a well-documented key region for virus fusogenic properties and immune recognition [8], prompted us to generate a three-dimensional model of the protein and thus achieve a more detailed mapping and assessment of the potential impact of these mutations. To date, structural information about a retroviral-encoded envelope glycoprotein is only available for the N-terminal RBD of Friend murine leukemia virus (Fr-MLV). The overall fold of RBD is hallmarked by an antiparallel β-sandwich core, with variable interstrand loops and helical regions (named VRA, VRB, and VRC), which determines the specific viral tropism. However, the lack of significant sequence identity between the proteins of BLV and Fr-MLV has hampered the reliability of models of gp51 RBD. To generate a 3D model of BLV-gp51, we followed a structural modelling approach relying on the reasoning that despite low identity, both proteins should share an overall common fold and polypeptide connectivity. We performed homology modelling with the program MODELLER using a structure-based alignment with Fr-MLV RBD restricted with a multiple sequence alignment of retroviral RBD sequences existing in databases (described in detail in “Material and methods”). Our model exhibits the typical RBD fold, with an antiparallel β-sheet core and loop-helix segments similar to variable regions VRA, VRB, and VRC (Fig. 4). A notable difference arises from the fact that the length of BLV RBD is shorter than that of Fr-MLV, resulting in a predicted small VRA-like segment. All of the natural amino acid substitutions found in the gp51 env protein of BLV South American isolates are exposed on the surface of the second neutralizing epitope, a location that suggests the possibility of directly facing neutralizing antibodies. This implies that differential immune recognition of BLV strains from different geographic locations may occur.

3D model of BLV gp51 env protein. a Three-dimensional model of the RBD of BLV-gp51. A cartoon representation is depicted, generated on the basis of sequence similarity with the X-ray structure of F-MLV RBD as template (see “Material and methods”). Positions where amino acid substitutions were found in BLV strains isolated in Uruguay are labelled, and their detailed location is shown as a dotted space-filling representation. b Predicted accessible surface of the putative BLV gp51-RBD. Mutations indicated in a are shown in shades of blue to highlight their exposure to solvent. Both residues are spatially contained in a continuous patch over the RBD accessible surface, the extension of which is indicated in yellow in previously identified second neutralizing domain (2 ND)
Discussion
It has been generally assumed that BLV exhibits high genetic stability. This low degree of genetic variability may be explained by the fact that BLV replicates its proviral DNA mainly by mitotic replication of infected B-cells, with a minor involvement of reverse transcription.
Earlier studies on the amino acid composition of the BLV gp51 env sequence showed highly conserved regions in the BLV env protein of different isolates [9, 24, 28]. However, more recent studies revealed that the degree of genetic variation of BLV was higher than previously anticipated, permitting different virus groups to be observed [12, 22, 29, 34]. Based on the analysis of the full-length BLV env gene, a new classification of BLV strains in seven genotypes has recently been proposed [29]. The results of these studies revealed the presence of seven BLV genetic groups circulating in the South American region (see Fig. 2), in agreement with recent results proposing a new classification of BLV strains in seven genotypes [29]. The presence of more than a single genotype of BLV in this geographic area of the world may be explained by modern cattle trading tendencies, as proposed previously [7, 34].
Due to the fact that the gp51 env gene codes for the surface envelope protein of BLV, which is subjected to immune pressures and selection processes [28], this gene has been found to be appropriate for phylogenetic analysis [34]. Interestingly, comparable phylogenetic relationships were obtained using full-length or partial gp51 env gene sequences, revealing the suitability of these partial sequences for rapid assignment and establishing phylogenetic relationships among BLV strains (compare Figs. 1, 2).
Different epidemiological situations have been found in different countries of South America (Fig. 1b). While BLV strains isolated in Uruguay belong to genotype 1, at least four different genotypes circulate in Brazil (1, 5, 6 and 7), three circulate in Argentina (1, 2 and 4) and two in Chile (4 and 7). No BLV strains included in these studies were assigned to genotype 3 (see Figs. 1, 2). Interestingly, all genotype 2 strains identified in these studies were isolated in Argentina (see Figs. 1, 2). This reveals a specific geographic cluster of BLV strains.
In the case of Chile, although some strains isolated in that country can be assigned to genotype 4, some Chilean strains have a close genetic relationship to BLV strain S83530, which was previously reported to be the only member of putative genotype 7 [29] (see Fig. 2, bottom). This supports the possibility that this genotype exists and it is present in the South American region.
An important number of substitutions found in BLV strains isolated in South America map to the second neutralization domain of gp51, in agreement with recent evolutionary analyses [29, 34] (see also Fig. 3). It has been reported that this domain could be involved in the interaction of gp51 with the receptor expressed on host cell membranes, a region that could affect viral fusion and infectivity in vivo [8, 14]. In particular, two amino acid changes at position 134 (D–N) and 146 (F–S), were shared between a Brazilian strain and most of the Uruguayan strains (see Fig. 3).
To gain further insight about the location of these mutations, we have generated a molecular model for the putative BLV gp51 RBD by using the crystallographic structure of the Fr-MLV RBD (PDB: 1AOL) as a template (Fig. 4). As shown in the model, the amino acid substitutions found in the second neutralization domain of the gp51 env protein of BLV strains isolated in the South American region are located on the surface of the molecule. In particular, the amino acid change at position 134 (D to N) changes the net charge of a loop (see Fig. 4).
Several immunoassays designed for the detection of anti-BLV antibodies use gp51 as antigen. However, it has been reported that several animals were positive by PCR but negative by serological analyses [26]. In particular, Fechner et al. [11] observed that some BLV variants were associated with the failure of antibody detection by standard serological methods and proposed that sequence variability of BLV strains may result in antigenic differences that could affect viral serological identification in infected cattle populations from different geographic areas. However, other studies reported that there is no correlation between the BLV genotype and the serological status of infected animals, proposing that the difference in the antiviral immune response may be related to the infection stage and other host factors [21]. More studies will be needed in order to address these issues.
The results of these studies revealed a higher degree of genetic variability than previously expected, with seven different BLV genotypes circulating in the South American region. This will be important for implementing appropriate cattle management policies and the development of appropriate anti-viral strategies and vaccines suitable for the South American region.
References
Anisimova M, Gascuel O (2006) Approximate likelihood ratio test for branchs: a fast, accurate and powerful alternative. Syst Biol 55:539–552
Asfaw Y, Tsuduku S, Konishi M, Murakami K, Tsuboi T, Wu D, Sentsui H (2005) Distribution and superinfection of bovine leukemia virus genotypes in Japan. Arch Virol 150:493–505
Ballagi-Pordány A, Klintevall K, Merza M, Klingeborn B, Belák S (1992) Direct detection of bovine leukemia virus infection: practical applicability of a double polymerase chain reaction. Zentralbl Veterinarmed B 39:69–77
Beier D, Blankestein P, Marquardt O, Kuzmak J (2001) Identification of different BLV proviruses isolates by PCR, RFLP, and DNA sequencing. Berl Munch Tieraztl Wschr 114:252–256
Burny A, Cleuter Y, Kettman R, Mammerickx M, Marbaix G, Portelle D, Vanden Broeke A, Willems L, Thomas R (1998) Bovine leukaemia: facts and hypotheses derived from the study of an infectious cancer. Vet Microbiol 17:197–218
Camargos MF, Stancek D, Rocha MA, Lessa LM, Reis JK, Leite RC (2002) Partial sequencing of env gene of bovine leukaemia virus from Brazilian samples and phylogenetic analysis. J Vet Med B Infect Dis Vet Public Health 49:325–331
Camargos MF, Pereda A, Stancek D, Rocha MA, Reis JK, Greiser-Wilke I, Leite RC (2007) Molecular characterization of the env gene from Brazilian field isolates of bovine leukaemia virus. Virus Genes 34:343–350
Callebaut I, Voneche V, Mager A, Fumiere O, Krchnak V, Merza M, Zavada J, Mammerickx M, Burny A, Portetelle D (1993) Mapping of B neutralizing and T-helper epitopes on the bovine leukaemia virus external glycoprotein gp51. J Virol 67:5321–5327
Coulson J, Naif H, Brandon R, Kumar S, Khan S, Daniel R, Lavin M (1990) Molecular cloning and sequencing of an Australian isolate of proviral bovine leukaemia virus DNA: comparison with other isolates. J Gen Virol 71:1737–1746
Fass D, Davey RA, Hamson CA, Kim PS, Cunningham JM, Berger JM (1997) Structure of a murine leukemia virus receptor-binding glycoprotein at 2.0 angstrom resolution. Science 277:1662–1666
Fechner H, Blankenstein P, Looman AC, Elwert J, Geue L, Albrecht C, Kurg A, Beier D, Marquardt O, Ebner D (1997) Provirus variants of the bovine leukemia virus and their relation to the serological status of naturally infected cattle. Virology 237:261–269
Felmer R, Muñoz G, Zuñiga J, Recabal M (2005) Molecular análisis of 444 bp fragment of the bovine leukaemia virus gp 51 env gene reveals a high frequency of non-silent point mutations and suggests the presence of two subgroups of BLV in Chile. Vet Microb 108:39–47
Finn RD, Mistry J, Schuster-Böckler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, Eddy SR, Sonnhammer EL, Bateman A (2006) Pfam: clans, web tools and services. Nucleic Acids Res 34:D247–D251
Gatot JS, Callebaut I, Van Lint C, Demonte D, Kerkhofs P, Portetelle D, Burny A, Willems L, Kettman R (2002) Bovine leukaemia Virus SU protein interacts with Zinc, and mutations within two interacting regions differently affect viral fusion and infectivity in vivo. J Virol 76:7956–7967
Guindon S, Lethiec F, Duroux P, Gascuel O (2005) PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res 33:W557–W559
Johnston ER, Albritton LM, Radke K (2002) Envelope proteins containing single amino acid substitutions support a structural model of the receptor-binding domain of bovine leukaemia virus surface protein. J Virol 76:10861–10872
Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McInerney JO (2006) Assessment of methods of amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol 6:29
Kettmann R, Portetelle D, Mammerickx M, Cleuter Y, Dekegel D, Galoux M, Ghysdael J, Burny A, Chantrenne H (1976) Bovine leukemia virus: an exogenous RNA oncogenic virus. Proc Natl Acad Sci USA 73:1014–1018
Kettmann R, Burny A, Callebaut I, Droogmans L, Mammerick M, Willens L, Portetelle D (1994) Bovine leukaemia virus. In: Levy JA (ed) The retroviridae. Plenum Press, New York, pp 39–81
Kumar S, Tamura K, Nei M (2007) MEGA 4: integrated software for Molecular Evolutionary Genetic Analysis and sequence alignment. Brief Bioinf 5:150–163
Licursi M, Inoshima Y, Wu D, Yokoyama T, González ET, Sentsui H (2002) Genetic heterogeneity among bovine leukemia virus genotypes and its relation to humoral responses in hosts. Virus Res 86:101–110
Licursi M, Inoshima Y, Wu D, Yokoyama T, Gonzalez E, Sentsui H (2003) Provirus variants of bovine leukaemia virus in naturally infected cattle from Argentina and Japan. Vet Microbiol 96:17–23
Llames L, Goyache J, Domenech A, Arjona A, Suarez G, Gomez-Lucia E (2001) Evaluation of virus excretion by cells persistently infected with the bovine leukaemia virus (BLV) using monoclonal antibodies. J Clin Virol 22:31–39
Mamoun R, Morison M, Rebeyrotte N, Busetta B, Couez D, Kettman R, Hospital M, Guillemain B (1990) Sequence variability of bovine leukaemia virus env gene and its relevance to structure and antigenicity of the glycoproteins. J Virol 64:4180–4188
Molteni E, Agresti A, Meneveri R, Marozzi A, Malcovati M, Bonizzi L, Poli G, Ginelli E (1996) Molecular characterization of a variant of proviral bovine leukaemia virus (BLV). Zentralbl Veterinarmed B 43:201–211
Monti G, Schrijver R, Beier D (2005) Genetic diversity and spread of Bovine leukaemia virus isolates in Argentine dairy cattle. Arch Virol 150:443–458
Picard L, Simmons G, Power CA, Meyer A, Weiss RA, Clapham PR (1997) Multiple extracellular domains of CCR-5 contribute to human immunodeficiency virus type 1 entry and fusion. J Virol 71:5003–5011
Portetelle E, Mammerickx M, Burny A (1989) Use of two monoclonal antibodies in an ELISA test for the detection of antibodies to bovine leukaemia virus envelope protein gp 51. J Virol Meth 23:211–222
Rodriguez SM, Golemba MD, Campos RH, Trono K, Jones LR (2009) Bovine leukemia virus can be classified into seven genotypes: evidence for the existence of two new clades. J Gen Virol. doi:10.1099/vir.0.011791-0
Shimodaira H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51:492–508
Shimodaira H, Hasegawa M (2001) CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17:1246–1247
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acid Res 22:4673–4680
Willems L, Thienpont E, Kerkhofs P, Burny A, Mamerickx M, Kettmann R (1993) Bovine leukaemia virus, an animal model for the study of intrastrain variability. J Virol 67:1086–1089
Zhao X, Buehring GC (2007) Natural genetic variations in bovine leukemia virus envelope genes: possible effects of selection and escape. Virology 15:150–165
Acknowledgments
We would like to thank Instituto Nacional de Investigación Agropecuaria (INIA, Uruguay) for support through Project No. FPTA-INIA 174 and the Centre National de la Recherche Scientifique (LIA-LPVL). Authors also acknowledge support by PEDECIBA and Agencia Nacional de Investigación e Innovación (ANII) (Uruguay).
Author information
Authors and Affiliations
Corresponding author
Additional information
G. Moratorio and G. Obal contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Moratorio, G., Obal, G., Dubra, A. et al. Phylogenetic analysis of bovine leukemia viruses isolated in South America reveals diversification in seven distinct genotypes. Arch Virol 155, 481–489 (2010). https://doi.org/10.1007/s00705-010-0606-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-010-0606-3