
Abstract
For the first time a competitive immunoassay was developed by employing T-2 antibody-functionalized magnetite nanoparticles and T-2 toxin-conjugated fluorescent quantum dots (QDs). Free T-2 and the T-2-modified QDs compete for binding to antibody-modified magnetic beads; the magnetic beads collected by magnetic separation were subjected to fluorescence intensity analysis (with excitation/emission wavelengths at 460/616 nm). This competitive immunoassay for T-2 toxin determination was applied both in a microcentrifuge tube and on a 96-well plate. The dynamic range of the immunoassay is 1–100 ng mL−1, the limit of detection (LOD) is 0.1 ng mL−1, and determination was completed in about 40 min and 30 min in the microcentrifuge tube and 96-well plate, respectively. Moreover, the biolayer interferometry (BLI) technique was employed for T-2 determination for the first time, in which the conjugate of T-2 toxin and bovine serum albumin (BSA) was immobilized on the sensors before detection. Its average recovery of T-2 toxin from barley sample ranged from 82.00 to 123.33%, and the relative standard deviation (RSD) was between 9.42 and 15.73%. The LOD of the BLI-based assay is 5 ng mL−1, and it only takes 10 min to finish the determination.

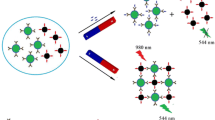
Graphical abstract
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As one of the most prevalent type A trichothecenes, T-2 toxin (T-2) is produced by several Fusarium species, such as F. acuinatum, F. poae, and F. soprotrichioides, which can be found in various cereal grains [1]. Because of its 12,13-epoxy ring, T-2 is considered as the most acutely toxic trichothecene. And it can cause alimentary toxicity and immunotoxicity by inhibiting the synthesis of DNA, RNA, and protein [1, 2]. T-2 is highly stable and non-volatile even under extreme conditions and thus has the potential for weaponization [3, 4]. There is currently no effective treatment for detoxication when poisoning occurs [1, 3]. A rapid, simple, and specific method for T-2 determination to prevent exposure in humans and animals is urgently needed.
In the past few years, researchers have attempted to develop a variety of analytical techniques for T-2 determination. These techniques have included thin-layer chromatography, high-performance liquid chromatography, gas chromatography, liquid chromatography-tandem mass spectrometry, and gas chromatography-tandem mass spectrometry [5, 6]. Although these instrument-dependent methods are reliable and sensitive, they cannot meet the needs of fast T-2 screening, as they typically require complicated sample pretreatment steps, costly equipment, and skilled technicians [7, 8]. Rapid achievements in nanotechnology have led to the development of nanoprobes for mycotoxin determination [9]. Various nanomaterials, including silver nanoclusters, gold nanoparticles, multi-walled carbon nanotubes, magnetic nanoparticles, and semiconductor quantum dots (QDs), have been used as catalytic tools, immobilization scaffolds, or fluorescent nanoprobes to achieve high specificity and sensitivity [5, 10,11,12,13,14,15]. Among the different types of nanomaterials, magnetic iron oxide nanoparticles have attracted increasing interest in the biomedical field because of their superparamagnetism, biocompatibility, and ease of synthesis [16]. The optical and electronic characteristics of QDs have enabled their application in cellular imaging and bioassays [17]. However, a T-2 determination nanoprobe combining the advantages of both magnetic nanoparticles and QDs has not been explored.
Biolayer interferometry (BLI) is an optical analytical technology for real-time determination of interactions between biological molecules. This method uses fiber-optic biosensors to measure changes in thickness on the sensor layer when binding and interaction occur between a ligand on the biosensor surface and analyte in solution [18]. BLI is ideally suited for detecting interactions between protein and nucleic acids, as well as proteins and small molecules [19]. Because of its simplicity, sensitivity, and specificity, BLI has been utilized to detect platelet-derived growth factor-BB [20], marine biotoxin palytoxin [21], ricin, and the gram-negative bacterium Francisella tularensis [19]. However, there have been no reports of the determination of T-2 using BLI.
In the present investigation, a competitive immunoassay for sensitive T-2 determination based on T-2-specific antibody-functionalized magnetic nanoparticles and T2-bovine serum albumin (BSA)-modified QDs is described (Scheme 1a). Importantly, this immunoassay is also applicable to the 96-well plate format (Scheme 1b). Further, we showed that BLI can act as a nanobiosensor for highly specific and rapid T-2 determination (Scheme 1c). Thus, magnetic/fluorescent nanoprobe and the BLI technique can be applied to detect other mycotoxins.

Schematic representation of the determination of T-2 toxin by competitive immunoassay based on engineered magnetic/fluorescent nanoparticles performed in microcentrifuge tubes (a) and 96-well plate (b). c Schematic representation of BLI-based determination procedures. BLI sensors were coated with T-2-BSA and immersed in a mixture of T-2 antibody and T-2 toxin, and the response signal was recorded
Materials and methods
Materials and apparatus
T-2 toxin and T-2 polyclonal antibody were purchased from Fermentek (Jerusalem, Israel; https://www.fermentek.com/) and Biosco Bio-Tek Company (Dalian China; http://www.iactest.com/), respectively. Aflatoxin B1 toxin (AFB1), fumonisin B1 toxin (FB1), and N-hydroxy-succinimide (NHS) were supplied by Sigma-Aldrich (St. Louis, MO, USA; https://www.sigmaaldrich.com/united-states.html). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and oleylamine were obtained from Fluka (Munich, Germany; http://www.fluka.org/fluka.php?id=courses&sub=ALL&mm2=1). Hydrophilic QDs (2 mg mL−1) were synthesized as we previously described [22]. The size and morphology of nanoparticles were determined by transmission electron microscopy (TEM) with a model H8100 instrument (Hitachi Ltd., Tokyo, Japan; http://www.hitachi.com/). The fluorescent spectra were recorded on a RF-5301 PC fluorescence spectrophotometer (Shimadzu, Kyoto, Japan; https://www.shimadzu.com/). BLI experiments were performed on an Octet Red system (ForteBio, Fremont, CA, USA; https://www.labx.com/item/octet-red96e-system-from-fortebio/10821001).
Functionalization of magnetic polystyrene nanospheres and QDs
The preparation of Fe3O4 nanoparticle-embedded magnetic polystyrene nanospheres was described in the Electronic Supporting Material (ESM). Magnetic polystyrene nanospheres and QDs were dissolved in phosphate-buffered solution (0.1 M, pH 7.4). Magnetic polystyrene nanospheres (1 mL, 1 mg mL−1) were incubated with EDC (0.4 mL, 2 mg mL−1) and NHS (0.4 mL, 4 mg mL−1) at 37 °C for 30 min to activate the carboxyl group. And QDs (2 mL, 2 mg mL−1) were incubated with EDC (0.6 mL, 2 mg mL−1) and NHS (0.6 mL, 4 mg mL−1) under the same condition. The activated magnetic polystyrene nanospheres and QDs were collected by magnetic separation and centrifugation (40,000 rpm, 20 min), respectively. And then they were re-dispersed in phosphate-buffered solution (0.1 M, pH 7.4, 2 mL) for further use. Carboxyl-activated magnetic polystyrene nanospheres and QDs were mixed with T-2-specific antibody (40 mg mL−1, 0.025 mL) and T-2-BSA (40 mg mL−1, 0.025 mL), respectively, and maintained at 37 °C for 5 h with gentle stirring. Next, 2 mL BSA solution (0.1 M in phosphate-buffered solution, pH 7.4) was injected into the two solutions and incubated at 37 °C for 0.5 h with constant shaking. The T-2 antibody-functionalized magnetic polystyrene nanospheres and T-2-BSA-conjugated QDs were purified by magnetic separation and centrifugation (40,000 rpm, 20 min), respectively, re-dissolved in phosphate-buffered solution, and stored at 4 °C until use.
Optimization of incubation time and amount of functionalized QDs by competitive immunoassay
The following parameters were optimized: (a) the incubation time in competitive immunoassay and (b) the amount of functionalized QDs in competitive immunoassay. Respective text and figures about the optimizations are given in the Electronic Supporting Material (ESM).
Specificity evaluation of competitive immunoassay
First, the standard plot of T-2 toxin was established in a microcentrifuge tube and 96-well plate, respectively. Next, 30 μL of different concentrations (1–100 ng mL−1) of T-2 toxin standard solution and 30 μL antibody-functionalized magnetic nanospheres were mixed in a microcentrifuge tube. After incubating at 37 °C for 15 min, 150 μL T-2-BSA-conjugated QDs was added, followed by incubation for another 15 min. The magnetic probes were collected by magnetic separation and dissolved in 400 μL phosphate-buffered solution. And the fluorescence intensity at 616 nm with excitation wavelength of 460 nm was measured with a fluorescence spectrophotometer, and a standard plot was established. To establish a T-2 toxin standard plot in the 96-well plate, the T-2 polyclonal antibody (25 μg mL−1 in phosphate-buffered solution) was added to each well and incubated for 24 h at 4 °C to block the 96-well plate. The antibody solution was replaced with 1% BSA solution and incubated for 2 h. The plate was washed with phosphate-buffered solution thrice and dried at 4 °C before use. Thirty microliter of different concentrations of T-2 standard solution (1–100 ng mL−1) were added to the T-2 antibody-coated 96-well plate and incubated for 15 min. T-2-BSA-conjugated QD solution was added and incubated for another 15 min. After removing the solution from the 96-well plate and washing three times with phosphate-buffered solution, 200 μL phosphate-buffered solution was added to each well. Fluorescence intensity was measured at excitation/emission wavelengths of 460/616 nm, and the standard plot was plotted.
To evaluate the specificity of the competitive immunoassay, 30 μL mixtures of different concentrations (5–100 ng mL−1) of T-2 toxin and 50 ng mL−1 AFB1 toxin or 50 ng mL−1 FB1 toxin were added to the microcentrifuge tubes. After mixing with functionalized magnetic nanospheres in microcentrifuge tubes, the mixture was incubated with shaking at 37 °C for 15 min. Next, 150 μL T-2-BSA-conjugated QDs were added to each tube, and the magnetic probes were collected, and the fluorescence was measured at excitation/emission wavelengths of 460/616 nm. For the competitive immunoassay in the 96-well plate, 30 μL of the same mixtures were mixed and added to the T-2 antibody-coated 96-well plate, rather than to the microcentrifuge tube.
Determination of T-2 toxin in barley with competitive immunoassay in microcentrifuge tube and 96-well plate
Barley was purchased from a local market and was previously shown to be free of T-2 toxin. Finely ground barley (25 g) was immersed in water (100 mL) for 48 h and filtered using filter paper. The filtrate was diluted to 100 mL with water, stirred for 2 min, filtered through filter paper three times, and diluted with phosphate-buffered solution (pH 7.4). Different concentrations of T-2 standard solution (5–100 ng mL−1), prepared with this barley extract instead of phosphate-buffered solution, was detected with the two methods, respectively. And the steps are the same as those described for establishing standard plots section.
Determination of T-2 toxin by biolayer interferometry (BLI)
Aminopropylsilane Dip and Read™ biosensors (for kinetics) were pre-wetted by dipping them into 0.01 M NaHCO3 for 10 min before use. And they were incubated with 0.2 mL T-2-BSA solution (25 μg mL−1 in 0.1 M NaHCO3, pH 8.5) at 37 °C to covalently immobilize the T-2-BSA on sensor tips. Then, the sensors were incubated in casein solution (1 μg mL−1 in 0.1 M NaHCO3, pH 8.5) for 3 h to reduce the interference of non-specific binding. To optimize the concentration of T-2 antibody for T-2 determination, different concentrations of T-2 antibody ranging from 10 to 60 μg mL−1 were added. And the optimal concentration was obtained by measuring and comparing the signal intensity. For BLI determination, the first step is equilibration; the sensors were incubated with casein solution to establish a baseline signal. The second step is binding and determination. In this step, the sensors were incubated with a mixture of T-2 antibody and T-2 toxin-containing samples. The equilibration step was carried out on an Octet Red system (ForteBio) to determine the optimal binding time. After the pre-experiment, this step was performed outside of the instrument to ensure the maximum response signal. Binding and determination were conducted immediately after equilibration. The signal intensity reached a plateau at a binding time of 160 s, and thus, this time was used in subsequent analyses.
Results and discussion
Characterization of functionalized QDs and magnetic polystyrene nanospheres
The fluorescence spectra of QDs before and after T-2-BSA functionalization are presented in Fig. 1a. The fluorescence peaks of bare QDs and T-2-BSA-modified QDs are 618 and 616 nm, respectively. The blue shift of the emission peak of T-2-BSA-modified QDs indicates the successful conjugation of T-2-BSA to QDs [23]. Thermal decomposition is a commonly used method to prepare high-quality magnetic nanoparticles [24]. Figure S1a shows a representative TEM image of the magnetite nanoparticles obtained by thermal decomposition of the iron (III) acetylacetonate. The slow evaporation of a dispersion of nanocrystals in hexane on the TEM grid results in the formation of an extended monolayer of nanoparticles. The monodisperse magnetite nanoparticles are nearly spherical in shape and show a uniform size of approximately 10 nm. As shown in Fig. S1b and Fig. 1b, the clusters of magnetite nanoparticles are encapsulated into the polystyrene nanospheres, and several nanospheres adjoined after T-2 antibody functionalization. The average diameter of the magnetic polystyrene nanospheres is approximately 160 nm, and there are no significant differences in particle size before and after T-2 antibody conjugation. From the energy dispersive spectroscopy results shown in Fig. S1c and S1d, C, O, Fe, Cu, and P chemical signals are detected in T-2 antibody-functionalized magnetic polystyrene nanospheres. This is different from the bare magnetic polystyrene nanospheres, where the P signal cannot be detected [25]. It is noteworthy that the T-2 antibody-functionalized magnetic polystyrene nanospheres and T-2-BSA-conjugated QDs are very stable, and there is no aggregation that can be observed after storage at 4 °C for 1 month.

Characterization of functionalized QDs and magnetic polystyrene nanospheres. a Fluorescence spectra of bare QDs and the T2-BSA-modified QDs (with excitation peaks at 460 nm). b TEM image of T-2 antibody-functionalized magnetic polystyrene nanospheres
Optimization of incubation time and amount of functionalized QDs in competitive immunoassay
To develop a fast and sensitive immunoassay method for T-2 toxin detection, we optimized the incubation time and volume of functionalized QDs. As illustrated in Fig. 2a and c, we fix the other conditions, and the incubation time is set to 5, 10, 15, 20, 25, and 30 min. The fluorescence intensity increases with increasing incubation time and tends to be stable after 15 min in the microcentrifuge tube (Fig. 2a) and 10 min in the 96-well plate (Fig. 2c). Figure 2b and d show the change in fluorescence intensity with increasing volumes of functionalized QDs when all other conditions are unchanged. Substantial increases in fluorescence intensity are observed when the volume of functionalized QD was increased from 90 to 150 μL in the microcentrifuge tube and 30 to 60 μL in the 96-well plate; there is a negligible change in fluorescence intensity when the volumes of functionalized QD are higher than 150 and 60 μL in the microcentrifuge tube (Fig. 2b) and 96-well plate (Fig. 2d), respectively.

Optimization of incubation time and amount of functionalized QDs in competitive immunoassay performed in microcentrifuge tube (a, b) and 96-well plate (c, d). a Change in fluorescence intensity against different incubation time in microcentrifuge tube (a) and 96-well plate (c). b Change in fluorescence intensity when different volume of functionalized QDs was added to microcentrifuge tube (b) and 96-well plate (d)
In short, the following experimental conditions were found to give best results: (a) the optimal incubation time and the amount of functionalized QD for the competitive immunoassay in microcentrifuge tube are determined as 15 min and 150 μL, respectively, and (b) the optimal incubation time and the amount of functionalized QD for the competitive immunoassay in 96-well plate are 10 min and 60 μL, respectively.
Specificity and accuracy of the competitive immunoassay
First, a standard plot for T-2 toxin in the microcentrifuge tube and 96-well plate was established, respectively. As shown in Fig. S2, fluorescence intensity shows a good linear relationship with the T-2 toxin concentrations in the range of 1–100 ng mL−1 both in the microcentrifuge tube and 96-well plate. The linear equation of the competitive immunoassay in microcentrifuge tube is y = − 1.99 × 102x + 4.88 × 104 (R2 = 0.9914), and the limit of detection (LOD) is 0.1 ng mL−1, as determined by estimating the variability in response from 20 measurements in the absence of T-2 toxin (Fig. S2a). For the immunoassay in the 96-well plate, the linear regression equation is y = − 1.62 × 102x + 4.64 × 104 (R2 = 0.9980), and the LOD is 0.1 ng mL−1 (Fig. S2b). To evaluate the specificity and accuracy of this competitive immunoassay for T-2 toxin determination, two toxins (AFB1 and FB1) with similar structures as T-2 toxin are used as interfering toxins. AFB1 and FB1 toxins were spiked into T-2 toxin solution at different concentrations (5–100 ng mL−1) and analyzed by competitive immunoassay in microcentrifuge tube and 96-well plate. The specificity and accuracy of the competitive immunoassay for T-2 toxin determination were assessed by calculating the recovery percentage of T-2 toxin in three replicates. As shown in Table S1, the average recoveries of T-2 toxin in AFB1 solution range from 88.30 to 113.90% in microcentrifuge tube and 89.10 to 116.40% in 96-well plate. The relative standard deviation (RSD) is 8.65–12.74% in microcentrifuge tube and 8.67–12.11% in 96-well plate. In comparison, the mean recovery of T-2 toxin in spiked FB1 solutions ranges from 88.17 to 115.24% in microcentrifuge tube and 88.30 to 115.10% in 96-well plate, respectively (Table S2). The RSD is smaller than 14.31% and 13.26% in microcentrifuge tube and 96-well plate, respectively. Thus, high T-2 toxin recoveries are obtained in AFB1 and FB1 solutions by competitive immunoassay at each spiked level in the microcentrifuge tube and 96-well plate. These results indicate that this method is accurate and specific and can detect T-2 toxins in real agricultural commodities.
Determination of T-2 toxin in barley sample by competitive immunoassay
T-2 toxin mainly contaminates some food crops, particularly barley, wheat, oats, and corn [26, 27]. Thus, barley was used as an actual sample to verify the efficacy of this competitive immunoassay. Barley extract was fortified with different concentrations (1–100 ng mL−1) of T-2 toxin and subjected to competitive immunoassay analysis in microcentrifuge tube and 96-well plate. As summarized in Table 1, the detected T-2 toxin concentrations are consistent with the spiked concentrations. The average recovery values of T-2 toxin range from 83.80 to 114.33% in microcentrifuge tube and from 93.30 to 113.17% in 96-well plate, with RSD values smaller than 9.47% and 8.54%, respectively (Table 1). This competitive immunoassay based on engineered magnetic/fluorescent nanoparticles is simple, highly specific, and can be finished in 30–40 min. And this assay does not require specially trained personnel once the sensors were prepared. Thus, it is reliable and can be applied for specific T-2 toxin determination in barley and other food crops.
Determination of T-2 toxin in barley sample by BLI
During BLI determination, the signal response is closely related to the number of molecules that bind the sensors during the reaction, and thus, the concentration of T-2 antibody is crucial for BLI determination. Therefore, the concentration of T-2 antibody is optimized by measuring the response and binding rate of sensors when different concentrations (10, 20, 30, 40, 50, and 60 μg mL−1) of T-2 antibody were added. The change in the response reflects the change in the sensor layer thickness and thus reflects the number of molecules that bind to the sensors. As shown in Fig. 3a, the response increases with increasing T-2 antibody concentrations from 10 to 50 μg mL−1, and there is a slight decrease at 60 μg mL−1. The change in the binding rate obtained from the response data also displays the same tendency (Fig. 3b). These results reveal that the number of T-2 antibodies attached to the sensors reached a plateau at a T-2 antibody concentration of 50 μg mL−1. This concentration is selected in subsequent experiments.

Optimization of the T-2 antibody concentration in BLI assay. a Effect of T-2 antibody concentration (10–60 μg mL−1) on the response of BLI assay. b Effect of T-2 antibody concentration (10–60 μg mL−1) on the binding rate. This was obtained from the response data. Every point is presented as the average ± standard errors of three independent measurements
To apply the BLI assay for T-2 toxin determination, a standard plot of T-2 toxin was established. Different concentrations (0.01–10 μg mL−1) of T-2 toxin were mixed with 50 μg mL−1 T-2 antibody solution, and then, the T-2-BSA-coated sensors were submerged into the mixture, and the response signal was recorded. As shown in Fig. 4a, the response signal strengthens with increasing T-2 concentrations. The standard plot of T-2 toxin is plotted as the concentrations of T-2 and binding rate obtained from the response data (Fig. 4b). The linear regression equation is y = 0.644 + 6.28 × 10−3x − 2.54E−4 × 2, (R2 = 0.9986). To determine the potential application of this BLI-based assay for T-2 toxin determination of food crops, barley samples were evaluated. The barley extract was spiked with different concentrations of T-2 toxin, and T-2 antibody solution (50 μg mL−1) was added. T-2-BSA-coated sensors were immersed into the mixture, and signals were collected for 160 s. The T-2 concentration in barley extract was calculated according to the standard plot equation, and the recovery percentage was determined. As summarized in Table 2, the recovery percentages of T-2 toxin in barley sample range from 82.00 to 123.33%, and the RSD is between 9.42 and 15.73%. Notably, only 10 min is required to finish the determination. The LOD of the BLI-based assay is 5 ng mL−1. BLI-based assay allows real-time monitoring of T-2 toxin due to its property of visualization and facilitates the error finding during determination process. These results indicate that the BLI-based assay can be used to rapidly detect T-2 toxin.

Establishment of the standard curve of T-2 toxin by BLI assay. a Responses of different concentrations of T-2 toxin ranging from 0.01 to 10 μg/mL obtained from BLI assay. b Calibration curve of T-2 toxin, plot of binding rate (obtained from response) against the T-2 toxin concentration. Points are the means ± standard errors of three independent measurements
Conclusions
A competitive immunoassay based on T-2 antibody-functionalized magnetite nanoparticles and T-2 toxin-conjugated fluorescent QDs was developed and employed for the determination of T-2 toxin. This competitive immunoassay was performed in both microcentrifuge tubes and 96-well plates and displayed good accuracy for T-2 toxin determination even in the presence of interfering toxins. In the following study, we will focus on improving accuracy by signal amplification under the premise of portability. This is the first time that label-free BLI-based assay is developed for T-2 toxin determination. The BLI-based assay can be completed in 10 min and shows relatively high selectivity. This time-saving assay facilitates rapid identification of life-threatening toxins and enables immediate initiation of medical treatment following exposure. Although only the capability of the competitive immunoassay and BLI-based assay for T-2 toxin determination was demonstrated, these two assays can be easily adapted to detect other toxins by substituting the corresponding antibodies. The methods in this study for T-2 determination are simple, rapid, cost-effective, and highly specific and provide a new platform for food safety supervision and clinical application to prevent T-2 toxin-related diseases.
References
Li Y, Luo X, Yang S, Cao X, Wang Z, Shi W, Zhang S (2014) High specific monoclonal antibody production and development of an ELISA method for monitoring T-2 toxin in rice. J Agric Food Chem 62:1492–1497. https://doi.org/10.1021/jf404818r
Lippolis V, Pascale M, Maragos CM, Visconti A (2008) Improvement of determination sensitivity of T-2 and HT-2 toxins using different fluorescent labeling reagents by high-performance liquid chromatography. Talanta 74:1476–1483. https://doi.org/10.1016/j.talanta.2007.09.024
Li Y, Zhang J, Mao X, Wu Y, Liu G, Song L, Li Y, Yang J, You Y, Cao X (2017) High-sensitivity chemiluminescent immunoassay investigation and application for the determination of T-2 toxin and major metabolite HT-2 toxin. J Sci Food Agric 97:818–822. https://doi.org/10.1002/jsfa.7801
Ler SG, Lee FK, Gopalakrishnakone P (2006) Trends in determination of warfare agents-determination methods for ricin. Staphylococcal enterotoxin B and T-2 toxin. J Chromatogr A 1133:1–12. https://doi.org/10.1016/j.chroma.2006.08.078
Zhang M, Wang Y, Yuan S, Sun X, Huo B, Bai J, Peng Y, Ning B, Liu B, Gao Z (2019) Competitive fluorometric assay for the food toxin T-2 by using DNA-modified silver nanoclusters. aptamer-modified magnetic beads. and exponential isothermal amplification. Microchim Acta 186:4. https://doi.org/10.1007/s00604-019-3322-z
Khan IM, Zhao S, Niazi S, Mohsin A, Shoaib M, Duan N, Wu S, Wang Z (2018) Silver nanoclusters based FRET aptasensor for sensitive and selective fluorescent determination of T-2 toxin. Sensors Actuators B Chem 277:328–335. https://doi.org/10.1016/j.snb.2018.09.021
Wang Y, Ning B, Peng Y, Bai J, Liu M, Fan X, Sun Z, Lv Z, Zhou C, Gao Z (2013) Application of suspension array for simultaneous determination of four different mycotoxins in corn and peanut. Biosens Bioelectron 41:391–396. https://doi.org/10.1016/j.bios.2012.08.057
Wang Y, Liu N, Ning B, Liu M, Lv Z, Sun Z, Peng Y, Chen C, Li J, Gao Z (2012) Simultaneous and rapid determination of six different mycotoxins using an immunochip. Biosens Bioelectron 34:44–50 https://doi.org/10.1016/j.bios.2011.12.057
Rai M, Jogee PS, Ingle AP (2015) Emerging nanotechnology for determination of mycotoxins in food and feed. Int J Food Sci Nutr 66:363–370. https://doi.org/10.3109/09637486.2015.1034251
Zhang M, Huo B, Yuan S, Ning B, Bai J, Peng Y, Liu B, Gao Z (2018) Ultrasensitive determination of T-2 toxin in food based on bio-barcode and rolling circle amplification. Anal Chim Acta 1043:98–106. https://doi.org/10.1016/j.aca.2018.09.007
Dong M, Si W, Wang W, Bai B, Nie D, Song W, Zhao Z, Guo Y, Han Z (2016) Determination of type A trichothecenes in coix seed by magnetic solid-phase extraction based on magnetic multi-walled carbon nanotubes coupled with ultra-high performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem 408:6823–6831. https://doi.org/10.1007/s00216-016-9809-0
Foubert A, Beloglazova NV, Saeger SD (2017) Comparative study of colloidal gold and quantum dots as labels for multiplex screening tests for multi-mycotoxin determination. Anal Chim Acta 955:48–57. https://doi.org/10.1016/j.aca.2016.11.042
Malhotra BD, Srivastava S, Ali MA, Singh C (2014) Nanomaterial-based biosensors for food toxin determination. Appl Biochem Biotechnol 174:880–896. https://doi.org/10.1007/s12010-014-0993-0
Reverté L, Prieto-Simón B, Campàs M (2016) New advances in electrochemical biosensors for the determination of toxins: nanomaterials. Magnetic beads and microfluidics systems. A review. Anal Chim Acta 908:8–21. https://doi.org/10.1016/j.aca.2015.11.050
Qie Z, Yan W, Gao Z, Meng W, Xiao R, Wang S (2019) Ovalbumin antibody-based fluorometric immunochromatographic lateral flow assay using CdSe/ZnS quantum dot beads as label for determination of T-2 toxin. Microchim Acta 186:12. https://doi.org/10.1007/s00604-019-3964-x
Ding G, Wang Y, Guo Y, Xu L (2014) Integrin αVβ3-targeted magnetic nanohybrids with enhanced antitumor efficacy, cell cycle arrest ability, and encouraging anti- cell- migration activity. ACS Appl Mater Interfaces 6:16643–16652. https://doi.org/10.1021/am503359g
Gao X, Cui Y, Levenson RM, Chung LWK, Nie S (2004) In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol 22:969–976. https://doi.org/10.1038/nbt994
Concepcion J, Witte K, Wartchow C, Choo S, Yao D, Persson H, Wei J, Li P, Heidecker B, Ma W, Varma R, Zhao LS, Perillat D, Carricato G, Recknor M, Du K, Ho H, Ellis T, Gamez J, Howes M, Phi-Wilson J, Lockard S, Zuk R, Tan H (2009) Label-free determination of biomolecular interactions using biolayer interferometry for kinetic characterization. Comb Chem High Throughput Screen 12:791–800. https://doi.org/10.2174/138620709789104915
Mechaly A, Cohen H, Cohen O, Mazor O (2016) A biolayer interferometry-based assay for rapid and highly sensitive determination of biowarfare agents. Anal Biochem 506:22–27. https://doi.org/10.1016/j.ab.2016.04.018
Gao S, Zheng X, Wu J (2018) A biolayer interferometry-based enzyme-linked aptamer sorbent assay for real-time and highly sensitive determination of PDGF-BB. Biosens Bioelectron 102:57–62. https://doi.org/10.1016/j.bios.2017.11.017
Gao S, Zheng X, Hu B, Sun M, Wu J, Jiao B, Wang L (2017) Enzyme-linked. Aptamer-based. Competitive biolayer interferometry biosensor for palytoxin. Biosens Bioelectron 89:952–958. https://doi.org/10.1016/j.bios.2016.09.085
Guo Y, Li C, Shen H, Zhang X, Li L, Yu Q, Xu L (2011) RGDC peptide modified quantum dots labelling and imaging of tumor cells. Chem Res Chin Univ 27:832–835. http://crcu.jlu.edu.cn/EN/Y2011/V27/I5/832
Abozaid RM, Lazarević ZŽ, Radović I, Gilić M, Šević D, Rabasović MS, Radojević V (2019) Optical properties and fluorescence of quantum dots CdSe/ZnS-PMMA composite films with interface modifications. Opt Mater 92:405–410. https://doi.org/10.1016/j.optmat.2019.05.012
Laurent S, Forge D, Port M, Roch A, Robic C, Elst LV, Muller RN (2008) Magnetic iron oxide nanoparticles: synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem Rev 108:2064–2110. https://doi.org/10.1021/cr068445e
Du P, Jin M, Zhang C, Chen G, Cui X, Zhang Y, Zhang Y, Zou P, Jiang Z, Cao X, She Y, Jin F, Wang J (2018) Highly sensitive determination of triazophos pesticide using a novel bio-bar-code amplification competitive immunoassay in a micro well plate-based platform. Sensors Actuators B Chem 256:457–464. https://doi.org/10.1016/j.snb.2017.10.075
Shi H, Schwab W, Yu P (2019) Natural occurrence and co-contamination of twelve mycotoxins in industry-submitted cool-season cereal grains grown under a low heat unit climate condition. Toxins 11:160. https://doi.org/10.3390/toxins11030160
Janssen EM, Liu C, Fels-Klerx HJVD (2018) Fusarium infection and trichothecenes in barley and its comparison with wheat. World Mycotoxin J 11:33–46. https://doi.org/10.3920/WMJ2017.2255
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81771965, 31571017, 81571791, 81271697), the National “Significant New Drug Creation” Science and Technology Major Program (No. 2012ZX09503001-003), the project of Science and Technology Department of Jilin Province, China (No. 20170204027NY, 20130206069GX), and the Special Project of Biological Medicine of Jilin Province, China (No. SXGJSF2017-1).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interest
The authors declare that they have no competing interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary data to this article can be found online at https://doi.org/xx.xxx
ESM 1
(DOC 4626 kb).
Rights and permissions
About this article

Cite this article
Li, Y., Xu, L., Fu, X. et al. A competitive immunoassay based on engineered magnetic/fluorescent nanoparticles and biolayer interferometry-based assay for T-2 toxin determination. Microchim Acta 187, 514 (2020). https://doi.org/10.1007/s00604-020-04493-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-020-04493-3