
Abstract
Southern Europe is home to two naturally occurring pear species: the European wild pear (Pyrus pyraster (L.) Burgsd.) and the almond-leaved pear (P. spinosa Forssk.). In addition to these two species, the cultivated pear (P. communis L.) is also grown throughout Europe. Since the cultivated pear is one of the most widespread fruits in Europe, gene flow between the cultivated species and their wild relatives is to be expected. The aim of this study was to determine the genetic diversity of natural populations and whether gene flow from the cultivated pear can alter the genetic composition of wild pear populations. We collected samples from 21 populations of P. pyraster and 22 populations of P. spinosa as well as 24 cultivars of P. communis. DNA fingerprinting based on nine microsatellite markers (SSR) was used to analyze the genetic diversity and structure of the wild pear populations and to assess the relationship between wild and cultivated pears. In general, we found a higher genetic diversity of P. pyraster compared to P. spinosa. In three Mediterranean P. pyraster populations, significant cultivated-to-wild gene flow was observed, whereas in P. spinosa it was rare and only observed in a few individuals. Furthermore, in regions where the ecological niches of the two wild pear species overlap, hybrids between them were also found. This study provides valuable insights into the genetic variability of these species and can significantly advance their use in sustainable forest management, conservation, and breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Pyrus L. (Rosaceae), pears, is a highly diverse genus composed of both cultivated and wild species (Bell and Itai 2010). The number of pear species varies according to different studies, ranging from 26 (Bell and Itai 2010) to 80 (Browicz 1993). Nowadays, centers of pear diversity are located in eastern Asia, Caucasus and the Mediterranean (Korotkova et al. 2018), prompting the popular division into Oriental (Asian) and Occidental (European) pears (Zheng et al. 2014). Pears are best known for their fruits, and owing to their nutritional value, have a long history of cultivation. The earliest mention of pear cultivation in Europe dates back to Ancient Greece, around 3000 years ago, when the Greek poet Homer referred to them as a “gift from God” (Layne and Quamme 1975). Today, pears are among the most important commercially grown fruits, with more than 3000 cultivars registered (Bielsa et al. 2021). In addition, species from the genus Pyrus have very high quality and valuable wood, used in the production of musical instruments and furniture (Parle 2016). Generally speaking, wild pear species are extremely important for the maintenance of biodiversity, as their fruits are food for many mammals such as foxes, badgers and martens, while their flowers are pollinated by different insects (Debussche and Isenmann 1989; Pigozzi 1992).
Among the most important wild pear species in Europe is the European wild pear (Pyrus pyraster (L.) Burgsd.). This species grows in deciduous forests, usually in a scattered distribution pattern individually or in smaller groups (Paganová 2011). Due to their poor competitive ability compared to other species, as well as pronounced heliophilia, they most commonly grow along the edges of forests, on neglected agricultural lands and in extreme, marginal habitats (Stephan et al. 2003). We usually find them in areas with cool-temperate climates, in lowlands and hills, and rarely in mountains and deciduous sub-Mediterranean forests characterized by somewhat longer rain periods. In southeast Europe, this pear species sometimes grows with another related species from the Circum-Mediterranean group of pears (Bell and Itai 2010), almond-leaved pear (P. spinosa Forssk.). Almond-leaved pear is a species adapted to more xerothermic habitats (Vidaković et al. 2021, 2024), mostly growing in discontinuous, widely spaced bush associations with open spaces in Mediterranean forests, usually called garrigue. Although the ecological niches of these two species seldom overlap, in the habitats where they grow together, one can also find their hybrids (Kumar et al. 2017).
Namely, as pears have been cultivated since ancient times and are one of the most widely grown fruit species in Europe (Deckers and Schoofs 2008), the vicinity of cultivated trees to natural populations is inevitable. In that case, when cultivated and wild taxa are sympatric, both are fertile and sexually compatible and there is flowering synchrony, interspecific gene flow can be expected (Oliveira et al. 2021). One of the most important cultivated pears, not just in this part of Europe but in other temperate regions around the world is the common pear (Pyrus communis L.) (Bound 2021). The majority of orchards in Europe are composed of cultivars of precisely this species, characterized by fruits of different shapes, sizes and colors, times of fruit ripening, etc. Spontaneous hybridization between cultivated and wild pears has already been confirmed on several occasions (Iketani et al. 2010; Jiang et al. 2016; Bisi et al. 2021), including the hybrids between common pear and European wild pear (Dolatowski et al. 2005; Bennici et al. 2018; Wagner and Büttner 2019), as well as the hybrids between common pear and almond-leaved pear (Bennici et al. 2018; Sau et al. 2020).
In previous studies, various molecular markers were used to explore the genetic diversity of natural pear populations (Dolatowski et al. 2005; Liu et al. 2012; Wolko et al. 2015) and cultivars (Sehić et al. 2012; Liu et al. 2015; Nishio et al. 2016; Ferradini et al. 2017; Kumar et al. 2017; Queiroz et al. 2019; Kocsisńe et al. 2020; Ouni et al. 2020; Bielsa et al. 2021), including amplified fragment length polymorphisms (AFLPs), random amplified polymorphic DNA (RAPD), restriction fragment length polymorphisms (RFLPs), simple sequence repeats (SSRs), and noncoding regions of chloroplast DNA (cpDNA). However, the majority of previous studies on the genetic diversity of wild and cultivated pears were performed using simple sequence repeat (SSR) markers or microsatellites (Wolko et al. 2015; Reim et al. 2017; Queiroz et al. 2019; Bielsa et al. 2021). Such studies provide data on a variety of evolutionary parameters, including genetic variability and its partitioning on variability within and between populations, levels of inbreeding, effective population sizes, and the dynamics of recent population bottlenecks. In general, the maintenance of wild populations’ genetic diversity is crucial for the long-term survival of a species and its adaptation (Liu et al. 2012), especially in populations in extreme climatic sites (Hampe and Petit 2005). Therefore, studies covering the issue provide useful information for the conservation and management of wild species, which is increasingly represented in national and international policies (Pearman et al. 2024).
With regard to the natural populations of European wild pear and almond-leaved pear, there have only been a few studies conducted to date that addressed their genetic (Wolko et al. 2015; Reim et al. 2017) and morphological (Paganová 2011; Vidaković et al. 2021, 2022, 2024; Vidaković and Poljak 2024) variability, and featuring only a small number of populations. However, more extensive genetic studies were performed on other wild pears, such as P. calleryana Decne (Liu et al. 2012; Sapkota et al. 2021, 2022), P. elaeagrifolia Pall. (Uzun et al. 2022), P. pyrifolia (Burm.f.) Nakai (Jiang et al. 2009) and P. ussuriensis Maxim. ex. Rupr. (Katayama et al. 2007, 2016), all of which have significant economic value in their respective regions. Nevertheless, the majority of studies carried out to date on species from the genus Pyrus were focused on the conservation and evaluation of pear cultivars.
The main objectives of this study were: (1) to assess genetic diversity and structure of two wild pear species, i.e., European wild pear and almond-leaved-pear, in the area that covers Eastern Adriatic coast and North-western part of the Balkan Peninsula; (2) to test whether neutral spatial genetic variation correlates with the geographic (isolation by distance—IBD) or environmental variation (isolation by environment—IBE); (3) to test for the occurrence of hybrid genotypes between European wild pear and almond-leaved pear in the wild were the ecological niches of these two species overlap; and (4) to test for the occurrence of hybrid genotypes between cultivated and wild species in the natural populations.
Material and methods
Study area and plant material
The research was conducted on three different Pyrus taxa, including two wild species, P. pyraster and P. spinosa, and one cultivated, P. communis. In total, 740 individuals form 21 wild populations of P. pyraster, 22 wild populations of P. spinosa and one orchard of P. communis were included in the research (Fig. 1, Suppl. Table 1). The research area covers the eastern Adriatic coast and the northwestern part of the Balkan Peninsula, where the distributions of these two wild species overlap in certain parts. Pyrus communis samples were collected from the Agro-Car orchard in Novaki Bistranski, in the vicinity of Zagreb, specialized in the production of planting material of fruit crops. The samples collected in this orchard included 24 registered cultivars (Croatian Agency for Agriculture and Food 2021; Suppl. Table 2).

Geographical location of studied Pyrus pyraster (P01-P21, blue circles) and P. spinosa (P22-P43, green circles) populations and P. communis orchard (P44, pink circle). Populations: P01–Kuberton; P02–Lupoglav; P03–Krk; P04–Rumin; P05–Kamešnica; P06–Studenci; P07–Perušić; P08–Plitvička jezera; P09–Brinje; P10–Ogulin; P11–Lukovdol; P12–Karlovac; P13–Žumberak; P14–Strahinščica; P15–Kalnik; P16–Hrvatska Kostajnica; P17–Moslavačka gora; P18–Lipovljani; P19–Okučani; P20–Donji Miholjac; P21–Vinkovci; P22–Tar; P23–Žminj; P24–Pula; P25–Vir; P26–Nin; P27–Obrovac; P28–Krka; P29–Drniš; P30–Radošić; P31–Klis; P32–Sinj; P33–Bisko; P34–Seoca; P35–Batinići; P36–Brač; P37–Hvar; P38–Kotišina; P39–Vrgorac; P40–Pelješac; P41–Mljet; P42–Slano; P43–Konavle; P44–orchard
To conduct DNA extraction, fully developed buds were collected from each tree in the autumn of 2022. Upon the collection, buds were immediately put in filter bags, which were then stored in plastic bags with silica gel. The samples were kept in silica gel-filled bags at room temperature and in the dark until further analysis. All plant material was collected according to relevant institutional, national, and international guidelines. Voucher specimens were identified by Antonio Vidaković and Igor Poljak and are deposited in the DEND Herbarium, Zagreb, Croatia.
DNA extraction and microsatellite characterization
Total genomic DNA was extracted from collected samples using the DNeasy Plant Mini Kit (Qiagen GmbH, Hilden, Germany), following instructions provided by the manufacturer. Additionally, 1% polyvinylpyrrolidone (PVP) and 1% β-mercaptoethanol in lysis buffer were added to the extraction process for better performance. Finally, DNA concentrations were measured with a NanoPhotometer P300 (Implen GmbH, Munich, Germany).
Nine microsatellite SSR markers were used for genetic analysis (Suppl. Table 3). These included EMPc10, EMPc104, EMPc105, EMPc111, EMPc115, EMPc117, NB105, NB113 and NH025, produced for P. communis (Fernández-Fernández et al. 2006; Lukoseviciute et al. 2013; Urrestarazu et al. 2015; Rana et al. 2015). For polymerase chain reactions (PCRs), a total volume of 20 μL containing 1 × PCR buffer, 1.5 mM MgCl2, 200 μM each of dNTPs, 5 μM forward and reverse primers, 0.5 U TaqHS polymerase (TaKaRa Bio Inc. Shiga, Japan) and 5 ng of template DNA were used. The resulting products were analyzed by capillary electrophoresis on an ABI 3730xL DNA analyzer (Applied Biosystems, Foster City, CA, USA) provided by Macrogen DNA service (Amsterdam, Netherlands), and alleles were scored using GeneMapper software version 4.0 (Applied Biosystems, Foster City, CA, USA).
Data analysis
The total number of alleles (Na), polymorphic information content (PIC) and probability of identity (PI) of each microsatellite locus were calculated separately for each species and for the entire dataset using Cervus v. 3.0 (Kalinowski et al. 2007). The microsatellite data of P. pyraster and P. spinosa populations were checked for the presence of null alleles and scoring errors using Micro-Checker v. 2.2.3 (van Oosterhout et al. 2004) and the frequencies of null-alleles were estimated using the expectation–maximization algorithm in FreeNA (Chapuis and Estoup 2007). The accessions of P. communis were excluded from the analysis of null alleles and scoring errors, as this is a group of cultivars for which the Hardy–Weinberg equilibrium (HWE) assumption cannot apply in principle. Allele frequencies adjusted for the presence of null alleles in FreeNA were used to recalculate the expected heterozygosity values [HE(null)] in each population of P. pyraster and P. spinosa and compare them with the original values (HE) using the Wilcoxon rank-sum test in SAS v. 9.4 (SAS Institute Inc. Cary, NC, USA).
Diversity within populations was determined by calculating the average number of alleles (Nav), allelic richness (Nar), number of private alleles (Npr) and private allelic richness (Npar) using HP-Rare v. 1.0 (Kalinowski 2005). Population genetic parameters such as the observed heterozygosity (HO), the expected heterozygosity (HE) and the inbreeding coefficient (FIS) were calculated using Genepop v. 4.7 (Rousset 2008). Possible deviations from Hardy–Weinberg equilibrium (HWE) were tested in Genepop and the significance level was adjusted after sequential Bonferroni corrections for multiple testing using SAS. Populations that showed significant deviation from Hardy–Weinberg equilibrium were further analyzed using INEST v. 2.2 (Chybicki and Burczyk 2009), which applies a Bayesian approach (individual inbreeding model; IIM) to simultaneously estimate the effects of null alleles and inbreeding. The full model (‘nfb’), which includes the effect of inbreeding and genotypic failures (FIS > 0), was tested against a random model (‘nb’) that assumes the presence of null alleles and genotypic failures (FIS = 0). The parameters used were set as follows: 500,000 MCMC cycles with an update every 200 cycles and a burn-in of 50,000. The deviance information criterion (DIC) was used to compare the models. Friedman’s non-parametric two-way ANOVA was used to assess differences in Nar, HO and HE among species and markers. When results were significant for species, Wilcoxon signed-rank tests were applied to detect significant differences among pairs of species. Both analyses were performed using SAS.
The signs of bottlenecks in the populations of P. pyraster and P. spinosa were examined with the heterozygosity-excess test (Cornuet and Luikart 1996) and the M-ratio test (Garza and Willimason 2001) using INEST. The test for heterozygosity excess, indicating a recent population reduction, was performed under the two-phase model (TMP) using the parameter values recommended by Peery et al. (2012): the proportion of multistep mutations, pg = 0.22 and the average multi-step mutation size, δg = 0.31. The M-ratio was calculated as the average ratio between the total number of alleles (k) and the overall allele size range (r) in each population. A significant value of the M-ratio test is strong evidence of a past bottleneck. The significance of both tests was determined using Wilcoxon’s signed-rank test P-values based on 1,000,000 permutations.
To evaluate the genetic differentiation among populations (P. pyraster, P. spinosa) and a group of cultivars (P. communis), the pairwise FST values and the respective P values (after 10,000 permutations) were estimated in FSTAT v. 2.9.3.2 (Goudet 1995).
Analysis of molecular variance (AMOVA; Excoffier et al. 1992) was performed in ARLEQUIN v. 3.5.2.2 (Excoffier and Lischer 2010) by partitioning total microsatellite diversity among species, among populations (including a group of cultivars of P. communis) within species, and within populations. Additional two-way AMOVAs were similarly performed for each pair of species. The one-way AMOVAs for partitioning total microsatellite diversity among and within populations were performed separately for P. pyraster and P. spinosa. The significance level of the parameters φ was determined after 10,000 permutations. A model-based clustering method was applied to multilocus microsatellite data to infer genetic structure and determine the number of clusters in the dataset using STRUCTURE (Pritchard et al. 2000). The analysis was performed for the entire dataset as well as for each Pyrus species separately. Thirty runs of STRUCTURE were performed, with the number of clusters (K) set from 1 to 11. Each run consisted of a burn-in period of 200,000 steps followed by 1,000,000 Monte Carlo Markov Chain (MCMC) steps, assuming an admixture model and correlated allele frequencies. The calculations were performed on the Padobran computer cluster at the University of Zagreb (Croatia), University Computing Center (SRCE). The choice of the most probable number of clusters (K) was determined by calculating an ad hoc ΔK statistic (Evanno et al. 2005) using the web-based software StructureSelector (Li and Liu 2018), which also allows clustering and merging of the resulting runs according to the CLUMPAK method described in Kopelman et al. (2015). The individuals with membership probabilities (Q) of less than 75% for each cluster were considered to be of mixed origin (Matsuoka et al. 2002).
Isolation by distance (IBD) (Rousset 1997) was analyzed by calculating the correlation between the matrix of the natural logarithm of geographic distances (in km) and the matrix of pairwise FST/(1 − FST) ratios between population pairs separately for P. pyraster and P. spinosa. Significance was determined by the Mantel test after 10,000 permutations using NTSYS-pc v. 2.21L (Rohlf 2009).
Environmental dissimilarity between populations was based on data of the average bioclimatic conditions for the period from 1970 to 2000, in the area of the studied populations obtained from the WorldClim 2 database with a spatial resolution close to a square kilometer (Fick and Hijmans 2017). The bioclimatic variables used in the analysis represent yearly, seasonal, and monthly averages and extremes in temperature and precipitation. All 19 bioclimatic variables (Suppl. Table 1) were included in the analysis for calculating the environmental dissimilarity matrix. Firstly, the PRINCOMP procedure in SAS was used to perform principal component analysis and the pairwise Euclidean distances between population scores on the first two principal components were used as a measure of environmental dissimilarity between populations. Accordingly, the isolation by environment (IBE) was analyzed by calculating the correlation between the matrix of environmental dissimilarity and the matrix of pairwise FST/(1 − FST) ratios between pairs of populations. Significance was determined by the Mantel test after 10,000 permutations using NTSYS-pc v. 2.21L (Rohlf 2009).
Results
Microsatellite diversity
Nine SSR markers were used to identify genetic diversity in all three species. In total, 214 alleles were revealed for Pyrus pyraster, 191 for P. spinosa and 94 for P. communis (Suppl. Table 3). The average number of alleles revealed in each species was 23.78, 21.22 and 10.44, respectively. In P. pyraster, the number of alleles varied between 17 (NB105) and 28 (EMPc105, EMPc117). A larger range was revealed in P. spinosa, from 12 (NB113) to 33 (EMPc10), while the number of alleles in P. communis varied between 6 (EMPc104) and 16 (EMPc115). The average Polymorphism Information Content (PIC) values for P. pyraster, P. spinosa and P. communis were 0.824, 0.768 and 0.768, respectively, indicating high informativeness of used markers (Suppl. Table 3). In P. pyraster, PIC values ranged from 0.582 (EMPc10) to 0.936 (EMPc117), in P. spinosa they ranged from 0.519 (NB113) to 0.875 (EMPc117) and in P. communis from 0.686 (EMPc10) to 0.838 (EMPc115). Across all three species, PIC values identified a limited number of moderately informative markers: EMPc10 in all three species, NB113 in P. spinosa and NH025 in P. communis.
Population diversity
Genetic diversity parameters for each population based on allelic frequencies are summarized in Table 1. The average number of alleles in P. pyraster varied from 7.111 (P07) to 11.889 (P21), while allelic richness ranged from 5.286 (P07) to 7.762 (P03). The number of private alleles was the highest in P14 (4), followed by P21 (3) and P06 and P09 (2). One private allele each was found in P02, P10, P18 and P19, while no private alleles were found in the remaining 13 populations of P. pyraster. Observed and expected heterozygosity were the highest in P04 (0.972) and P03 (0.870), respectively, while both values were the lowest in P07 (0.627 and 0.681). After Bonferroni correction, significant deviation from Hardy–Weinberg equilibrium (HWE) was detected only in P04. However, the DIC value of the model assuming the presence of null alleles and genotyping failures (DICnb = 720.60) was slightly lower than the full model including the effect of inbreeding (DICnfb = 720.95). A total number of detected null alleles across all of 189 microsatellite loci × population combinations in P. pyraster was 12, with the frequency from 0.093 (EMPc115 in P09) to 0.190 (EMPc105 in P02). The largest number of null alleles in P. pyraster was found in EMPc105 (Supplementary Table 4). The expected heterozygosity values adjusted for the presence of null alleles [HE(null)] did not differ significantly from the original values in any population (PWilcoxon = 0.62–0.95), indicating that null alleles had no effect on the results.
Overall lower allelic diversity was found in P. spinosa, where the average number of alleles varied from 5.000 (P37) to 8.889 (P25, P30, P35), while allelic richness was within the range of 4.426–6.426, with the lower limit of the range characteristic of P37, and the upper limit of P35 (Table 1). Furthermore, a number of private alleles was notably lower as well, with only five populations with one private allele each (P35, P38, P39, P42, P43), and the rest of populations without any private alleles. Expected and observed heterozygosity were very similar across studied populations of P. spinosa and ranged from 0.683 (P38) to 0.806 (P24), and from 0.688 (P22) to 0.810 (P43), respectively. In two populations, P36 and P38, a significant deviation from the Hardy–Weinberg equilibrium (HWE) was found. In both populations, the model that excludes the effect of inbreeding fitted the data better [DICnb(P36) = 781.09, DICnb(P38) = 1109.78] than the full model [DICInfb(P36) = 781.09, DICnfb(P38) = 1112.04], suggesting that the deviation from HWE is due to the presence of null alleles and not to inbreeding. Compared to P. pyraster, a larger number of null alleles was detected in P. spinosa. The total number of null alleles across all 198 microsatellite loci × population combinations in P. spinosa was 29, with the frequency ranging from 0.074 (EMPc104 × P28) to 0.316 (EMPc104 × P40) (Suppl. Table 4). The highest number of null alleles was found in EMPc104, in which they were found in 14 out of 22 populations. As with P. pyraster, no significant differences were found between HE and HE(null) in any P. spinosa population (PWilcoxon = 0.25–0.95).
The average number of alleles in P. communis was 10.22, with an allelic richness of 6.186. Within P. communis samples only one private allele was found. Observed heterozygosity and expected heterozygosity were both higher than the average values in the other two species, being 0.857 and 0.812, respectively.
Allelic richness differed significantly among species, with P. pyraster having a significantly higher value than P. spinosa and P. communis. The observed heterozygosity was also found to be significantly different among species according to the Friedman test, but subsequent pairwise comparisons between species were not significant. All three species had the same level of expected heterozygosity. The heterozygosity-excess method utilizing the Wilcoxon signed rank test assuming a two-phased model (TPM), followed by Bonferroni correction, identified a significant recent bottleneck only in one population of P. spinosa, P37. On the other hand, the M-ratio test detected strong signals of population size reduction in the past in six populations of P. pyraster (P04, P06, P08, P09, P14, P17) and six populations of P. spinosa (P22, P23, P27, P30, P38, P43) (Table 1).
Population genetic differentiation and structure
Genetic differentiation values (FST) were calculated between all pairs of populations within each species (Suppl. Table 5). In P. pyraster, the genetic differentiation ranged from 0.003 (between P12 and P13) to 0.126 (between P05 and P07). Out of 210 possible pairs, statistically significant differences were found in 160 cases, or in 76.2%. Population P06 showed significant differentiation from all of the other populations, while on the other hand, population P16 was differentiated only from P06. Furthermore, among the most differentiated populations were also P05 and P14, which showed significant differences from all but one population (P16), and P04, P15 and P21, which showed significant differences from all but two populations each.
The opposite results were found for P. spinosa. Namely, its populations were revealed to be much more similar than the populations of P. pyraster, and significant differences were found only in 76 out of a possible 231 cases, which is 32.9% (Suppl. Table 6). The genetic differentiation between P. spinosa populations ranged from 0 (between P36 and P42) to 0.117 (between P22 and P41). The most differentiated populations were P40 and P37, which showed significant differences from all but one population (P35) and four populations (P26, P29, P32 and P41), respectively. On the other hand, the least differentiated populations were P26, which had only one population it was significantly differentiated from (P40), and P29 and P31, which were significantly differentiated from two populations each.
The results of the two-way AMOVAs conducted for all three species showed statistically significant differences between species, among populations within species and within populations (Table 2). The two-way AMOVAs revealed that most of the total variation (85.83%) could be attributed to within-population variation, while between-species and between-population differences within-species accounted for 10.51% and 3.66% of the total variability, respectively. However, both sources of variation were highly significant (P < 0.0001). Pairwise two-way AMOVAs between species showed that the highest percentage of total variation in the between-species variance component was between P. spinosa and P. communis (11.27%) and the lowest was between P. pyraster and P. communis (4.27%). The results of one-way AMOVAs among and within populations of P. pyraster and P. spinosa showed that only a limited percentage (4.75% and 3.58%) of the total variance could be attributed to among-population variation, although the φST values were very highly significant in both cases (P < 0.0001).
For the purpose of classifying individuals into genetically differentiated groups assuming Hardy–Weinberg equilibrium as well as linkage equilibrium within each group, a Bayesian analysis of the population structure was performed. The highest ΔK (4671.80) value was obtained at K = 2, which revealed the most probable division of studied individuals into two genetic clusters, followed by ΔK of 116.89 at K = 3 (Suppl. Figure 1). At K = 2, populations P01-P21 and P44 were assigned to Cluster K2_A including all the populations belonging to P. pyraster and P. communis, while all the populations belonging to P. spinosa (P22-P43) were assigned to Cluster K2_B (Fig. 2A). In Cluster K2_A, 17 individuals were of a mixed origin (Q < 0.75), all of which were present in populations P01-P06, with the exception of one sample in P16. In Cluster K2_B, 11 individuals proved to be of a mixed origin.

The results of structure analysis at K = 2 (A) and K = 3 (B) depicted as barplots with posterior probabilities of membership of each individual into each genetic cluster. Populations acronyms as in Fig. 1
The second most probable division of populations was at K = 3. The three clusters correspond to the three species studied, P. pyraster (Cluster K3_A), P. spinosa (Cluster K3_B) and P. communis (Cluster K3_C) (Fig. 2B). At the individual tree level, 59 individuals initially collected as P. pyraster were assigned to Cluster K3_C, i.e. P. communis, and two individuals to Cluster K3_B, or P. spinosa. In total, 58 individuals of P. pyraster were of mixed origin, most of which had significant admixture with P. communis. With the exception of two individuals being assigned to Cluster K3_C, all of the individuals of P. spinosa were assigned to Cluster K3_B. Within P. spinosa, 15 individuals were found to be of a mixed origin. Attribution of P. communis individuals to Cluster K3_C proved to be complete for all individuals. In total, 45 hybrids between P. pyraster and P. communis were found, the majority of which were located in P02, P03, P14, P20 and P21. Furthermore, 14 cases of hybrids between P. spinosa into P. pyraster were detected, all of which were present in the areas of overlapping distributions of these two species. Hybridization between P. spinosa and P. communis was also confirmed, as 10 individuals showed such genetic structuring.
A model-based clustering method was also performed for each species separately. In P. pyraster, the most probable division of individuals was at K = 2, followed by K = 3 (Suppl. Figure 2). At K = 2, the majority of individuals in sub-Mediterranean populations P01-P06 and continental populations P16, P20 and P21 were assigned to Cluster K2_A, while all of the remaining populations had individuals belonging to Cluster K2_B (Fig. 3A). At K = 3, populations P01, P02 and P16, which correspond to those with the highest number of hybrid trees with cultivated P. communis, had the greatest proportion of Cluster K3_A. Cluster K3_B consisted mostly of individuals in populations P03, P06, P14, P15, P20 and P21, while the proportion of the Cluster K3_C was the highest in central populations P07-P13 (Fig. 3B).

The results of structure analysis for each species, depicted as barplots with posterior probabilities of membership of each individual into each genetic cluster. Pyrus pyraster at K = 2 (A) and K = 3 (B) and P. spinosa at K = 2 (C) and K = 4 (D)
In P. spinosa, the most probable division of individuals was into four clusters, followed by division into two clusters (Suppl. Figure 3). At K = 2, the majority of studied individuals showed mixed ancestry between clusters K2_A and K2_B, with the exceptions of P36, P40, P42 and P43, which had a somewhat greater proportion of Cluster K2_B (Fig. 3C). At K = 4, most of the studied individuals had even proportion of four clusters. However, the northernmost populations P22-P24, and southern populations P36-P42 stand out as the ones with the greatest proportion of clusters K4_C and K4_D (Fig. 3D).
Association between genetic, geographical and environmental variation
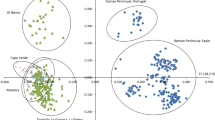
The results of Mantel tests identified significant correlations between geographical and genetic distances in both wild pear species, P. pyraster (r = 0.436, PMantel < 0.0001) and P. spinosa (r = 0.378, PMantel < 0.0001). Therefore, the genetic variability of the studied populations showed significant dependence on geographical distances between populations, indicating greater genetic similarity in geographically adjacent populations. On the other hand, genetic distances did not correlate significantly with environmental distances (Fig. 4).

Isolation by geographical distance (IBD) and isolation by environmental distance (IBE) in Pyrus pyraster (A) and P. spinosa (B) populations. Scatter plots of Mantel’s tests showing the relationships between geographic and genetic distances on the left, and between environmental and genetic distances on the right
Discussion
Our results confirmed the high informativeness of all SSR markers used in describing the genetic diversity of P. pyraster, P. spinosa and P. communis. Overall, the microsatellite markers used in this research had the highest informativeness in P. pyraster, followed by lower values in P. spinosa and P. communis. Overall, all of the markers except EMPc10 showed high informativeness across all three species, with PIC values above 0.8. Such results are in accordance with previously reported PIC values for these markers (Wolko et al. 2010, 2015; Rana et al. 2015; Ouni et al. 2020). The average number of alleles in P. pyraster ranged from 17 to 28, which is higher than previously reported values for comparable microsatellites (Rugienius et al. 2013; Wolko et al. 2015; Reim et al. 2017). With the average number of alleles ranging from six to 16, P. communis had a similar or somewhat higher number of alleles per marker, compared to values reported by numerous authors (Fernández-Fernández et al. 2006; Wolko et al. 2010; Sehić et al. 2012; Lukoseviciute et al. 2013; Urresterazu et al. 2015; Rana et al. 2015; Reim et al. 2017; Ouni et al. 2020). Genetic diversity of wild P. spinosa has not been investigated so far. This study has thus proved that microsatellite markers can be successfully used for assessing its genetic diversity.
Our study revealed that P. pyraster contains high and roughly uniform genetic diversity throughout the study area. With the average expected heterozygosity of 0.80, observed heterozygosity of 0.81 and average allelic richness of 6.55, P. pyraster stands out as a genetically very diverse species, compared to other ecologically and biologically similar species, like Prunus avium (L.) L. (Schueler et al. 2006), Sorbus aucuparia L. (Yousefzadeh et al. 2021), S. torminalis (L.) Crantz (Bednorz et al. 2006) and Malus sylvestris (L.) Mill. (Coart et al. 2003; Reim et al. 2020). In addition, the genetic diversity of P. pyraster populations included in our research was higher compared to populations in Poland (Wolko et al. 2015) and Germany (Reim et al. 2017). This greater diversity was expected, given that the Balkan Peninsula served as a refugial area for many plant species during the last glaciation (Hewitt 2004), probably including P. pyraster. It is well known that populations from Northern Europe have a much lower genetic diversity than populations from Southern Europe (Pearman et al. 2024), as they resulted from recolonization events from the Balkan, Apennine and Iberian peninsulas, where in general populations have retained a high genetic diversity (Bennett et al. 1991; Taberlet and Cheddadi 2002). The recolonization of former habitats usually involves a founder effect leading to decreased genetic diversity (Stewart et al. 2010). Therefore, higher genetic diversity is to be expected at low to intermediate latitudes, where many temperate species have survived glaciation periods (Lawrence and Fraser 2020) since genetic diversity had time to accumulate over a longer period of evolution (Adams and Hadly 2013). Furthermore, even at the local scale of our research, southern populations showed greater differentiation, containing unique alleles and have experienced significant historical bottleneck, which may indicate that a refugial population of European wild pear was indeed located nearby (Petit et al. 2003). According to Cornille et al. (2013), a refugial population of European crabapple (Malus sylvestris) was located in the Balkan Peninsula, in close proximity to populations P04-P06. Considering major similarities in biology and ecology between European crabapple and European wild pear, it is safe to assume they survived in the same area during the last glaciation. Nowadays, however, P. pyraster is rather rare in this area, with very low population density, suggesting these individuals might be the remnants of a large historical population, which lost its size due to low competitiveness with other species and selective pressures of climate change (Yousefzadeh et al. 2021).
Significant inbreeding was detected in a single population of P. pyraster, P04. It is a small population at high altitude and a strained ecological niche for this species. Despite a small number of individuals in this populations, relatively high allelic richness was found, indicating high mate availability and high S-allele diversity. This suggests that small populations of self-incompatible species such as P. pyraster do not necessarily suffer from limitations in mating within the population (Holderegger et al. 2008). However, reproductive isolation from neighboring populations seems to cause significant inbreeding in this population. Namely, this population is located in the high mountain area of the Dinaric Alps, where the terrain configuration is limiting gene flow, as it is separated from the nearest other population (P05) by mountain peaks and valleys. Since it is an animal-dispersed species, gene flow between such populations is very difficult, and the number of possible seed transfer vectors is limited. In addition, due to heterogeneous habitat conditions, divergence in reproductive phenology might also play an important role in gene flow limitation (Bonner et al. 2019).
The genetic diversity in P. spinosa was somewhat lower than in P. pyraster. To the best of our knowledge, this is the first study on population genetics of wild P. spinosa. To date, only a few genetic studies have been conducted on this species, all related to its interaction with local cultivars in Italy (Bennici et al. 2018; Sau et al. 2020). The genetic diversity reported here is similar to that of other wild fruit species in the area, such as P. avium (Mariette et al. 2010) and M. sylvestris (Kišek et al. 2021), but also various landraces of Pyrus (Ferradini et al. 2017). However, the obtained values of genetic diversity for P. spinosa are higher than those of Sorbus domestica L. in the same region (George et al. 2015). Genetic diversity was very uniform throughout all the studied populations, and no population stands out as the most genetically diverse. Furthermore, FST values indicated there were no significant pairwise differences between most of the studied populations, especially in the central part of the study area (P25-P36). Their similarity suggests a natural continuum between neighboring populations, due to which it might be considered as one large population. Unlimited gene flow between these populations can be attributed to the abundance of animals that serve as vectors of P. spinosa seed dispersal, such as foxes, badgers and martens (Debussche and Isenmann 1989; Pigozzi 1992), and even wild boars, whose settling onto Adriatic islands is increasing (Tomljanović and Grubešić 2014). Apart from animals, human impact on the homogenous genetic diversity of P. spinosa populations in this area must be considered, as it is commonly used as a rootstock for grafting commercial pear cultivars (Matsumoto et al. 2006). On the other hand, populations P37, P40 and P43, and to some extent P22, P23 and P24 stood out with greater differences compared to other populations. Their distinctiveness was also confirmed by the STRUCTURE analysis. These populations are geographically the most isolated ones. Populations P22, P23 and P24 are located in the northernmost part of the study area, separated by the large area where P. spinosa was not found. Population P37 is located on the highest peak of the island Hvar (Sveti Nikola, 626 m.a.s.l.), while population P40 is located in the northernmost part of the steep Pelješac peninsula, with no other populations nearby, which makes it a de facto insular population. Increased differentiation of the populations is expected with the increasing distance of the island population from the mainland (Frankham 1997) and was previously confirmed for various plant species (Hufford et al. 2014). Nevertheless, despite the geographical isolation, these populations showed significant genetic diversity, equaling that of mainland populations. Similar levels of genetic diversity between mainland and island populations were previously confirmed in another woody species, Periploca laevigata Aiton (García-Verdugo et al. 2015). The authors explained such pattern of genetic diversity with high dispersal ability and potential for population establishment and persistence, and by the general hypothesis that locally widely distributed species are associated with moderate to high levels of genetic diversity (Gitzendanner and Soltis 2000).
According to AMOVAs, a small fraction of the total variability of both wild species could be attributed to the inter-population variability, with more than 95% of the total variability associated with intra-population variability. The same was previously confirmed for P. pyraster populations in Poland (Wolko et al. 2015) and Germany (Reim et al. 2017). In addition, the distribution of total variability was the same as in other woody species, i.e. intra-population variability was much higher compared to inter-population variability (Wolko et al. 2015; Li et al. 2020). As expected, significant differences were also confirmed between the three species studied. However, the differentiation between P. pyraster and P. communis was weaker than that between P. spinosa and P. communis. This result is most likely prompted by the close relation between P. pyraster and P. communis, as shown by the STRUCTURE analysis as well. Namely, the most probable division of the entire dataset was into two distinct groups, the first one corresponding to P. pyraster and P. communis, and the second one to P. spinosa. This result may be attributed to a close phylogenetic relationship between P. pyraster and P. communis, as the former is considered to be one of the main wild ancestors of the latter (Heywood and Zohary 1995; Volk et al. 2006; Asanidze et al. 2011).
Natural hybridization is one of the major contributors to plant evolution and can cause a wide range of evolutionary consequences, such as adaptation and speciation (Wong et al. 2022; Wu et al. 2024). Hybrids usually display intermediate and transgressive morphological characters of their parents (Wang et al. 2021), often resulting in taxonomic complexity within the group (Wu et al. 2024). One of the families with a large number of interspecies hybrids is Rosaceae (Fritz 2004). Within this family, natural interspecific hybridization has been confirmed in Crataegus L. (Vaškova and Kolarčik 2019), Prunus L. (Szymajda et al. 2022), Rosa L. (Mercure and Bruneau 2008), Sorbus L. (Wu et al. 2024) and Pyrus (Bell and Hough 1986), to name a few. Genus Pyrus is known to have no major incompatibility barriers to interspecific hybridization (Bell and Hough 1986; Kumar et al. 2017). Therefore, natural hybrids can occur in the areas of overlapping distributions (Sadeghnejad et al. 2014). Indeed, our study confirmed hybridization between two wild species, P. pyraster and P. spinosa, which was, to the best of our knowledge, only reported by Dostálek (1980) in Bulgaria. However, the mentioned report described the hybrid solely based on morphological characters. In total, our study confirmed 14 hybrid individuals, all of which were present in a very limited area of overlapping distributions of these two species. Furthermore, although not tested in this research, based on the research by Westwood and Bjornstad (1971) conducted on various Pyrus taxa, fertile offspring are expected to result from these hybrids.
In addition, hybridization between cultivated and wild pear populations was detected in our research. In total, 55 individuals were found to have a significant admixture of cultivated P. communis, 45 of which were P. pyraster, and only 10 were P. spinosa. This disparity probably stems from the fact that P. communis is much more common in the continental part, often grown in commercial plantations, while on the coast it is present mostly as individual trees in private gardens. Due to this, hybridization between P. communis and P. spinosa in the area is hampered, despite their compatibility. Hybridization between the cultivated pears and their wild relatives has been described on multiple occasions (Dostálek 1989; Dolatowski et al. 2004; Bennici et al. 2018; Wagner and Büttner 2019; Sau et al. 2020) and is recognized as the major threat to the genetic diversity of wild species (Wagner and Büttner 2019), which may result in the increased risk of their extinction (Todesco et al. 2016). As cultivated pears became one of the most widespread fruit crops in Europe, the likelihood of contact between them and wild species has been increased, leading to a higher frequency of gene flow between cultivated forms and their wild relatives. This might lead to genetic swamping, by which wild pear could be replaced by hybrids, or demographic swamping, which may lead to reduced population growth rates due to wasteful production of maladaptive hybrids (Todesco et al. 2016). On the other hand, wild relatives of cultivated pear may be valuable for improving its gene pool through alleles providing greater tolerance to abiotic or biotic threats (Cornille et al. 2013). In addition, the presence of feral individuals, i.e. generative descendants of cultivated pears, may not be excluded. They usually retain some domesticated traits but also contain valuable adaptive features (Pisias et al. 2022), usually resulting in higher morphological similarity to wild individuals (Kišek et al. 2021). In general, gene flow between cultivated and wild species is not unexpected and has so far been proven for many economically important fruit species, like apples (Cornille et al. 2013; Ha et al. 2021; Kišek et al. 2021), grapevine (di Vecchi-Staraz et al. 2009; De Andrés et al. 2012), Japanese chestnut (Nishio et al. 2021) and pears (Iketani et al. 2010).
In both P. pyraster and P. spinosa, a significant correlation between genetic and geographical distances, i.e. isolation by distance (IBD), was found. This implies greater similarities between geographically proximate populations, and vice versa (Sexton et al. 2014; Morente-Lopez et al. 2018). Isolation by distance arises from evolutionary mechanisms such as genetic drift and natural selection, which, at greater distances, occur faster than the homogenization of populations by dispersal (Twyford et al. 2020). Both P. pyraster and P. spinosa are mammal-dispersed species, mostly by foxes, badgers, martens and wild boars (Debussche and Isenmann 1989; Pigozzi 1992), all of which are highly territorial. Therefore, possibility of long-distance seed dispersal is highly reduced and can be considered a sporadic event facilitated by birds and humans. A similar pattern of genetic structure has been found in other wild, mammal-dispersed fruit species such as Sorbus domestica (Armbruster et al. 2022), Malus sylvestris (Chen et al. 2023) and Pyrus ussuriensis (Wujun et al. 2015), as opposed to bird-dispersed species like S. torminalis (Angelone et al. 2007) and Prunus mahaleb L. (Jordano and Godoy 2000) where IBD is weak or insignificant. On the other hand, isolation by environment (IBE) was also tested but proved non-significant. In our previous research on morphological variability of P. pyraster and P. spinosa, leaf phenotypic diversity proved to be under significant influence of environment and geographical distances in both species (Vidaković et al. 2021, 2022). This indicated the high phenotypic plasticity and adaptability of these species, which are the key mechanisms in species responses to environmental heterogeneity (Stotz et al. 2021), as they act faster than evolutionary mechanisms (Vázquez et al. 2017).
Conclusions
Overall, our results indicate high genetic diversity in both wild species, P. pyraster and P. spinosa, with the former having somewhat higher values of genetic diversity. Significant differences were confirmed between studied species, and in both wild species notably higher intra-population compared to inter-population variability was found. However, P. spinosa proved to have higher gene flow between populations and therefore genetically more similar populations, probably due to higher population density and lack of geographical barriers. Significant inbreeding and historical bottleneck were found in southern P. pyraster populations, which might indicate the refugial character of these populations as they are located in geographical proximity of the known refugial area of Malus sylvestris, a species ecologically and biologically very similar to P. pyraster. In areas of overlapping distributions of the two wild species studied, interspecific hybrids were found. However, their frequency was rather low. In addition, interspecific hybridization was detected between both wild species and cultivated P. communis, with greater occurrence in P. pyraster. This comes as no surprise, considering there are no major incompatibility barriers to interspecific hybridization in pears. Finally, our results suggest that the main driver of the genetic structure of the studied wild populations is their mutual geographical distance, as a significant correlation between genetic and geographical distances has been found. This study provided insight into the genetic diversity and structure of the two most common wild pear species in Europe and will serve as the baseline data for the creation of guidelines for their protection and conservation, as well as in afforestation programs.
Data availability
The allelic datasets analyzed during the current study are available in the Zenodo repository (https://zenodo.org/doi/https://doi.org/10.5281/zenodo.11185761).
References
Adams RI, Hadly EA (2013) Genetic diversity within vertebrate species is greater at lower latitudes. Evol Ecol 27:133–143. https://doi.org/10.1007/s10682-012-9587-x
Angelone S, Hilfiker K, Holderegger R, Bergamini A, Hoebee SE (2007) Regional population dynamics define the local genetic structure in Sorbus torminalis. Mol Ecol 16:1291–1301. https://doi.org/10.1111/j.0962-1083.2006.03202.x
Armbruster GFJ, Lucek K, Willi Y (2022) Cryptic population structure at the northern range margin of the service tree Sorbus domestica. PeerJ 10:e14397. https://doi.org/10.7717/peerj.14397
Asanidze Z, Akhalkatsi M, Gvritishvili M (2011) Comparative morphometric study and relationships between the Caucasian species of wild pear (Pyrus spp.) and local cultivars in Georgia. Flora 206:974–986. https://doi.org/10.1016/j.flora.2011.04.010
Bednorz L, Myczko L, Kosiński P (2006) Genetic variability and structure of the wild service tree (Sorbus torminalis (L.) Crantz) in Poland. Silvae Genet 55:197–202. https://doi.org/10.1515/sg-2006-0027
Bell RL, Hough LF (1986) Interspecific and intergeneric hybridization of Pyrus. Hortic Sci 21:62–64
Bell RL, Itai A (2010) Pyrus. In: Kole C (ed) Wild crop relatives: genomic and breeding resources. Springer, Berlin/Heidelberg, pp 147–177
Bennett KD, Tzedakis PC, Willis KJ (1991) Quaternary refugia of north European trees. J Biogeogr 18:103–115. https://doi.org/10.2307/2845248
Bennici S, Las Casas G, Distefano G, Di Guardo M, Continella A, Ferlito F, Gentile A, La Malfa S (2018) Elucidating the contribution of wild related species on autochthonous pear germplasm: a case study from Mount Etna. PLoS ONE 13:e0198512. https://doi.org/10.1371/journal.pone.0198512
Bielsa FJ, Irisarri P, Errea P, Pina A (2021) Genetic diversity and structure of local pear cultivars from mountainous areas from Aragon (northeastern Spain). Agronomy 11:1778. https://doi.org/10.3390/agronomy11091778
Bisi RB, Pio R, Locatelli G, da Hora FD, Silva Botelho FB (2021) General and specific combining ability in the selection of polliniser cultivars of hybrid pear trees (Pyrus communis x P. pyrifolia). Sci Hortic 277:109797. https://doi.org/10.1016/j.scienta.2020.109797
Bonner C, Sokolov NA, Westover SE, Ho M, Weis AE (2019) Estimating the impact of divergent mating phenology between residents and migrants on the potential for gene flow. Ecol Evol 9:3770–3783. https://doi.org/10.1002/ece3.5001
Bound SA (2021) Managing crop load in European pear (Pyrus communis L.)—a review. Agriculture 11:637. https://doi.org/10.3390/agriculture11070637
Browicz K (1993) Conspect and chorology of the genus Pyrus L. Arboretum Kórnickie Rocznik 38:17–33
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Chen X, Avia K, Forler A, Remoué C, Venon A, Rousselet A, Lucas G, Kwarteng AO, Rover R, Le Guilloux M, Belcram H, Combes V, Corti H, Olverà-Vazquez S, Falque M, Alins G, Kirisits T, Ursu TM, Roman A, Volk GM, Bazot S, Cornille A (2023) Ecological and evolutionary drivers of phenotypic and genetic variation in the European crabapple [Malus sylvestris (L.) Mill.], a wild relative of the cultivated apple. Ann Bot 131:1025–1037. https://doi.org/10.1093/aob/mcad061
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113. https://doi.org/10.1093/jhered/esn088
Coart E, Vekemans X, Smulders MJ, Wagner I, Van Huylenbroeck J, Van Bockstaele E, Roldán-Ruiz I (2003) Genetic variation in the endangered wild apple (Malus sylvestris (L.) Mill.) in Belgium as revealed by amplified fragment length polymorphism and microsatellite markers. Mol Ecol 12:845–857. https://doi.org/10.1046/j.1365-294x.2003.01778.x
Cornille A, Giraud T, Bellard C, Tellier A, Le Cam B, Smulders MJ, Kleinschmit J, Roldan-Ruiz I, Gladieux P (2013) Postglacial recolonization history of the European crabapple (Malus sylvestris Mill.), a wild contributor to the domesticated apple. Mol Ecol 22:2249–2263. https://doi.org/10.1111/mec.12231
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:1119–1127. https://doi.org/10.1093/genetics/144.4.2001
Croatian agency for agriculture and food (2021) Popis sorti voćnih vrsta. HAPIH. https://www.hapih.hr/wp-content/uploads/2021/07/2.1.-Popis-sorti-vocnih-vrsta.pdf
De Andrés MT, Benito A, Pérez-Rivera G, Ocete R, Lopez MA, Gaforio L, Muñoz G, Cabello F, Martínez Zapater JM, Arroyo-García R (2012) Genetic diversity of wild grapevine populations in Spain and their genetic relationships with cultivated grapevines. Mol Ecol 21:800–816. https://doi.org/10.1111/j.1365-294X.2011.05395.x
Debussche M, Isenmann P (1989) Fleshy fruit characters and the choices of bird and mammal seed dispersers in a Mediterrenean region. Oikos 56:327–338. https://doi.org/10.2307/3565617
Deckers T, Schoofs H (2008) Status of the pear production in Europe. Acta Hortic 800:95–106
Di Vecchi-Staraz M, Laucou V, Bruno G, Lacombe T, Gerber S, Bourse T, Boselli M, This P (2009) Low level of pollen-mediated gene flow from cultivated to wild grapevine: consequences for the evolution of the endangered subspecies Vitis vinifera L. subsp. silvestris. J Hered 100:66–75. https://doi.org/10.1093/jhered/esn084
Dostálek J (1980) Pyrus spinosa und ihre Hybriden in Südwestbulgarien. Folia Geobot Phytotax 15:59–73
Dostálek J (1989) Pyrus × amphigenea, seine Taxonomie und Nomenklatur. Folia Geobot Phytotax 24:103–108
Dolatowski J, Nowosielski J, Podyma W, Szymańska M, Zych M (2005) Molecular studied on the variability of Polish semi-wild pears (Pyrus) using AFLP. J Fruit Ornam Plant Res 12:331–337
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491. https://doi.org/10.1093/genetics/131.2.479
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Fernández-Fernández F, Harvey NG, James CM (2006) Isolation and characterization of polymorphic microsatellite markers from European pear (Pyrus communis L.). Mol Ecol Notes 6:1039–1041. https://doi.org/10.1111/j.1471-8286.2006.01422.x
Ferradini N, Lancioni H, Torricelli R, Russi L, Dalla Ragione I, Cardinali I, Marconi G, Gramaccia M, Concezzi L, Achilli A, Veronesi F, Albertini E (2017) Characterization and phylogenetic analysis of ancient Italian landraces of pear. Front Plant Sci 8:751. https://doi.org/10.3389/fpls.2017.00751
Fick SE, Hijmans RJ (2017) WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int J Climatol 37:4302–4315. https://doi.org/10.1002/joc.5086
Frankham R (1997) Do island populations have less genetic variation than mainland populations? Heredity 78:311–327. https://doi.org/10.1038/hdy.1997.46
Fritz RS (2004) Plant hybrids. In: Levin AS (ed) Encyclopedia of biodiversity. Elsevier, Amsterdam, pp 659–675
García-Verdugo C, Sajeva M, La Mantia T, Harrouni C, Msanda F, Caujapé-Castells J (2015) Do island plant populations really have lower genetic variation than mainland populations? Effects of selection and distribution range on genetic diversity estimates. Mol Ecol 24:726–741. https://doi.org/10.1111/mec.13060
Garza J, Williamson E (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318. https://doi.org/10.1046/j.1365-294X.2001.01190.x
George JP, Konrad H, Collin E, Thevenet J, Ballian D, Idzojtic M, Kamm U, Zhelev P, Geburek T (2015) High molecular diversity in the true service tree (Sorbus domestica) despite rareness: data from Europe with special reference to the Austrian occurrence. Ann Bot 115:1105–1115. https://www.jstor.org/stable/26525695
Gitzendanner MA, Soltis PS (2000) Patterns of genetic variation in rare and widespread plant congeners. Am J Bot 87:783–792. https://doi.org/10.2307/2656886
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486. https://doi.org/10.1093/oxfordjournals.jhered.a111627
Ha Y-H, Oh S-H, Lee S-R (2021) Genetic admixture in the population of wild apple (Malus sieversii) from the Tien Shan mountains. Kaz Genes 12:104. https://doi.org/10.3390/genes12010104
Hampe A, Petit RJ (2005) Conserving biodiversity under climate change: the rear edge matters. Ecol Lett 8:461–467. https://doi.org/10.1111/j.1461-0248.2005.00739.x
Hewitt GM (2004) Genetic consequences of climatic oscillations in the quaternary. Philos Trans R Soc Lond B Biol Sci 359:183–195. https://doi.org/10.1098/rstb.2003.1388
Heywood V, Zohary D (1995) A catalogue of the wild relatives of cultivated plants native to Europe. Flora Mediterr 5:375–415
Holderegger R, Häner R, Csencsics D, Angelone S, Hoebee SE (2008) S-allele diversity suggests no mate limitation in small populations of a self-incompatible plant. Evolution 62:2922–2928. https://doi.org/10.1111/j.1558-5646.2008.00498.x
Hufford KM, Mazer SJ, Hodges SA (2014) Genetic variation among mainland and island populations of a native perennial grass used in restoration. AoB Plants 6:plt055. https://doi.org/10.1093/aobpla/plt055
Iketani H, Yamamoto T, Katayama H, Uematsu C, Mase N, Sato Y (2010) Introgression between native and prehistorically naturalized (archaeophytic) wild pear (Pyrus spp.) populations in Northern Tohoku. Northeast Jpn Conserv Genet 11:115–126. https://doi.org/10.1007/s10592-009-0009-8
Jiang Z, Tang F, Huang H, Hu H, Chen Q (2009) Assessment of genetic diversity of Chinese sand pear landraces (Pyrus pyrifolia Nakai) using simple sequence repeat markers. Hort Sci 44:619–626
Jiang S, Zheng X, Yu P, Yue X, Ahmed M, Cai D, Teng Y (2016) Primitive genepools of Asian pears and their complex hybrid origins inferred from fluorescent sequence-specific amplification polymorphism (SSAP) markers based on LTR retrotransposons. PLoS ONE 11:e0149192. https://doi.org/10.1371/journal.pone.0149192
Jordano P, Godoy JA (2000) RAPD variation and population genetic structure in Prunus mahaleb (Rosaceae), an animal-dispersed tree. Mol Ecol 9:1293–1305. https://doi.org/10.1046/j.1365-294x.2000.01009.x
Kalinowski ST (2005) HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189. https://doi.org/10.1111/j.1471-8286.2004.00845.x
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106. https://doi.org/10.1111/j.1365-294X.2007.03089.x
Katayama H, Adachi S, Yamamoto T, Uematsu C (2007) A wide range of genetic diversity in pear (Pyrus ussuriensis var. aromatica) genetic resources from Iwate, Japan revealed by SSR and chloroplast DNA markers. Genet Resour Crop Evol 54:1573–1585. https://doi.org/10.1007/s10722-006-9170-9
Katayama H, Amo H, Wuyun T, Uematsu C, Iketani H (2016) Genetic structure and diversity of the wild Ussurian pear in east Asia. Breed Sci 66:90–99. https://doi.org/10.1270/jsbbs.66.90
Kišek M, Jarni K, Brus R (2021) Hybridisation of Malus sylvestris (L.) Mill. with Malus × domestica Borkh. and implications for the production of forest reproductive material. Forests 12:367. https://doi.org/10.3390/f12030367
Kocsisné GM, Bolla D, Anhalt-Brüderl UCM, Forneck A, Taller J, Kocsis L (2020) Genetic diversity and similarity of pear (Pyrus communis L.) cultivars in central Europe revealed by SSR markers. Genet Resour Crop Evol 67:1755–1763. https://doi.org/10.1007/s10722-020-00937-0
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Korotkova N, Parolly G, Khachatryan A, Ghulikyan L, Sargsyan H, Akopian J, Borsch T, Gruenstaeudl M (2018) Towards resolving the evolutionary history of Caucasian pears (Pyrus, Rosaceae)—phylogenetic relationships, divergence times and leaf trait evolution. J Systemat Evol 56:35–47. https://doi.org/10.1111/jse.12276
Kumar S, Kirk C, Deng C, Wiedow C, Knaebel M, Brewer L (2017) Genotyping-by-sequencing of pear (Pyrus spp.) accessions unravels novel patterns of genetic diversity and selection footprints. Hortic Res 4:17015. https://doi.org/10.1038/hortres.2017.15
Lawrence ER, Fraser DJ (2020) Latitudinal biodiversity gradients at three levels: linking species richness, population richness and genetic diversity. Global Ecol Biogeogr 29:770–788. https://doi.org/10.1111/geb.13075
Layne REC, Quamme HA (1975) Pears. In: Janick J, Moore JN (eds) Advances in fruit breeding. Purdue University Press, West Lafayette, pp 38–70
Li YL, Liu JX (2018) StructureSelector: a web-based software to select and visualize the optimal number of clusters using multiple methods. Mol Ecol Resour 18:176–177. https://doi.org/10.1111/1755-0998.12719
Li W, Liu L, Wang Y, Zhang Q, Fan G, Zhang S, Wang Y, Liao K (2020) Genetic diversity, population structure, and relationships of apricot (Prunus) based on restriction site-associated DNA sequencing. Hortic Res 7:69. https://doi.org/10.1038/s41438-020-0284-6
Liu J, Zheng X, Potter D, Hu C, Teng Y (2012) Genetic diversity and population structure of Pyrus calleryana (Rosaceae) in Zhejiang province, China. Biochem Syst Ecol 45:69–78. https://doi.org/10.1016/j.bse.2012.06.027
Liu Q, Song Y, Liu L, Zhang M, Sun J, Zheng S, Wu J (2015) Genetic diversity and population structure of pear (Pyrus spp.) collections revealed by a set of core genome-wide SSR markers. Tree Genet Genomes 11:128. https://doi.org/10.1007/s11295-015-0953-z
Lukoseviciute V, Gelvonauskienė D, Baniulis D, Frercks B, Sasnauskas A, Stanys V, Rugienius R (2013) Characterization of pear (Pyrus communis) cultivars from Lithuania using microsatellite markers. Acta Hortic 976:257–263
Mariette S, Tavaud M, Arunyawat U, Capdeville G, Millan M, Salin F (2010) Population structure and genetic bottleneck in sweet cherry estimated with SSRs and the gametophytic self-incompatibility locus. BMC Genet 11:77. https://doi.org/10.1186/1471-2156-11-77
Matsumoto K, Tamura F, Chun J-P, Tanabe K (2006) Native Mediterranean Pyrus rootstock, P. amygdaliformis and P. elaeagrifolia, present higher tolerance to salinity stress compared with Asian natives. J Jpn Soc Hortic Sci 75:450–457. https://doi.org/10.2503/jjshs.75.450
Matsuoka Y, Vigouroux Y, Goodman MM, Sanchez JG, Buckler E, Doebley J (2002) A single domestication for maize shown by multilocus microsatellite genotyping. Proc Natl Acad Sci USA 99:6080–6084. https://doi.org/10.1073/pnas.052125199
Mercure M, Bruneau A (2008) Hybridization between the escaped Rosa rugosa (Rosaceae) and native R. blanda in eastern north America. Am J Bot 95:597–607. https://doi.org/10.3732/ajb.2007385
Morente-López J, García C, Lara-Romero C, García-Fernández A, Draper D, Iriondo JM (2018) Geography and environment shape landscape genetics of Mediterranean alpine species Silene ciliata Poiret. (Caryophyllaceae). Front Plant Sci 9:1698. https://doi.org/10.3389/fpls.2018.01698
Nishio S, Takada N, Saito T, Yamamoto T, Iketani H (2016) Estimation of loss of genetic diversity in modern Japanese cultivars by comparison of diverse genetic resources in Asian pear (Pyrus spp.). BMC Genet 17:81. https://doi.org/10.1186/s12863-016-0380-7
Nishio S, Takada N, Terakami S, Takeuchi Y, Kimura MK, Isoda K, Saito T, Iketani H (2021) Genetic structure analysis of cultivated and wild chestnut populations reveals gene flow from cultivars to natural stands. Sci Rep 11:240. https://doi.org/10.1038/s41598-020-80696-1
Oliviera GCX, Vieira ICG, Tremblay RL (2021) Gene flow among wild and domesticated plant species in the neotropical region. Front Ecol Evol 9:799071. https://doi.org/10.3389/fevo.2021.799071
Ouni R, Zborowska A, Sehic J, Choulak S, Iñaki Hormaza J, Garkava-Gustavsson L, Mars M (2020) Genetic diversity and structure of Tunisian local pear germplasm as revealed by SSR markers. Hortic Plant J 6:61–70. https://doi.org/10.1016/j.hpj.2020.03.003
Paganová V (2011) Taxonomic reliability of leaf and fruit morphological characteristics of the Pyrus L. taxa in Slovakia. Hortic Sci 30:98–107
Parle M (2016) Why pear is so dear. Int J Re Ayurveda Pharm 7:108–113. https://doi.org/10.7897/2277-4343.07139
Pearman PB, Broennimann O, Aavik T, Albayrak T, Alves PC, Aravanopoulos FA, Bertola LD, Biedrzycka A, Buzan E, Cubric-Curik V, Djan M, Fedorca A, Fuentes-Pardo AP, Fussi B, Godoy JA, Gugerli F, Hoban S, Holderegger R, Hvilsom C, Iacolina L, Kalamujic Stroil B, Klinga P, Konopiński MK, Kopatz A, Laikre L, Lopes-Fernandes M, McMahon BJ, Mergeay J, Neophytou C, Pálsson S, Paz-Vinas I, Posledovich D, Primmer CR, Raeymaekers JAM, Rinkevich B, Rolečková B, Ruņģis D, Schuerz L, Segelbacher G, Kavčič Sonnenschein K, Stefanovic M, Thurfjell H, Träger S, Tsvetkov IN, Velickovic N, Vergeer P, Vernesi C, Vilà C, Westergren M, Zachos FE, Guisan A, Bruford M (2024) Monitoring of species’ genetic diversity in Europe varies greatly and overlooks potential climate change impacts. Nat Ecol Evol 8:267–281. https://doi.org/10.1038/s41559-023-02260-0
Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Bëer E, Robinson S, Vásquez-Carrillo C, Pauli JN, Palsbøll PJ (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418. https://doi.org/10.1111/j.1365-294X.2012.05635.x
Petit R, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Müller-Starck G, Demesure-Musch B, Palmé A, Martín JP, Rendell S, Vendramin GG (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565. https://doi.org/10.1126/science.1083264
Pigozzi G (1992) Frugivory and seed dispersal by the European badger in a Mediterrenean habitat. J Mammal 73:630–639. https://doi.org/10.2307/1382035
Pisias MT, Bakala HS, McAlvay AC, Mabry ME, Birchler JA, Yang B, Pires JC (2022) Prospects of feral crop de novo redomestication. Plant Cell Physiol 63:1641–1653. https://doi.org/10.1093/pcp/pcac072
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Queiroz Á, Bagoin Guimarães J, Sánchez C, Simões F, Maia de Sousa R, Viegas W, Veloso MM (2019) Genetic diversity and structure of the Portuguese pear (Pyrus communis L.) germplasm. Sustainability 11:5340. https://doi.org/10.3390/su11195340
Rana JC, Chahota RK, Sharma V, Rana M, Verma N, Vermna B, Sharma TR (2015) Genetic diversity and structure of Pyrus accessions of Indian Himalayan region based on morphological and SSR markers. Tree Genet Genomes 11:821. https://doi.org/10.1007/s11295-014-0821-2
Reim S, Lochschmidt F, Proft A, Wolf H, Wolf H (2017) Species delimitation, genetic diversity and structure of the European indigenous wild pear (Pyrus pyraster) in Saxony, Germany. Genet Resour Crop Evol 64:1075–1085. https://doi.org/10.1007/s10722-016-0426-8
Reim S, Lochschmidt F, Proft A, Höfer M (2020) Genetic integrity is still maintained in natural populations of the indigenous wild apple species Malus sylvestris (Mill.) in Saxony as demonstrated with nuclear SSR and chloroplast DNA markers. Ecol Evol 10:11798–11809. https://doi.org/10.1002/ece3.6818
Rohlf FJ (2009) Exeter software (Firm) NTSYS-Pc: numerical taxonomy and multivariate analysis system. Applied Biostatistics Inc., Setauket
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228. https://doi.org/10.1093/genetics/145.4.1219
Rousset F (2008) Genepop’007: a complete re-implementation of the Genepop software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Rugienius R, Blažyté A, Lukoševičiūtė V, Bronė Šikšnianienė J, Frercks B, Gelvonauskienė D, Gelvonauskius B, Sasnauskas A, Baniulis D, Stanys V (2013) Genetic polymorphism of wild pear accessions collected in Lithuania. Balt For 19:13–21
Sadeghnejad S, Abdollahi H, Nikzad A, Hassani M (2014) Interspecies hybridization of Pyrus species along the silk road, detected by using species-specific s-alleles. Acta Hortic 1032:169–172
Sapkota S, Boggess SL, Trigiano RN, Klingeman WE, Hadziabdic D, Coyle DR, Olukolu BA, Kuster RD, Nowicki M (2021) Microsatellite loci reveal genetic diversity of Asian Callery pear (Pyrus calleryana) in the species native range and in the North American cultivars. Life 11:531. https://doi.org/10.3390/life11060531
Sapkota S, Boggess SL, Trigiano RN, Klingeman WE, Hadziabdic D, Coyle DR, Nowicki M (2022) Microsatellite loci reveal high genetic diversity, mutation, and migration rates as invasion drivers of Callery pear (Pyrus calleryana) in the Southeastern United States. Front Genet 13:861398. https://doi.org/10.3389/fgene.2022.861398
Sau S, Pastore C, D’hallewin G, Dondini L, Bacchetta G (2020) Characterisation of microsatellite loci in Sardinian pears (Pyrus communis L. and P. spinosa Forssk). Sci Hortic 270:109443. https://doi.org/10.1016/j.scienta.2020.109443
Schueler S, Tusch A, Scholz F (2006) Comparative analysis of the within-population genetic structure in wild cherry (Prunus avium L.) at the self-incompatibility locus and nuclear microsatellites. Mol Ecol 15:3231–3243. https://doi.org/10.1111/j.1365-294X.2006.03029.x
Sehić J, Garkava-Gustavsson L, Fernández-Fernández F, Nybom H (2012) Genetic diversity in a collection of European pear (Pyrus communis) cultivars determined with SSR markers chosen by ECPGR. Sci Hortic 145:39–45. https://doi.org/10.1016/j.scienta.2012.07.023
Sexton JP, Hangartner SB, Hoffmann AA (2014) Genetic isolation by environment or distance: which pattern of gene flow is most common? Evolution 68:1–15. https://doi.org/10.1111/evo.12258
Stephan BR, Wagner I, Kleinschmit J (2003) EUFORGEN Technical guidelines for genetic conservation and use for wild apple and pear (Malus sylvestris and Pyrus pyraster). International Plant Genetic Resources Institute, Rome
Stewart JR, Lister AM, Barnes I, Dalen L (2010) Refugia revisited: individualistic responses of species in space and time. Proc R Soc B 277:661–671. https://doi.org/10.1098/rspb.2009.1272
Stotz GC, Salgado-Luarte C, Escobedo VM, Valladares F, Gianoli E (2021) Global trends in phenotypic plasticity of plants. Ecol Lett 24:2267–2281. https://doi.org/10.1111/ele.13827
Szymajda M, Studnicki M, Kuras A, Zurawicz E (2022) Cross-compatibility in interspecific hybridization between three Prunus species. S Afr J Bot 146:624–633. https://doi.org/10.1016/j.sajb.2021.11.036
Taberlet P, Cheddadi R (2002) Ecology. Quaternary refugia and persistence of biodiversity. Science 297:2009–2010. https://doi.org/10.1126/science.297.5589.2009
Todesco M, Pascual MA, Owens GL, Ostevik KL, Moyers BT, Hübner S, Heredia SM, Hahn MA, Caseys C, Bock DG, Rieseberg LH (2016) Hybridization and extinction. Evol Appl 9:892–908. https://doi.org/10.1111/eva.12367
Tomljanović K, Grubešić M (2014) Wild boar dynamics in Croatia. In: Poličnik H (ed) Abstract book of 10th international symposyum on wild boar and other suids. Ljubljana
Twyford AD, Wong ELY, Friedman J (2020) Multi-level patterns of genetic structure and isolation by distance in the widespread plant Mimulus guttatus. Heredity 125:227–239. https://doi.org/10.1038/s41437-020-0335-7
Urrestarazu J, Royo JB, Santesteban LG, Miranda C (2015) Evaluating the influence of the microsatellite marker set on the genetic structure inferred in Pyrus communis L. PLoS ONE 10:e0138417. https://doi.org/10.1371/journal.pone.0138417
Uzun A, Pinar H, Yaman M, Yigit MA, Cakiroglu Y, Karakaya A, Uysal M, Ozturk G, Yilmaz KU, Gurcan K, Ercisli S (2022) Identification of genetic diversity in wild pear (Pyrus elaeagrifolia Pall.) genotypes collected from different regions of Turkey with SSR marker system. Genetika 54:109–118. https://doi.org/10.2298/GENSR2201109U
van Oosterhout C, Hutchinson WF, Wills DPM, Shipely P (2004) Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Vašková D, Kolarčik V (2019) Breeding systems in diploid and polyploid hawthorns (Crataegus): evidence from experimental pollinations of C. monogyna, C. subsphaerica, and natural hybrids. Forests 10:1059. https://doi.org/10.3390/f10121059
Vázquez DP, Gianoli E, Morris WF, Bozinovic F (2017) Ecological and evolutionary impacts of changing climatic variability. Biol Rev Camb Philos Soc 92:22–42. https://doi.org/10.1111/brv.12216
Vidaković A, Liber Z, Šatović Z, Idžojtić M, Volenec I, Zegnal I, Pintar V, Radunić M, Poljak I (2021) Phenotypic diversity of almond-leaved pear (Pyrus spinosa Forssk.) along Eastern Adriatic coast. Forests 12:1630. https://doi.org/10.3390/f12121630
Vidaković A, Šatović Z, Tumpa K, Idžojtić M, Liber Z, Pintar V, Radunić M, Runjić TN, Runjić M, Rošin J, Gaunt D, Poljak I (2022) Phenotypic variation in European wild pear (Pyrus pyraster (L.) Burgsd.) populations in the north-western part of the Balkan Peninsula. Plants 11:335. https://doi.org/10.3390/plants11030335
Vidaković A, Poljak I (2024) Fruit morphological variability and chemical composition in European wild pear (Pyrus pyraster (L.) Burgsd.) natural populations. Genet Resour Crop Evol. https://doi.org/10.1007/s10722-024-01912-9
Vidaković A, Radunić M, Poljak I (2024) Variation in chemical composition and fruit morphometric traits of almond-leaved pear (Pyrus spinosa Forssk.) natural populations. Genet Resour Crop Evol. https://doi.org/10.1007/s10722-024-02059-3
Volk GM, Richards CM, Henk AD, Reilley AA, Bassil NV, Postman JD (2006) Diversity in wild Pyrus communis based on microsatellite analyses. J Am Soc Hortic Sci 131:408–417
Wagner I, Büttner R (2019) Hybridization in wild pear (Pyrus pyraster) from various regions in Germany and from Luxembourg with respect to Pyrus × communis. Acta Hortic 1242:427–434
Wang Z, Jiang Y, Bi H, Lu Z, Ma Y, Yang X, Chen N, Tian B, Liu B, Mao X, Ma T, DiFazio SP, Hu Q, Abbott RJ, Liu J (2021) Hybrid speciation via inheritance of alternate alleles of parental isolating genes. Mol Plant 14:208–222. https://doi.org/10.1016/j.molp.2020.11.008
Westwood MN, Bjornstad HO (1971) Some fruit characteristics of interspecific hybrids and extent of self-sterility in Pyrus. Bull Torrey Bot Club 98:22–24. https://doi.org/10.2307/2483493
Wolko Ł, Antkowiak W, Lenartowicz E, Bocianowski J (2010) Genetic diversity of European pear cultivars (Pyrus communis L.) and wild pear (Pyrus pyraster (L.) Burgsd.) inferred from microsatellite markers analysis. Genet Resour Crop Evol 57:801–806. https://doi.org/10.1007/s10722-010-9587-z
Wolko Ł, Bocianowski J, Antkowiak W, Słomski R (2015) Genetic diversity and population structure of wild pear (Pyrus pyraster (L.) Burgsd.) in Poland. Open Life Sci 10:19–29. https://doi.org/10.1515/biol-2015-0003
Wong ELY, Hiscock SJ, Filatov DA (2022) The Role of interspecific hybridisation in adaptation and speciation: Insights from studies in Senecio. Front Plant Sci 13:907363. https://doi.org/10.3389/fpls.2022.907363
Wu Y, Yu X, Tang W, Yang W, Fu Q, Zheng Y, Zhang C (2024) Morphological and molecular evidence for natural hybridization between Sorbus pohuashanensis and S. discolor (Rosaceae). J for Res 35:25. https://doi.org/10.1007/s11676-023-01659-6
Wuyun T, Amo H, Xu J, Ma T, Uematsu C, Katayama H (2015) Population structure of and conservation strategies for wild Pyrus ussuriensis Maxim. in China. PLoS ONE 10:e0133686. https://doi.org/10.1371/journal.pone.0133686
Yousefzadeh H, Raeisi S, Esmailzadeh O, Jalali G, Nasiri M, Walas Ł, Kozlowski G (2021) Genetic diversity and structure of rear edge populations of Sorbus aucuparia (Rosaceae) in the Hyrcanian forest. Plants 10:1471. https://doi.org/10.3390/plants10071471
Zheng X, Cai D, Potter D, Postman J, Liu J, Teng Y (2014) Phylogeny and evolutionary histories of Pyrus L. revealed by phylogenetic trees and networks based on data from multiple DNA sequences. Mol Phylogenet Evol 80:54–65. https://doi.org/10.1016/j.ympev.2014.07.009
Acknowledgements
This research was funded by the project KK.01.2.2.03.0017 CEKOM 3LJ, and the University of Zagreb financial support. We would like to extend our heartfelt thanks to our dear colleagues, Prof. Marilena Idžojtić and Ida Volenec, for their efforts in collecting samples on the islands of Vir and Hvar. We are also grateful to the employees of the Croatian Forests branches for providing valuable information and assistance during the sample collection. Finally, special thanks to our dedicated students, Daniel Gaunt, Toni Knatek, and Zvonimir Opačak, for their help in collecting samples from the populations Vinkovci and Lipovljani.
Funding
CEKOM, KK.01.2.2.03.0017, Mira Radunić
Author information
Authors and Affiliations
Contributions
Conceptualization and study design: A.V. and I.P. Sampling: A.V., M.R., M.S.B. and I.P. Molecular analysis: A.V. and Z.L. Data analysis: Z.Š. Writing—original draft preparation: A.V. and I.P. Review and editing: Z.Š., Z.L., M.R. and M.S.B.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
The collection of plant material was carried out in accordance with relevant institutional, national, and international guidelines and legislation.
Additional information
Communicated by R. Guy .
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vidaković, A., Šatović, Z., Liber, Z. et al. Genetic diversity of Pyrus pyraster (L.) Burgsd. and P. spinosa Forssk.: evidence of introgression from cultivated into wild pear populations. Trees 38, 1297–1314 (2024). https://doi.org/10.1007/s00468-024-02553-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00468-024-02553-2