
Abstract
The authors describe a screen-printed electrode (SPE) modified with a composite consisting of Mn3O4 microcubes and thin sheets of graphene oxide for use in amperometric determination of nitrite. The composite was prepared by a hydrothermal method, and its morphology, elemental composition, diffraction, impedance and electrochemical properties were studied. The modified SPE displays excellent electrocatalytic activity towards nitrite, and the oxidation peak current (measured typically at 0.70 V vs. Ag/AgCl) is related to the concentration of nitrite in the range between 0.1 and 1300 μM, with a 20 nM detection limit. The method was successfully applied to the determination of nitrite in spiked samples of beef and water.

Electrochemical determination of nitrite using graphene oxide nanosheets/Mn3O4 microcubes composite modified screen-printed electrode
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nitrite is the source of non-enzymatic nitric oxide production in mammalian cells [1] and also it is a signalling molecule in mammalian tissues [2]. Nitrite has been widely used as food preservative owing to its good antibacterial property [3]. However, nitrite can react with amines to form carcinogenic N-nitrosamines either inside the body or during the meat curing process [4]. The International Agency for Research on Cancer (IARC) had listed nitrite as Group 2A type carcinogenic, i.e., ‘there is strong evidence that nitrite can cause cancer in humans, but at present it is not conclusive’ [5]. In the circulatory system, nitrite irreversibly oxidizes hemoglobin into methemoglobin which causes methemoglobinemia [6]. Continuous uptake of meat with high content of nitrite has the possibility to increase the risk of cancer. On the other hand, nitrite is an inorganic pollutant to the environment and its increasing distribution and accumulation in water bodies is huge threats to environmental safety [6]. As per the Environmental Protection Agency (EPA) regulation, the allowable amount of nitrite in drinking water is 1 mg L−1 [7]. Thus, the development of portable, cheaper and reliable sensor devices is necessary for rapid and accurate detection of nitrite in food and water samples. In comparison with other bulky analytical methods, electroanalytical methods are more reliable as they are low-cost, portable, easy-to-use, and offer quick responses. Compton et al. reported a nitrite sensor using bare electrode, however, sonoelectrochemical cleaning is required in order to maintain the activity of electrode surface. Nevertheless, fabricating suitable modified electrodes have considerably minimized the fouling effect and reduced the overpotential as well [8]. Most of the previous reports are focused on the detection of nitrite in drinking water; herein a robust modified electrode was described for the nitrite detection in meat samples and water samples as well.
Graphene and graphene oxide (GO) based composites are likely to remain one of the leading subjects in electrochemistry for many years owing to their interesting catalytic properties [9, 10]. GO, an oxygenated derivative of graphene is an amphiphilic molecule possessing aliphatic and aromatic regions. It contains abundant oxygen functionalities; epoxy and hydroxyl groups on the plane, while carboxyl, carbonyl, ester, ether, diol, ketone, phenol, quinone and lactone groups present at the edges. Graphene supported manganese oxide (MnOx) composites have been reported to exhibit prominent electrochemical properties. MnOx materials are low cost, abundant on earth crust and highly stable, while various shaped MnOx (needles, tubes, particles, plates and wires) in a multi-valent states have been reported [11]. Mn3O4 incorporated graphene or GO based composites can be prepared by many methods [12,13,14]. Virtually, all reports had been focused on energy applications; nonetheless, GO/Mn3O4 microcubes (MC) composite has never been explored in electrochemical sensing applications. By tuning the synthetic protocol, nanocubes structured materials with six surfaces can be prepared. These cube structured materials encompassed with sharp edges and corners and they have shown excellent catalytic activity over nanoparticles [15].
Herein, the preparation of Mn3O4 MC networks embedded in thin sheets of GO as a composite is described and applied in nitrite sensing (Fig. 1). The preparation of GO/Mn3O4 MC composite does not require any hazardous reducing agents. Screen-printed carbon electrode (SPCE) was adopted as working electrode because of its low-cost, easy fabrication, flexibility, and reproducibility [16]. The GO/Mn3O4 MC film modified SPCE exhibited large electrochemical active area, excellent electrocatalytic ability and outstanding nitrite sensing characteristics.

Synthesis of GO/Mn3O4 MC for the determination of nitrite in beef and water samples. EtOH = ethanol
Experimental
Materials and instrumentation
Graphite (powder, <20 μm), sodium acetate, manganese(II) sulfate mono hydrate (MnSO4·H2O) and urea were purchased from Sigma-Aldrich (http://www.sigmaaldrich.com/taiwan.html) and used as received. The SPCEs were purchased from Zensor R&D Co., Ltd., Taipei, Taiwan (http://www.zensor.com.tw/index_en.html). All the reagents used were of analytical grade and used without any further purification. The supporting electrolyte used for the electrochemical studies was 0.1 M phosphate buffer (PB), prepared using Na2HPO4 and NaH2PO4 and the pH was adjusted either using H2SO4 or NaOH. All assays were performed in triplicate, and the results reported are the average of at least three experiments. The error is calculated from standard deviation of these three readings.
Field Emission Scanning electron microscopy (FESEM) studies were performed with Hitachi S-3000 H scanning electron microscope. Energy-dispersive X-ray (EDX) spectra and mapping were recorded using HORIBA EMAX X-ACT (Sensor +24 V = 16 W, resolution at 5.9 keV). Powder X-ray diffraction (XRD) studies were performed in a XPERT-PRO (PANalytical B.V., The Netherlands) diffractometer using Cu Kα radiation (k = 1.54 Å). FTIR spectra were carried out using a Perkin-Elmer IR spectrometer. X-ray photoelectron spectra (XPS) were obtained by PerkinElmer PHI-5702. Raman spectra were acquired using Micro-Raman spectrometer (RENISHAW in via system, U.K) by a 514.4 nm He/Ne laser. EIM6ex Zahner (Kronach, Germany) was used for electrochemical impedance spectroscopy (EIS) studies.
Synthesis of GO/Mn3O4 micro cubes
GO was prepared through modified Hummers method [17]. Mn3O4 microcubes (MCs) were synthesized via hydrothermal method followed by calcination (Fig. 1). Briefly, solutions of urea (3 g) and MnSO4·H2O (0.01 M) were prepared separately in 40 mL water via magnetic stirring followed by ultrasonication for 15 min. Next, the MnSO4 solution was slowly added dropwise into urea solution. The mixture was stirred for 30 min to acquire a homogeneous solution and the resulting solution was transferred to a Teflon–lined stainless steel autoclave and heated at 180 °C for 10 h. After hydrothermal treatment, the resulting reddish product was collected and washed with ethanol (2 × 50 mL), and then dried at 40 °C for overnight. Then the solid was transferred to a tube furnace and heated to 400 °C for 2 h at a heating rate of 2 °C min−1. Finally, the product Mn3O4 MC was collected. In order to prepare, GO/Mn3O4 MC, 2 mg of Mn3O4 MC was added into 2 mL GO aqueous dispersion (1 mg mL−1) and stirred for 15 min. The resulting mixture was ultrasonicated for another 15 min to ensure sufficient interfacial self-assembly of negatively charged GO sheets on the positively charged Mn3O4 MC through electrostatic interaction, thus yielding stable GO/Mn3O4 MC network [18, 19]. The GO/Mn3O4 MC aqueous dispersion is stable for about 1 h without any sediment formation. The dispersion is ultrasonicated for 10 min prior to its usage in order to obtain homogenous dispersion.
Preparation of modified electrode
The surface of SPCE was pre-cleaned by cycling between −1.0 V and 1.2 V (vs. Ag/AgCl), in 0.1 M PB (pH 7) for 10 cycles. Next, 5 μL of GO/Mn3O4 MC was dropped at SPCE and dried at room temperature for 30 min. As control, GO and Mn3O4 MC modified SPCEs were prepared.
Experimental parameters
The cyclic voltammograms (CVs) were carried out in 0.1 M PB (pH 7.0). The potential range was = −0.10 to 0.90 V and scan rate = 50 mV s−1 unless otherwise specified. EIS experiments were performed in 0.1 M KCl containing 5 mM Fe(CN)6 3−/4− and the optimized parameters: amplitude 5 mV and Frequency 0.1 Hz to 100 kHz. Each measurement above was conducted in triplicate.
The electrochemical measurements were performed using CHI 1205A workstation. The electrochemical studies were carried out in a conventional three-electrode cell using BAS SPCE as a working electrode (area 0.20 cm2), saturated Ag/AgCl as a reference electrode and Pt wire as a counter electrode. Amperometric measurements were performed with analytical rotator AFMSRX (PINE instruments, USA) with a rotating disc electrode (RDE, glassy carbon) having working area of 0.20 cm2.
Assay procedure
The supporting electrolyte is 5 mL aliquots of 0.1 M PB (pH 7.0). Prior to each experiment, the electrolyte solutions were deoxygenated with pre-purified nitrogen gas for 10 min unless otherwise specified. Appropriate amounts of nitrite was injected and stirred for 2 min before electrochemical experiments were performed. In the case of interference study, other compounds were added along with nitrite in the detection mixture. Each measurement above was conducted in triplicate.
Results and discussions
Choice of materials
Literature studies revealed that graphene/metal oxide composites have excellent sensing ability towards nitrite. Graphene oxide (GO) possess large surface area, abundant oxygen functionalities, ample sites for nitrite adsorption, high edge density and rich edge defects and hence GO is the choice of supporting material. Mn3O4 is well known for its excellent electrocatalytic property to many important reactions and it is low-cost, earth abundant and highly stable. Therefore, we have designed and prepared GO/Mn3O4 MC. The important sensor parameters such as, sensitivity, detection limit, selectivity and reproducibility are significantly improved with GO/Mn3O4 MC over existing modifiers.
Surface morphological and elemental characterization
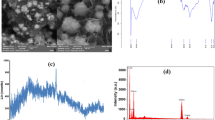
The FESEM image of Mn3O4 MC is given in Fig. 2a that clearly revealed the observation of numerous microcubes structured particles. The magnified image displays high crystalline single cube and the size was in micrometer range (Fig. 2b). With the help of XRD, we identified these cubes as Mn3O4 MC. The EDX profile of Mn3O4 MC displays the expected elements Mn and O (Fig. 2e) and corresponding mapping analysis presents the distribution of O and Mn in the cubes with weight percentages of 58.47 and 41.53, respectively (Fig. S1). The FESEM image, EDX profile, and mapping of GO are given in supporting information and the results are consistent with previous reports [20] (Fig. S2). The image of GO/Mn3O4 MC shows 3D structure, which composed of thin GO sheets and Mn3O4 MC (Fig. 2c, d). The microcubes are wrapped and covered by ultrathin sheets of GO and each cubes and bricks were interconnected under the bed of GO sheets. EDX profile of GO/Mn3O4 MC shows signals corresponding to C, O, and Mn with weight percentages of 41.72, 23.61 and 34.67, respectively (Fig. 2f). The mapping of GO/Mn3O4 MC reveals the distribution of expected elements C, O and Mn (Fig. S3).

FESEM images of Mn3O4 MC (a, b) and GO/Mn3O4 MC (c, d). EDX profile of Mn3O4 MC (e) and GO/Mn3O4 MC (f)
XRD, Raman, XPS and FT-IR spectral studies
Figure 3a displays the XRD patterns of GO, Mn3O4 MC, and GO/Mn3O4 MC. The pattern of GO featured with characteristic sharp peak at 11.02 Å (001). The curve of Mn3O4 MC displayed peaks at 18.6° (101), 29.5o (112), 31.8o (200), 32.9o (103), 35.8o (211), 39.1o (004), 43.5o (220), 50.7o (105), 53.6o (513), 57.1o (303), 59.3o (321), 61.4o (224), and 65.7o (440) consistent with standard pattern of Mn3O4 (JCPDS no. 24–0734). The XRD pattern of GO/Mn3O4 MC contains all the aforementioned peaks of Mn3O4 MC (star symbol) and GO (square symbol). The peak positions were not altered in the composite which indicating that the crystal structures of individual components were not damaged during composite formation.

a XRD, b Raman, and c FT-IR spectra of GO, Mn3O4 MC and GO/Mn3O4 MC. d XPS profile of GO/Mn3O4 MC
Figure 3b shows the Raman spectra of GO, Mn3O4 MC and GO/Mn3O4 MC. The spectrum of GO displayed D band at 1407 cm−1 (related to defects) and G band at 1594 cm−1 (originates from stretching of in-plane sp2 atoms) which are characteristic bands expected for GO [8, 21]. The spectrum of Mn3O4 MC exhibited a well-defined peak at 643.2 cm−1, which can be assigned to the Raman active mode of Mn3O4 NC. The spectrum of GO/Mn3O4 MC displays active modes corresponding to both Mn3O4 NC and GO. The level of disorder can be elucidated by analyzing the D/G band intensity, I D/I G. The I D/I G of GO/Mn3O4 MC (0.98) which is comparatively higher than that of GO (0.85) revealing that the level of disorder was considerably increased. In addition, the D band is slightly red shifted which indicating the alteration in defect sites of GO.
In order to track the changes in functional groups, FT-IR spectra of GO, Mn3O4 MC and GO/Mn3O4 MC were studied (Fig. 3c). The FT-IR spectra were performed using ethanol as solvent; as a result, the characteristic vibration modes of ethanol were observed in all the FT-IR curves. For clarity, we have ignored the modes of ethanol solvent. The spectrum of GO displays peaks at wavenumbers of 3417, 1763, 1682, 1391, 1254, and 1087 cm−1 which are correlated to stretching vibrations of O − H, C = O, C = C, O − H, C − O − C and C − O, respectively. However, the spectrum of GO/Mn3O4 MC displays additional peaks at 624 cm−1 and 563 cm−1 that were originated from the incorporated Mn3O4.
Next, the surface chemical compositions of GO/Mn3O4 MC composite were examined by XPS (Fig. 3d). The XPS curve of GO/Mn3O4 MC shows characteristic signals of C 1 s, O 1 s, 2p1/2 and 2p3/2 at binding energies of 286.5, 532.5, 642.6 and 641.0 eV, respectively which are consistent with previous reports [19]. The higher-resolution deconvoluted spectra corresponding to C 1 s, O 1 s, 2p1/2 and 2p3/2 were given in supporting information (Fig. S4 ) which added additional evidence for the successful formation of composite.
Impedance and electrochemical properties of the composite
Fig. 4a displays the EIS curves of GO/SPCE (a), Mn3O4/SPCE (b), and GO/Mn3O4 MC/SPCE (c) in 0.1 M KCl containing 5 mM Fe(CN)6 3−/4−. Randles equivalent circuit model has been used to fit the experimental data (inset to Fig. 4a), in which, R ct, R s, C dl and W were depicting charge transfer resistance, electrolyte resistance, double layer capacitance and Warburg element, respectively. The diameter of semicircles (i.e., R ct) were in the following order; GO/SPCE (96.23 ± 1.52 Ω) > Mn3O4 MC (17.51 Ω ± 0.92)/SPCE > GO/Mn3O4 MC/SPCE (28.35 ± 0.41 Ω). The results indicate that the resistance at GO/Mn3O4 MC/SPCE was smallest over other electrodes.

(a) EIS curves of GO (a), Mn3O4 MC (b) and GO/Mn3O4 MC (c) obtained in 0.1 M KCl containing 5 mM Fe(CN)6 3−/4−. Amplitude: 5 mV, Frequency: 0.1 Hz to 100 kHz. Inset: Randles equivalent circuit used to fit the data; R s, R ct, C dl, and Z w are electrolyte resistance, charge transfer resistance, double layer capacitance and Warburg impedance, respectively. (b) CVs of GO (a), Mn3O4 MC (b), and GO/Mn3O4 MC (c) films modified SPCEs in 0.1 M NaOH at scan rate of 2 mV s−1. (c) Cyclic voltammograms of unmodified (a), GO (b), Mn3O4 MC (c), and GO/Mn3O4 MC (d) films modified SPCEs in 0.1 M PB buffer (pH 7) containing 50 μM nitrite, scan rate = 50 mV s−1. (d) Cyclic voltammograms of GO/Mn3O4 MC/SPCE in 0.1 M PB (pH 7) containing nitrite (a to j; 50 to 500 μM), scan rate = 50 mV s−1. Inset [nitrite]/μM vs. current/μA. (e) Cyclic voltammograms obtained at GO/Mn3O4 MC/SPCE in 0.1 M PB (pH 7) containing 50 μM nitrite at different scan rates (a to k; 20 to 200 mVs−1). Insert: (scan rate)1/2 (V.s−1)1/2 vs. peak currents (μA). (f) Cyclic voltammograms performed in 0.1 M PB (pH 7) containing 50 μM nitrite at different pH (a = 3.0, b = 5.0, c = 7.0 and d = 11.0), scan rate = 50 mV s−1. Inset: Plot between peak current (μA) vs. pH
Electrochemical Activity of the prepared composite was investigated. The cyclic voltammograms (CVs) of GO/SPCE (a), Mn3O4 MC/SPCE (b), and GO/Mn3O4 MC/SPCE (c) over a potential range of – 0.4 to 0.5 V (vs. Ag/AgCl) at a scan rate of 2 mV.s−1 is illustrated in Fig. 4b. The CV of GO/Mn3O4 MC/SPCE exhibited enhanced background current over controls. Two redox couples, − 0.25 V/− 0.3 V (I/II) and – 0.05 V/− 0.14 V (III/IV) corresponding to reversible reactions of Mn3O4/MnOOH and MnOOH/MnO2 were observed and those are characteristic voltammetric behaviour of electrochemically active Mn3O4 MC [22]. The peak currents were linearly increased as the scan rate increased, and their corresponding plot exhibited good linearity which indicating surface-confined redox process (Fig. S5).
The electrochemically effective surface areas of the GO/Mn3O4 MC/SPCE and control electrodes have been calculated using K3[Fe(CN)6] as a model complex (Fig. S6) and by following Randles–Sevcik equation; i p = (2.69 × 105)n 3/2 AcD 1/2 ν 1/2. Here, i p, n, A, c, D and n are representing the peak current, the number of electrons involved in the reaction (n = 1), the effective surface area (cm2), the concentration of the reactant (M), the diffusion coefficient (7.6 × 10−6 cm2 s−1) and the scan rate (in V s−1), respectively [23]. Using this formula, the effective surface areas of Mn3O4/SPCE, GO/SPCE and GO/Mn3O4 MC/SPCE were estimated to be 0.083 (± 0.008) cm2, 0.107 (± 0.016) cm2, and 0.138 (± 0.01) cm2.
Electrocatalysis of nitrite
Fig. 4c displays the cyclic voltammograms (CVs) obtained at bare SPCE (a), GO/SPCE (b), Mn3O4 MC/SPEC (c), and GO/Mn3O4 MC/SPCE (d) in PB (pH 7) containing 50 μM nitrite. The scan rate was applied as 50 mV s−1. The electrocatalytic ability of these modified was GO/Mn3O4 MC > Mn3O4 MC > GO/SPCE > GO/Mn3O4 MC > bare SPCE. The GO/Mn3O4 MC exhibited highly enhanced electrocatalytic ability and fast electron transfer as revealed by highly enhanced anodic peak current at minimized overpotential (0.70 V, Ag/AgCl). The net faradic current obtained at GO/Mn3O4 MC/SPCE was 1.5, 3.3, and 6.2 fold higher than those obtained at Mn3O4 MC/SPCE, GO/SPCE and unmodified SPCE, respectively. The improved electrocatalytic ability can be manifested to the great synergetic effect between GO and Mn3O4 MC in terms of increasing surface area and conductivity. Thus, the electrocatalytic properties of GO can be upgraded by tailoring it with Mn3O4 MC as composite. The residual functional groups located at the edges of GO can provide additional sites for nitrite adsorption. Fig. 4d presents the CVs obtained at GO/Mn3O4 MC/SPCE in PB (pH 7.0) towards different concentrations of nitrite. The anodic peak current increased as the concentration of nitrite increased (inset to Fig. 4d). The plot between oxidation peak current and square root of scan rate exhibits good linearity which indicating diffusion controlled electrocatalytic process (Fig. 4e). The influence of buffer pH on the electrocatalytic response of nitrite was investigated (Fig. 4f). CVs were performed in supporting electrolyte of different pH containing 50 μM nitrite and the changes in oxidation peak current with respect to pH were given as plot (inset to Fig. 4f). The current increased as the pH increased, reached maxima at pH 7 and followed decreased trend at basic pH.
Amperometric determination of nitrite
Figure 5a shows the amperometric i-t curve obtained at GO/Mn3O4 MC composite modified electrode (Rotation speed at 1200 RPM) upon following additions of 0.1, 0.5, 0.3, 1, 10, 50 and 75 μM nitrite into PB (pH 7) at regular intervals of 50 s (applied potential, E app = + 0.70 V, vs. Ag/AgCl). Well-defined and stable responses were observed for each addition and the response currents were increased linearly as the concentrations increased. Two linear ranges were obtained; (1) 0.1 μM to 420 μM with sensitivity of 2.37 μAμM−1 cm−2, (inset to Fig. 5a) and (2) 490 μM to 1318 μM with sensitivity of 1.23 μAμM−1 cm−2 (Fig. 5b). Notably, Sensitivity at higher concentration range is lower than that at higher concentration range, which is due to the occurrence of substrate inhibition effects at higher concentration of H2O2.

(a) Amperometric response of GO/Mn3O4 MC film modified RDE for each sequential additions of nitrite (0.1, 0.5, 10, 50, and 75 μM) into 0.1 M PB (pH 7). The rotation speed = 1200 RPM. (b) Calibration plot between [nitrite]/μM and current (μA); working potential (E app) = + 0.70 V (vs. Ag/AgCl). (c) Amperometric responses of GO/Mn3O4 MC/SPCE towards 5 μM of nitrite (a) and 0.5 mM of uric acid (b), dopamine (c), ascorbic acid (d), NADH (e), cysteine (f), folic acid (g), epinephrine (h), guanine (i) and pyridoxine (j). E app = + 0.70 V (vs. Ag/AgCl). (d) Stability of the sensor as its continuous use for one month. The CV response of GO/Mn3O4 MC film modified electrode towards 10 μM nitrite in PB (pH 7.0) was monitored for the given number of days
The limit of detection (LOD) was calculated to be 20 nM (± 1.15) using the formula, LOD = 3 s b/S, where, s b = standard deviation of blank signal and S = sensitivity [8]. The low detection limit at nanomolar level illustrated the outstanding performance of the electrode and the sensor parameters were compared with previous reports. As can be seen from Table 1, our electrode presented better performance over existing nitrite sensors. Interestingly, the performance of our sensor was superior over previously reported sensors.
Selectivity, stability and reproducibility
The selectivity was evaluated by performing analysis in presence of likely interfering agents. Figure 5c shows the amperometric responses of GO/Mn3O4 MC/SPCE towards 5 μM nitrite (a) and 0.5 mM of uric acid (b), dopamine (c), ascorbic acid (d), NADH (e), cysteine (f), folic acid (g), epinephrine (h), guanine (i) and pyridoxine (j). The electrode quickly responded to nitrite, but it was insensitive to other species; thus, the electrode capable to recognize nitrite specifically in the pool of many species.
The sensor response had been monitored every day to find out the storage stability. The electrode retained 97.8% of its initial response still after 30 days of its continuous use (Fig. 5d), validated the good storage stability of the sensor. For the reproducibility studies, CVs at five individual electrodes were recorded in 0.1 M PB (pH 7.0) containing10 μM nitrite; R.S.D. of 4.82% was obtained.
Real sample analysis
The practical feasibility of the method was demonstrated in beef and water samples. In order to perform analysis in beef, first nitrite free beef sample was immersed in PB and stirred for 30 min. Then, the beef pieces were removed and the solution was spiked with known amounts of nitrite and amperometry was performed (Fig. 6a). The sensor was delivered quick signals as lab samples. The linear range was 0.15 to 532 μM and LOD was 62 (± 1.32) nM (Fig. 6c). Government agencies and food manufacturers need analytical device to provide appropriate information about the amount of nitrite used for food preservation. Meeting the demand for cost-effective, robust and portable analytical device, the electrochemical sensor fabricated herein has enormous potential as useful sensing tool for real-time online monitoring of nitrite in meat samples (Fig. 6b). Similarly, our method showed good practical applicability in spiked drinking water sample. The linear range was 0.12 μM to 775.44 μM and LOD was 50 (± 1.59) nM (Fig. 6d), thus the sensor is also applicable in water analysis.

Amperometric response of GO/Mn3O4 MC/SPCE for each sequential addition of real samples containing spiked nitrite into 0.1 M PB (pH 7.0). (a) Beef sample and, (b) drinking water sample. Calibration plots for beef (c) and drinking water (d), E app = 0.70 V (vs. Ag/AgCl)
Conclusions
A highly sensitive, selective, stable, and durable nitrite sensor was demonstrated using a robust composite, GO/Mn3O4 MCs. The successful formation of the composite was revealed by FESEM, EDX, mapping, XRD, Raman, XPS, EIS and electrochemical methods. The electrochemical studies revealed that the modified electrode possesses outstanding electrocatalytic ability towards nitrite oxidation. At optimized working condition, the sensor performance was either superior or comparable to the previous works. The assay procedure was simple, fast, reproducible and suitable for real-time applications. Although, the preparation method was unable to produce nanocubes, the material still showed good sensing attributes. The method was successful in the determination of nitrite spiked in beef and water samples, thus holds great potential in food safety and water analysis.
References
Allen JD, Gow AJ (2009) Nitrite, NO and hypoxic vasodilation. Br J Pharmacol 158(7):1653–1654
Bryan NS, Fernandez BO, Bauer SM, Garcia-Saura MF, Milsom AB, Rassaf T, Maloney RE, Bharti A, Rodriguez J, Feelisch M (2005) Nitrite is a signaling molecule and regulator of gene expression in mammalian tissues. Nat Chem Biol 1(5):290–297
Silva MM, Lidon FC (2016) Food preservatives-an overview on applications and side effects. Emirates J Food Agri 28(6):366
Liu P, Zhang X, Feng L, Xiong H, Wang S (2011) Direct electrochemistry of hemoglobin on graphene nanosheet-based modified electrode and its electrocatalysis to nitrite. Am J Biomed Sci 3(1):69–76
Humans IWGotEoCRt (2010) IARC monographs on the evaluation of carcinogenic risks to humans. Ingested nitrate and nitrite, and cyanobacterial peptide toxins. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans 94:v
Yue R, Lu Q, Zhou Y (2011) A novel nitrite biosensor based on single-layer graphene nanoplatelet–protein composite film. Biosens Bioelectron 26(11):4436–4441
Council NR (1995) Nitrate and nitrite in drinking water. National Academies Press,
Mani V, Periasamy AP, Chen S-M (2012) Highly selective amperometric nitrite sensor based on chemically reduced graphene oxide modified electrode. Electrochem Commun 17:75–78
Geim AK, Grigorieva IV (2013) Van der Waals heterostructures. Nature 499(7459):419–425
Govindasamy M, Mani V, Chen S-M, Karthik R, Manibalan K, Umamaheswari R (2016) MoS2 flowers grown on graphene/carbon nanotubes: a versatile substrate for electrochemical determination of hydrogen peroxide. Int J Electrochem Sci 11:2954–2961
Gangaraju D, Sridhar V, Lee I, Park H (2016) Graphene–carbon nanotube–Mn 3 O 4 mesoporous nano-alloys as high capacity anodes for lithium-ion batteries. J Alloys Compounds 699:106–111
Ejigu A, Edwards M, Walsh DA (2015) Synergistic catalyst–support interactions in a graphene–Mn3O4 Electrocatalyst for vanadium redox flow batteries. ACS Catal 5(12):7122–7130. doi:10.1021/acscatal.5b01973
Li Y, Ni X (2017) The enhanced Supercapacitive performance of the hybrid material integrating doped-polymer with the composite of graphene oxide and Mn 3 O 4. Electrochimica Acta 227:162–169
Yuan Z, Chen S, Liu B (2017) Nitrogen-doped reduced graphene oxide-supported Mn3O4: an efficient heterogeneous catalyst for the oxidation of vanillyl alcohol to vanillin. J Mater Sci 52(1):164–172
Sayle TX, Caddeo F, Zhang X, Sakthivel T, Das S, Seal S, Ptasinska S, Sayle DC (2016) Structure–activity map of ceria nanoparticles, Nanocubes, and mesoporous architectures. Chem Mater 28(20):7287–7295
Barton J, García MBG, Santos DH, Fanjul-Bolado P, Ribotti A, McCaul M, Diamond D, Magni P (2016) Screen-printed electrodes for environmental monitoring of heavy metal ions: a review. Microchim Acta 183(2):503–517
Marcano DC, Kosynkin DV, Berlin JM, Sinitskii A, Sun Z, Slesarev A, Alemany LB, Lu W, Tour JM (2010) Improved synthesis of graphene oxide. ACS Nano 4(8):4806–4814
Özkaya T, Baykal A, Toprak MS (2008) 2-pyrrolidone-capped Mn3O4 nanocrystals. Cent Eur J Chem 6(3):465
Wang D, Li Y, Wang Q, Wang T (2012) Facile synthesis of porous Mn3O4 nanocrystal–graphene nanocomposites for electrochemical supercapacitors. Eur J Inorg Chem 2012(4):628–635
Mani V, Huang S-T, Devasenathipathy R, Yang TC (2016) Electropolymerization of cobalt tetraamino-phthalocyanine at reduced graphene oxide for electrochemical determination of cysteine and hydrazine. RSC Adv 6(44):38463–38469
Mani V, Dinesh B, Chen S-M, Saraswathi R (2014) Direct electrochemistry of myoglobin at reduced graphene oxide-multiwalled carbon nanotubes-platinum nanoparticles nanocomposite and biosensing towards hydrogen peroxide and nitrite. Biosens Bioelectron 53:420–427
Si P, Dong X-C, Chen P, Kim D-H (2013) A hierarchically structured composite of Mn 3 O 4/3D graphene foam for flexible nonenzymatic biosensors. J Mater Chem B 1(1):110–115
Liu C, Zhang H, Tang Y, Luo S (2014) Controllable growth of graphene/cu composite and its nanoarchitecture-dependent electrocatalytic activity to hydrazine oxidation. J Mater Chem A 2(13):4580–4587
Zhang S, Liu X, Huang N, Lu Q, Liu M, Li H, Zhang Y, Yao S (2016) Sensitive detection of hydrogen peroxide and nitrite based on silver/carbon nanocomposite synthesized by carbon dots as reductant via one step method. Electrochim Acta 211:36–43
Lin P, Chai F, Zhang R, Xu G, Fan X, Luo X (2016) Electrochemical synthesis of poly (3, 4-ethylenedioxythiophene) doped with gold nanoparticles, and its application to nitrite sensing. Microchim Acta 183(3):1235–1241
Chen D, Jiang J, Du X (2016) Electrocatalytic oxidation of nitrite using metal-free nitrogen-doped reduced graphene oxide nanosheets for sensitive detection. Talanta 155:329–335
Mehmeti E, Stanković DM, Hajrizi A, Kalcher K (2016) The use of graphene nanoribbons as efficient electrochemical sensing material for nitrite determination. Talanta 159:34–39
Li Z, An Z, Guo Y, Zhang K, Chen X, Zhang D, Xue Z, Zhou X, Lu X (2016) Au-Pt bimetallic nanoparticles supported on functionalized nitrogen-doped graphene for sensitive detection of nitrite. Talanta 161:713–720
Shen Y, Rao D, Bai W, Sheng Q, Zheng J (2017) Preparation of high-quality palladium nanocubes heavily deposited on nitrogen-doped graphene nanocomposites and their application for enhanced electrochemical sensing. Talanta 165:304–312
Chen L, Liu X, Wang C, Lv S, Chen C (2017) Amperometric nitrite sensor based on a glassy carbon electrode modified with electrodeposited poly (3, 4-ethylenedioxythiophene) doped with a polyacenic semiconductor. Microchimica Acta 184(7):2073–2079
Wang G, Han R, Feng X, Li Y, Lin J, Luo X (2017) A glassy carbon electrode modified with poly (3, 4-ethylenedioxythiophene) doped with nano-sized hydroxyapatite for amperometric determination of nitrite. Microchim Acta 184(6):1721–1727
Li S-J, Zhao G-Y, Zhang R-X, Hou Y-L, Liu L, Pang H (2013) A sensitive and selective nitrite sensor based on a glassy carbon electrode modified with gold nanoparticles and sulfonated graphene. Microchim Acta 180(9–10):821–827
Meng Z, Liu B, Zheng J, Sheng Q, Zhang H (2011) Electrodeposition of cobalt oxide nanoparticles on carbon nanotubes, and their electrocatalytic properties for nitrite electrooxidation. Microchim Acta 175(3–4):251–257
Haldorai Y, Kim JY, Vilian ATE, Heo NS, Huh YS, Han Y-K (2016) An enzyme-free electrochemical sensor based on reduced graphene oxide/Co3O4 nanospindle composite for sensitive detection of nitrite. Sensors Actuators B Chem 227:92–99. doi:10.1016/j.snb.2015.12.032
Radhakrishnan S, Krishnamoorthy K, Sekar C, Wilson J, Kim SJ (2014) A highly sensitive electrochemical sensor for nitrite detection based on Fe 2 O 3 nanoparticles decorated reduced graphene oxide nanosheets. Appl Catal B Environ 148:22–28
Pandikumar A, Yusoff N, Huang NM, Lim HN (2015) Electrochemical sensing of nitrite using a glassy carbon electrode modified with reduced functionalized graphene oxide decorated with flower-like zinc oxide. Microchim Acta 182(5–6):1113–1122
Wang Q, Yun Y (2012) A nanomaterial composed of cobalt nanoparticles, poly (3, 4-ethylenedioxythiophene) and graphene with high electrocatalytic activity for nitrite oxidation. Microchim Acta 177(3–4):411–418
Huang S-S, Liu L, Mei L-P, Zhou J-Y, Guo F-Y, Wang A-J, Feng J-J (2016) Electrochemical sensor for nitrite using a glassy carbon electrode modified with gold-copper nanochain networks. Microchim Acta 183(2):791–797
Wang H, Wen F, Chen Y, Sun T, Meng Y, Zhang Y (2016) Electrocatalytic determination of nitrite based on straw cellulose/molybdenum sulfide nanocomposite. Biosens Bioelectron 85:692–697
Rabti A, Aoun SB, Raouafi N (2016) A sensitive nitrite sensor using an electrode consisting of reduced graphene oxide functionalized with ferrocene. Microchim Acta 183(12):3111–3117
Wu W, Li Y, Jin J, Wu H, Wang S, Ding Y, Ou J (2016) Sensing nitrite with a glassy carbon electrode modified with a three-dimensional network consisting of Ni7S6 and multi-walled carbon nanotubes. Microchim Acta 183(12):3159–3166
Acknowledgements
This work was supported by the National Science Council and the Ministry of Education of Taiwan (Republic of China) and National Taipei University of Technology, Taipei, Taiwan.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The author(s) declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOCX 3.40 mb)
Rights and permissions
About this article

Cite this article
Muthumariappan, A., Govindasamy, M., Chen, SM. et al. Screen-printed electrode modified with a composite prepared from graphene oxide nanosheets and Mn3O4 microcubes for ultrasensitive determination of nitrite. Microchim Acta 184, 3625–3634 (2017). https://doi.org/10.1007/s00604-017-2379-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-017-2379-9