
Abstract
Background
Proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) is a glomerular disease defined by non-organized glomerular deposits of heavy and light chain–restricted immunoglobulin and is rarely reported in children.
Methods
We characterized a series of nine pediatric patients from two academic centers with biopsy-proven PGNMID and additionally describe two patients with monotypic IgG in the setting of IgM deposition.
Results
Each patient presented with hematuria and/or proteinuria; however, only five had elevated serum creatinine. Prodromal or concurrent infection was identified in six patients, low C3 in five, and alternate complement pathway gene variants in two. No monoclonal serum proteins were identified in five tested patients. Seven patients had monotypic deposits composed of IgG3-λ, two showed IgG3-κ, and one each IgG1 and IgG3 with lambda dominance in the setting of IgM deposition. The glomerular pattern was predominantly mesangial proliferative or membranoproliferative glomerulonephritis (MPGN). Treatment and outcomes were variable; four patients have recent PGNMID diagnoses and therefore minimal follow up, one had relatively stable kidney function for over a decade, and six experienced kidney failure, with four receiving transplants. Recurrent deposits of the same isotype were identified in five of six transplanted kidneys, corresponding to three of four transplanted patients. One of these patients developed PGNMID recurrences in three separate kidney allografts over a 20-year disease course.
Conclusions
Our study emphasizes the need for upfront IgG subclass investigation in pediatric mesangial or MPGN with IgG deposition and monotypic or biased light-chain staining. Furthermore, this pediatric experience suggests expanded pathogenic considerations in PGNMID.

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) was first described in ten adult patients in 2004 by Nasr and colleagues and subsequently in a larger cohort [1, 2]. The disease was defined by glomerular deposits composed of light and heavy chain–restricted immunoglobulins with a propensity for monotypic IgG3 deposition. In adults, clinical presentation is generally characterized by kidney insufficiency, proteinuria, and hematuria [2]. On light microscopy, the findings range from mesangial to membranoproliferative glomerulonephritis and occasionally mimic membranous nephropathy. Electron microscopy reveals granular non-organized deposits primarily within the mesangium and subendothelium, while rare cases show predominantly subepithelial deposits. Clinical outcomes in adults vary with 38% experiencing complete or partial recovery, 38% experiencing persistent kidney dysfunction, and 22% progressing to kidney failure [2]. In adults with kidney failure and transplantation, PGNMID was found to recur in 89% of patients with a median graft survival of 92 months [3].
In 2018, Torrealba and colleagues reported the first case of PDMID in a teenager, and Xing and colleagues subsequently described a series of five pediatric PGNMID patients [4, 5]. In the Xing study, clinical presentations varied but were characterized mainly by hematuria, proteinuria, and low C3 [4]. Four patients had partial response to treatment with stabilization of kidney function and decreased proteinuria but showed progressive tubular atrophy and interstitial fibrosis. None of these patients developed kidney failure during 8 to 56 months of follow-up. However, the fifth patient initially presented in kidney failure but did not receive a kidney transplant.
In adults, PGNMID has been grouped with monoclonal gammopathy of renal significance [6]. Nevertheless, only 20 to 30% of adult patients have detectable serum monoclonal immunoglobulins despite monotypic glomerular deposits [3]. However, when a serum paraprotein is found, it is generally of the same light and heavy chain isotype as the glomerular deposits. A documented hematologic malignancy is rare in adult patients, and in the subset of patients who underwent bone marrow biopsies, a clone was identified in only 10% of the biopsies [2]. Treatment for PGNMID in the reported adult series has been highly variable and has mostly included immunosuppression or treatment directed at an identified or hypothetical B cell or plasma cell “clone,” with partial efficacy. Likewise, there has been no consistent effective treatment for PGNMID in pediatric patients. In the small number of reported pediatric cases, there have been none with an identifiable paraprotein or B cell clone.
We present the largest pediatric series of PGNMID to date, derived from two academic medical centers, and describe in detail the clinicopathologic course of nine patients, including post-transplant course in four patients; two patients with monotypic IgG in the setting of IgM deposition are also cataloged. This series provides expanded insights into PGNMID in the pediatric population.
Methods
Patients and cases
This study was performed with IRB approval. Computerized kidney pathology archives at two academic medical centers (Stanford-LPCH, Cedars-Sinai) were searched for patients age 18 or less with a diagnosis of proliferative glomerulonephritis with monoclonal deposits (PGNMID, n = 6) since 2007. PGNMID was defined as glomerular immune deposits with staining for IgG, restricted to a single IgG subclass (IgG1, IgG2, IgG3, or IgG4), generally with single light chain isotype staining (κ, λ), and predominantly granular electron-dense deposits resembling immune deposits by electron microscopy [1, 2]. Cases with minimal IgA, IgM heavy-chain reactivity were included as long as IgG subclass staining was monotypic [2, 7]. One adult patient with PGNMID recurrences in allografts since childhood was included as confirmed by retrospective IgG subclass and light-chain staining.
Additional cases were retrospectively identified from pediatric membranoproliferative or mesangial proliferative glomerulonephritis cases with unequal light-chain staining (e.g., κ 2+/λ 3+; κ 3+/λ 1+; excluding patients with confirmed systemic lupus erythematosus, IgA-vasculitis/Henoch-Schönlein Purpura, and C3 glomerulopathy). Archival frozen tissue was evaluated with IgG subclass immunofluorescence staining as described below. Of the 19 tested cases from the same time frame, three additional cases with monoclonal IgG heavy chains fitting the above PGNMID definition were identified. Two cases with monoclonal IgG but moderate-strong IgM deposition and lambda light chain dominance despite staining for both light chains are described as cases no. 10–11. Reports of initial native biopsies were obtained for patient no. 4 and no. 9, but the original histologic slides and images could not be obtained; the tissue specimens from their subsequent biopsies were available for study review. Thus, a cohort of 9 pediatric patients with PGNMID and 2 IgM-rich patients were available for study.
Clinical, laboratory, and outcome data were abstracted from the medical record. Statistical calculations were performed with Excel (Microsoft, Redmond WA), or online at https://www.socscistatistics.com.
Pathologic analysis
Kidney core biopsy tissue was processed and evaluated with standard clinical methods for light, immunofluorescence, immunohistochemistry (C4d), and electron microscopy. Light microscopic slides were reviewed for study purposes, along with photomicrographs of immunofluorescence and electron microscopy. Scoring of interstitial fibrosis, tubular atrophy, and interstitial inflammation was modeled after the Banff schema: interstitial fibrosis and tubular atrophy scoring: 0 = 0%, min = 0–5%; 1 = 6–25%; 2 = 26–50%; 3 ≥ 50%.
In brief, IgG subclass staining was performed by direct immunofluorescence on frozen tissue with fluorescein-conjugated antibodies from The Binding Site (San Diego, CA) at 1:10–1:20 dilution per local validated protocol. Immunohistochemical staining for Membrane Attack Complex (MAC) was performed on acetone fixed frozen sections using primary monoclonal antibody aE11 (Dako M0777) at 1:80 and developed with Dako Envision+ (K4001) and Vector VIP SK-4100 substrate kit.
Results
Representative case report
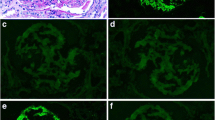
Patient no. 7 was a 7-year-old Hispanic female who initially presented to the Emergency Department with hypertension, hematuria, and proteinuria and was diagnosed with nephritic syndrome. Her past medical history was significant for multiple emergency room visits for abdominal pain of unknown etiology and an episode of streptococcal pharyngitis 2 months prior to presentation. Her family history was significant for kidney failure on the maternal side. Her initial kidney biopsy diagnosis was MPGN, type I pattern, and she was treated with prednisone, followed by mycophenolate mofetil (MMF) and cyclosporine. Two subsequent native biopsies demonstrated progressive MPGN with increasing interstitial fibrosis and tubular atrophy and numerous crescents on the third biopsy. Less than 1 year after her initial presentation, she developed kidney failure requiring peritoneal dialysis while awaiting transplant. She received a deceased donor kidney transplant and was placed on MMF, prednisone, and tacrolimus. Her 6-month protocol biopsy revealed focal tubular atrophy and interstitial fibrosis and early PGNMID with IgG3-κ deposition. Her 1-year biopsy showed persistent IgG3-κ deposits with minimal histologic progression of glomerulonephritis and features suggestive of calcineurin inhibitor nephrotoxicity. Her clinical follow up has been unremarkable, and she has been receiving routine care. Retrospective staining of the native biopsies demonstrated IgG3-κ monotypia (Figs. 1 and 2).

Native and transplant kidney biopsy findings from patient no. 7; native and 1-year transplant biopsies in left and right columns, respectively. a The native biopsy reveals a membranoproliferative pattern glomerulonephritis characterized by global mesangial sclerosis and proliferation with segmental endocapillary hypercellularity (PAS × 400); b The 1-year transplant biopsy reveals mild mesangial expansion and subtle hypercellularity with mild juxtaglomerular cell hypertrophy (H&E × 400 digitally enlarged); c A representative native electron micrograph reveals large electron-dense deposits in the mesangium with extension along the subendothelial aspect of the glomerular basement membranes and podocyte foot process effacement (left). d A representative 1-year transplant electron micrograph reveals granular electron-dense deposits in the mesangium, and patchy epithelial cell foot process effacement. e–f Representative immunofluorescence staining of native (retrospective) and transplant kidneys reveal IgG3 monotypia (negative IgG1, IgG2, IgG4; staining not shown; × 400). g–h Representative immunofluorescence staining of native (retrospective) and transplant kidneys reveal κ monotypia (negative λ; staining not shown; × 400 digitally enlarged)

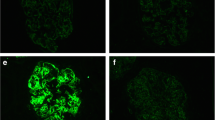
Native kidney immunofluorescence staining for light-chain isotypes, complement components, and IgG subclasses in patient no. 7. a, b Light-chain isotype staining reveals κ monotypia (a) with negative λ staining (b). c, d Complement component staining reveals strong staining for C3 (c) and weak staining for C1q (d). e–h IgG subclass staining reveals strong staining for IgG3 (g) with negative staining for IgG1 (e), IgG2 (f), and IgG4 (h) (× 400)
Case series: demographic and clinical features
Nine pediatric patients (three females and six males) with PGNMID, including the patient above, were identified from the pathology archives of two academic medical centers. Two additional patients, both female, had monoclonal IgG, but also had abundant IgM deposition and polytypic but lambda-dominant light chains (nos. 10–11; “IgM-rich” for the purposes of the paper). Five of these patients were identified after retrospective IgG subclass staining. Clinical data at presentation are listed in Table 1. Patient age at presentation ranged from 5 to 18 years (average 10 years PGNMID, 13 for IgM-rich). Nearly all patients had hematuria and/or proteinuria at presentation, yet only five patients had elevated serum creatinine. All patients were negative for ANA, and all the patients tested for ANCA, hepatitis B, hepatitis C, and HIV had negative results. Five PGNMID and one IgM-rich patient had evidence of concurrent or prodromal infection such as influenza B or an unspecified febrile illness, with three having elevated ASO, suggesting a prior or current group A streptococcal infection.
Four PGNMID and one IgM-rich patient had low serum C3 levels. Detailed complement panels were performed in three patients with mixed results (Table 1). Patient nos. 1 and 9 had genetic variants of unknown significance accompanied by significant dysregulation on functional studies (Table 1), while patient no. 6 had no significant mutations identified.
Treatment was typically immunosuppression, especially mycophenolate mofetil and/or prednisone; three patients also received a calcineurin inhibitor pre-transplant. Six PGNMID patients progressed to kidney failure, four of whom received kidney transplants. Follow-up was available for all nine PGNMID patients (Table 1); at last follow-up, including transplants, five had elevated serum creatinine. Eight PGNMID patients had hematuria, and seven had persistent proteinuria. One patient (no. 4) has had relatively stable kidney function for 11 years. One IgM-rich patient with available follow-up had elevated creatinine, hematuria, and mild proteinuria.
Case series: biopsy pathology
Native: LM
Index biopsy data are presented in Table 2. The patterns of glomerular injury ranged from mild mesangial widening (MesW) to mesangial hypercellularity/proliferation (MesHP), to endocapillary proliferative (EPGN), to membranoproliferative glomerulonephritis (MPGN; six PGNMID and one IgM-rich). Crescents were identified in three patients (nos. 2, 7, 11). The degree of interstitial fibrosis and tubular atrophy (IFTA) reflected the kidney function and ranged from none (0) to severe (3) (Table 2). Pathologic findings for index and all available native and transplant biopsies are presented in Online Resource 1.
Native: IF
By definition, immunofluorescence microscopy revealed granular IgG deposits in the glomeruli in all biopsies (Figs. 1 and 2). These deposits were primarily localized to the mesangium and along the glomerular peripheral capillary walls. Of the four IgG subclasses, seven native biopsies stained exclusively for IgG3. Five of those biopsies stained for IgG3-λ, while the other two were IgG3-κ. Two initial native biopsies were not available for IgG subclass staining; patient no. 4 had a subsequent native biopsy monotypic for IgG3-λ, while patient no. 9 had three transplants with IgG3-λ in each (see below). In biopsies with moderate to strong IgM, one native biopsy had monotypic IgG1 while another had monotypic IgG3, both with dominant, though not exclusive, λ staining.
All native biopsies revealed glomerular deposition of C3, and C1q was positive in nine in a similar distribution to IgG. C3 was scored as stronger than IgG in four PGNMID biopsies and both IgM-rich biopsies. Based on glomerular immunostaining, several of the biopsies demonstrated predominantly alternative complement pathway activation (C3-strong/C1q and C4d negative-weak; nos. 1, 3, 5, 6). The majority of biopsies had evidence of both classical and alternate complement activation (C3-positive/C1q and C4d moderate-strong). MAC (C5b-9) staining was performed in three native biopsies and showed strong staining in the same distribution as immune complexes in each (Online Resource 1).
Native: EM
Native kidney biopsies demonstrated granular electron-dense deposits (Fig. 1) in a global distribution in the mesangium in all 11 biopsies, in the subendothelium in ten biopsies, and in the subepithelium in six biopsies. Podocyte foot process effacement was diffuse in seven biopsies and partial in four biopsies.
Transplant: LM
Four PGNMID patients received a kidney transplant from either deceased or living donors with documented recurrence of immune deposits in three patients. On light microscopy, the earliest histologic changes were MesW followed by MesHP (Figs. 1 and 3).

Biopsy patterns, creatinine values, and proteinuria in transplanted kidneys in four patients (patient no. 6–no. 9). The length of each bar graph represents the length of time for each transplant to last available follow-up. The increased creatinine and MPGN pattern in no. 6 corresponded to an episode of cellular and antibody-mediated rejection, as described in the text. Patient no. 9 has received three kidney transplants over two decades
Following transplant, patient no. 6’s pattern of glomerular injury progressed from MesW (6-month protocol biopsy) to MesHP (12-month protocol biopsy) to MPGN at the time of C4d-positive active antibody-mediated and active cellular rejection (Banff grade IIA) following medication non-compliance (Fig. 3). Interestingly, the pattern of glomerular injury in her transplanted kidney returned to MesHP following treatment for and resolution of her acute rejection. It is not entirely clear whether the glomerular changes and response to immunosuppression reflect allograft glomerulitis/glomerulopathy, recurrent PGNMID, or both.
Patient no. 9 received three kidney transplants over a span of 20 years, each with recurrent glomerular disease (Fig. 3). Cellular crescents and recurrent MPGN were identified in biopsies from the first two transplants. Biopsies from the third transplant have demonstrated increasing mesangial deposits but no crescents or proliferation in the 2 years post-transplant while receiving eculizumab and immunosuppression (tacrolimus, prednisone, MMF); rituximab was added subsequent to the latest biopsy given increasing proteinuria.
Transplant: IF
Transplant biopsies with PGNMID recurrence maintained the IgG subclass and light chain staining profile seen in the native kidney. Patient no. 9 had monotypic IgG3-λ in biopsies of his third transplant, and we were able to document weak-moderate IgG3-λ staining in an archival frozen specimen from each of his prior transplants (there was κ reactivity in a different sclerosis pattern in one such biopsy). We were unable to obtain native biopsy tissue for retrospective study. MAC staining was performed in five transplant biopsies and was positive with a pattern similar to immune complexes and complement, as seen in native biopsies (Online Resource 1).
Transplant: EM
Electron microscopy was performed in ten transplant biopsies from three patients. Mesangial deposits were identified in all three 6-month protocol biopsies with subendothelial deposits also identified in one biopsy. Similar patterns were seen in the 12-month protocol and later biopsies (Online Resource 1).
Discussion
To our knowledge, this study represents the largest pediatric PGNMID series to date and the first study to describe and characterize the progression of PGNMID in pediatric transplant kidneys. In our cohort, children with PGNMID presented with hematuria and proteinuria and showed MesHP or MPGN on light microscopy. No serum paraproteins were identified in any of the five patients tested, which is consistent with the paucity of identifiable paraprotein or B cell clones in adult PGNMID case series. Clinical outcomes in our pediatric PGNMID patients were highly variable. Three PGNMID patients (nos. 1–3) had recent diagnoses of PGNMID and have had stable kidney function with limited clinical follow-up over 0.7 to 1.7 years. Six patients progressed to kidney failure, and four were transplanted. Interestingly, one patient experienced only gradual decline in kidney function over an extended follow-up of more than a decade (no. 4). As has been published in adults, we demonstrated recurrent monotypic glomerular deposits in three pediatric transplant recipients, including recurrences in multiple transplants in one patient.
For the purposes of this study, our PGNMID definition included immunoglobulin G (IgG) heavy-chain monotypia with concomitant light-chain monotypia. In this pediatric series, as well as in our clinical experience with adult biopsies, we observed some variation in staining for the non-dominant light chain in biopsies over time (Online Resource 1) with completely negative staining at some time points and trace-weak or rarely moderate reactivity in prior or follow-up biopsies. We separately report the two patients with monotypic IgG, in the setting of moderate to strong IgM, with lambda dominance and appreciable kappa deposition (no. 10–11). Hemminger et al. hypothesized that non-specific IgM could be accompanied by polyclonal light chains in PGNMID, potentially obscuring light-chain monotypia [7].
In three patients, PGNMID was initially recognized in transplant biopsies, and retrospective staining of prior native biopsies revealed similar monotypic glomerular deposits. Based on these findings, we suggest that light chain studies should be performed routinely on all pediatric kidney biopsies, similar to adult biopsies. Further, IgG subclass studies should be performed on pediatric biopsies when IgG deposition is accompanied by skewed light-chain staining.
The pathophysiology, etiology, and treatment of PGNMID remain enigmatic in adults and children. Our pediatric cohort reveals intriguing similarities and differences compared with adult studies that could provide clues to the pathophysiology of PGNMID (Table 3). Adult patients with PGNMID in Nasr’s and Hemminger’s studies have primarily monoclonal κ light-chain deposits in glomeruli. In contrast, our pediatric study and others had mostly monotypic or dominant λ light chains; this difference in light chain distribution between adult and pediatric patients is statistically significant (p value = 0.0077), which may reflect differences in the pathophysiology of the PGNMID in these populations.
In our pediatric PGNMID cohort, at least half of the children had low C3 and/or an infection prior to presentation with glomerulonephritis. In the largest adult PGNMID study, hypocomplementemia was reported in < 20% of patients (Table 3) [2]. These observations raise several possibilities as to pediatric PGNMID pathogenesis, including complement dysregulation, immune dysregulation, and/or glomerulotropic and nephrotoxic antibodies arising from an immune response to intrinsic or extrinsic antigens. Infection has been implicated as a trigger of various autoimmune disorders, and IgG3 antibodies are elicited during responses to infectious agents [8]. In 2012, PGNMID with IgG3-κ glomerular deposits was described in association with a parvovirus B19 infection in two adult patients, one with hypocomplementemia [9]. In these two cases, PGNMID resolved alongside the infection, suggesting that the virus had induced a transient IgG3 response that resulted in IgG3-κ accumulation in glomeruli. In our experience, five pediatric PGNMID patients and one IgM-rich patient presented with associated infections, but their kidney disease did not resolve with the clinical resolution of infection. Two case reports in adults have documented PGNMID evolving to or from C3 glomerulopathy, one of whom had a heterozygous CFH mutation [10, 11]. In our series, two of three tested patients have alternate complement pathway genetic alterations. Patient no. 1 has heterozygous missense variants in CFH. Similarly, patient no. 9 has a novel intronic variant in CFHR3, and heterozygosity for a large CFHR1–CFHR3 deletion. Although cataloged as variants of unknown significance, they were associated with documented serum complement activation in both patients.
Nasr and colleagues proposed that during a normal immune response, one or more B cell clones may proliferate and continue to express IgG with the ability to deposit in the kidneys. While IgG3 is a minor component of total serum IgG, it has several properties that make it a potent antibody that can induce an inflammatory response. IgG3 is the most positively charged of the four human IgG subclasses and thus has an affinity for the anionic glomerular membrane [12]. In addition, IgG3 has the highest molecular weight, can self-aggregate through Fc-Fc interactions, and has the greatest complement fixing capacity [12]. These properties may explain a propensity to deposit within the glomerulus, activate complement, and damage the kidney. The high affinity for glomerular constituents may render the offending monoclonal immunoglobulin undetectable by SPEP and other conventional methods [1]. It is possible that particularly nephrotoxic or nephrotropic antibodies may arise during a normal immune response to intrinsic and/or extrinsic antigens and, in a subset of patients, these polyclonal or oligoclonal antibodies may persist despite resolution of the offending trigger. This response may be amplified in those individuals with underlying complement or immune dysregulation (trigger and multiple hits).
Alternatively, a reactive or oligoclonal response could evolve to a monoclonal or neoplastic proliferation analogous to Type II cryoglobulin, with the caveat that evidence of circulating clonal immunoglobulin and clonality is lacking in the majority of PGNMID cases. Pediatric PGNMID shares some features with the recently described “membranous like glomerulopathy with masked IgG-k deposits” (MGMID), in that both have monotypic glomerular deposits without identifiable paraprotein, and may be associated with immune or complement dysregulation [13]. MGMID has not been grouped with monoclonal gammopathy of renal significance (MGRS), while PGNMID generally has been considered MGRS, even when no circulating protein or lymphoplasmacytic clone is identified [6]. We propose that the pathophysiology of PGNMID in adults with monoclonal gammopathy differs from PGNMID in younger patients without monoclonal gammopathy, although all patients should be carefully investigated clinically.
Effective treatment of PGNMID in children and adults has not been established due to its enigmatic pathogenesis. Immunosuppression or therapies directed at the identified or hypothetical “clone,” including steroids, mycophenolate mofetil, tacrolimus, cyclophosphamide, bortezomib, rituximab, or even in case reports, daratumumab (anti-CD38, with ongoing phase 2 trial: NCT03095118), have shown promise in a subset of adult patients [3, 14] but are met with frequent PGNMID recurrence. The anti-C5 antibody eculizumab has also been used to target downstream complement-mediated glomerular injury in PGNMID; however, data to support this approach are lacking [3].
Outcome in our series of 11 pediatric patients has been varied. While one patient had stable disease for over a decade with only prednisone treatment, six of the children progressed to kidney failure. As in adults, PGNMID disease frequently recurred after transplantation and followed the previously illustrated progression from mesangial matrix expansion to mesangial and/or membranoproliferative pattern [3, 15]. Our study remains limited by both its small numbers and retrospective nature. The diagnosis of PGNMID was made retrospectively in all transplanted patients, and the prospective diagnoses have been made recently with only short-term outcomes available.
In conclusion, we present the largest pediatric series of PGNMID to date. Based on our experience, we suggest that light-chain immunofluorescence should be performed on pediatric native kidney biopsies, as is routine for adults. Additionally, we recommend IgG subclass studies in the setting of moderate-strong IgG deposition if accompanied by monotypic or biased light-chain staining. PGNMID in our pediatric series was characterized by a propensity for monotypic IgG3-λ deposition in glomeruli despite negative serum paraprotein studies. PGNMID led to kidney failure in about half of the children and has recurred in three of the four transplanted patients. We highlight similarities and differences in PGNMID presenting in adults and children, but further study is needed to elucidate the causes and develop effective targeted treatments.
Data availability
Detailed histopathologic data from all biopsies are provided as supplementary table (online resource 1).
References
Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, Appel GB, Aucouturier P, D'Agati VD (2004) Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-comple glomerulonephritis. Kidney Int 65:85–96. https://doi.org/10.1111/j.1523-1755.2004.00365.x
Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, Stokes MB, Nadasdy T, D'Agati VD (2009) Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol 20:2055–2064. https://doi.org/10.1681/asn.2009010110
Said SM, Cosio FG, Valeri AM, Leung N, Sethi S, Salameh H, Cornell LD, Fidler ME, Alexander MP, Fervenza FC, Drosou ME, Zhang D, D'Agati VD, Nasr SH (2018) Proliferative glomerulonephritis with monoclonal immunoglobulin G deposits is associated with high rate of early recurrence in the allograft. Kidney Int 94:159–169. https://doi.org/10.1016/j.kint.2018.01.028
Xing G, Gillespie R, Bedri B, Quan A, Zhang P, Zhou XJ (2018) Proliferative glomerulonephritis with monoclonal IgG deposits in children and young adults. Pediatr Nephrol 33:1531–1538. https://doi.org/10.1007/s00467-018-3949-8
Torrealba J, Gattineni J, Hendricks AR (2018) Proliferative glomerulonephritis with monoclonal immunoglobulin G lambda deposits: report of the first pediatric case. Case Rep Nephrol Dial 8:70–75. https://doi.org/10.1159/000488641
Leung N, Bridoux F, Batuman V, Chaidos A, Cockwell P, D'Agati VD, Dispenzieri A, Fervenza FC, Fermand J-P, Gibbs S, Gillmore JD, Herrera GA, Jaccard A, Jevremovic D, Kastritis E, Kukreti V, Kyle RA, Lachmann HJ, Larsen CP, Ludwig H, Markowitz GS, Merlini G, Mollee P, Picken MM, Rajkumar VS, Royal V, Sanders PW, Sethi S, Venner CP, Voorhees PM, Wechalekar AD, Weiss BM, Nasr SH (2019) The evaluation of monoclonal gammopathy of renal significance: a consensus report of the international kidney and monoclonal gammopathy research group. Nat Rev Nephrol 15:45–59. https://doi.org/10.1038/s41581-018-0077-4
Hemminger J, Nadasdy G, Satoskar A, Brodsky SV, Nadasdy T (2016) IgG subclass staining in routine renal biopsy material. Am J Surg Pathol 40:617–626. https://doi.org/10.1097/pas.0000000000000605
Davis CW, Boyd SD, Ahmed R (2019) Longitudinal analysis of the human B cell response to Ebola virus infection. Cell 177:1566–1582. https://doi.org/10.1016/j.cell.2019.04.036
Fujita E, Shimizu A, Kaneko T, Masuda Y, Ishihara C, Mii A, Higo S, Kajimoto Y, Kanzaki G, Nagasaka S, Iino Y, Katayama Y, Fukuda Y (2012) Proliferative glomerulonephritis with monoclonal immunoglobulin G3κ deposits in association with parvovirus B19 infection. Hum Pathol 43:2326–2333. https://doi.org/10.1016/j.humpath.2012.04.004
Takehara E, Mandai S, Shikuma S, Akita W, Chiga M, Mori T, Oda T, Kuwahara M, Uchida S (2017) Post-infectious proliferative glomerulonephritis with monoclonal immunoglobulin G deposits associated with complement factor H mutation. Intern Med 56:811–817. https://doi.org/10.2169/internalmedicine.56.7778
Zhang F, Gao E, Liang S, Yang N, Wang J (2018) A case of switch from C3 glomerulonephritis to proliferative glomerulonephritis with monoclonal IgG deposits. Ann Clin Lab Sci 48:528–533 0091-7370/18/0400-528
Guiard E, Karras A, Plaisier E, Van Huyen J-PD, Fakhouri F, Rougier J-P, Noel L-H, Callard P, Delahousse M, Ronco P (2011) Patterns of Noncryoglobulinemic glomerulonephritis with monoclonal Ig deposits: correlation with IgG subclass and response to rituximab. Clin J Am Soc Nephrol 6:1609–1616. https://doi.org/10.2215/cjn.10611110
Larsen C, Boils C, Cossey L, Sharma S, Walker P (2016) Clinicopathologic features of membranous-like glomerulopathy with masked IgG kappa deposits. Kidney Int Rep 1:299–305. https://doi.org/10.1016/j.ekir.2016.08.012
Gumber R, Cohen JB, Palmer MB, Kobrin SM, Vogl DT, Wasserstein AG, Nasta SD, Bleicher MB, Bloom RD, Dember L, Cohen A, Weiss BM, Hogan JJ (2018) A clone-directed approach may improve diagnosis and treatment of proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Kidney Int 94:199–205. https://doi.org/10.1016/j.kint.2018.02.020
Katsuno T, Kato M, Fujita T, Tsuboi N, Hattori R, Ito Y, Maruyama S (2019) Chronological change of renal pathological findings in the proliferative glomerulonephritis with monoclonal IgG deposits considered to have recurred early after kidney transplantation. CEN Case Rep 8:151–158. https://doi.org/10.1007/s13730-019-00384-6
Author information
Authors and Affiliations
Contributions
Megan Troxell, Neeraja Kambham, Colin Lenihan, and Cynthia Nast contributed to the study conception and design. All the authors contributed to the data collection. Data tabulation and analysis was performed by Paul Miller, Andrew Xiao, Vanderlene Kung, and Megan Troxell. The first draft of the manuscript was written by Paul Miller, Andrew Xiao, and Megan Troxell. All the authors commented on previous versions of the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Dr. Amanda Uber is a Paul and Yuanbi Ramsay Endowed Fellow, Maternal and Child Health Research Institute at Stanford.
None of the other authors have conflicts of interest to declare.
Ethics approval
This study received Institutional Review Board approval at Stanford and Cedars-Sinai, and was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Consent to participate
Not applicable (HIPAA waiver for retrospective study).
Consent for publication
Not applicable (HIPAA waiver for retrospective study).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Miller, P., Xiao, A.Y., Kung, V.L. et al. Progression of proliferative glomerulonephritis with monoclonal IgG deposits in pediatric patients. Pediatr Nephrol 36, 927–937 (2021). https://doi.org/10.1007/s00467-020-04763-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-020-04763-5