
Abstract
Proliferative glomerulonephritis with monoclonal immunoglobulin G (IgG) deposits (PGNMID) is a rare disease that recently became recognized. Its pathological findings are characterized by the deposition of a single heavy chain subclass and a single light chain isotype. PGNMID has been proven to recur in renal allografts. Herein, the authors describe the case of a 46-year-old man who presented with nephrotic syndrome and progressive kidney injury following kidney transplantation. One month after transplantation, his clinical condition stabilized; however, the protocol biopsy showed depositions of IgG and complement on the glomeruli by immunofluorescence staining. Electron microscopy (EM) revealed granular electron-dense deposits (EDD) in the mesangium. Thereafter, renal biopsy was repeated because his proteinuria level increased. Proliferative glomerulonephritis, mainly in the mesangium, with IgG and complement deposits and mesangial and subendothelial EDD were observed; however, the pathological diagnosis was difficult. Renal dysfunction then became apparent, and renal biopsy was performed again 4 years and 10 months after kidney transplantation. Glomerular deposits on a single IgG subclass and a single light chain isotype (IgG3 kappa) with membranoproliferative features were observed. Abundant subendothelial EDD were detected on EM. Finally, the patient was diagnosed with PGNMID. Since it seemed that PGNMID had already developed at 1 month after transplantation, we considered recurrent PGNMID case in the allograft. The treatment for PGNMID has not been established yet, and even in this case, the graft function was eventually lost. For improving renal prognosis, early diagnosis and further investigation on the treatment are necessary.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Various recurrent glomerulonephritis cases after renal transplantation have been reported [1, 2]. Proliferative glomerulonephritis with monoclonal immunoglobulin G (IgG) deposits (PGNMID) is a recently described and rare disease entity [3, 4]. Recently, the novel concept of monoclonal gammopathy of renal significance (MGRS) has been reported [5]. PGNMID is one category of MGRS. PGNMID may also recur in the allograft; a small number of case reports or case series have been reported [6, 7]. Pathological diagnosis is essential for PGNMID, especially if characterized by a glomerular deposition of a single heavy-chain subclass and a single light-chain isotype on immunofluorescence (IF) staining. As a pattern of deposition, the IgG3 kappa is predominant. In addition, complement components, such as C3 and C1q, are often deposited in the glomeruli. On light microscopy (LM), the histological findings of PGNMID include membranoproliferative glomerulonephritis (MPGN), endocapillary proliferative glomerulonephritis, and membranous glomerulonephritis. Electron microscopy (EM) reveals mainly mesangial and subendothelial non-organized deposits. The majority of patients diagnosed with PGNMID exhibit proteinuria and progressive renal dysfunction. On standard electrophoresis or immunofixation electrophoresis, the monoclonal spike in the serum or urine is not detected in most patients. A previous case series reported that an administration of rituximab and aggressive immunosuppressive agents were successful [6]; however, there is no established treatment for PGNMID yet. PGNMID is a less well-known disease, and its diagnosis is difficult when IgG subclasses and light chains are not stained. As a result, treatment interventions will be delayed, which would consequently lead to a poor renal prognosis.
In this article, we report the clinical course and transition of the renal pathological findings of a PGNMID case, which recurred early after living, related-donor kidney transplantation.
Case report
A 46-year-old man with nephrotic syndrome and progressive renal impairment was admitted to our hospital. His medical history was as follows. Several years prior, proteinuria was detected, and a medical treatment was performed conservatively. Renal biopsy was not performed, and the cause of proteinuria was unknown. His renal function has deteriorated gradually; thus, hemodialysis was started. Six months after the initiation of hemodialysis, the patient received ABO-incompatible living related-donor kidney transplantation, with her mother as the donor. For the blood type incompatibility transplantation, rituximab was administered preoperatively. The maintenance immunosuppressive regimen consisted of prednisolone (PSL), tacrolimus, and mycophenolate mofetil. After transplantation, the serum creatinine levels were stable from 1.4 to 1.6 mg/dL. One month after transplantation, protocol renal biopsy was performed. LM showed no remarkable changes; however, the results of IF and EM indicated the deposition of immune complexes in the mesangium. Two years and 7 months after transplantation, the urinalysis showed a proteinuria level of over 1 g/24 h. The serum creatinine level temporarily increased to 2.3 mg/dL; therefore, kidney biopsy was performed 3 years and 4 months after transplantation. On LM, a mesangial proliferative change was observed. In the findings of IF and EM, immunocomplexes were deposited subendothelial area in addition to the mesangium. For proliferative glomerulonephritis, which was difficult to diagnose histologically, methylprednisolone pulse therapy (500 mg/day) was initiated, followed by oral PSL at a dose of 30 mg/day. However, the proteinuria level did not decrease. Thereafter, the patient presented with nephrotic syndrome and further deterioration of renal function. At 4 years and 10 months after transplantation, he was admitted to our institution for a detailed examination.
On admission, the patient had a blood pressure of 181/95 mmHg, height of 172.4 cm, and weight of 67.4 kg. Physical examination revealed bilateral lower leg edema. No abnormal signs were observed in the lungs, heart, or abdomen. Urinalysis showed proteinuria with a protein excretion of 4.85 g/24 h and 1–2 red blood cells per high power field. Hematologic tests indicated a white blood cell count of 1.1 × 103/µL, hemoglobin level of 8.0 g/dL, and platelet count of 226 × 103/µL. Laboratory investigation revealed a serum creatinine concentration of 2.77 mg/dL, blood urea nitrogen level of 36 mg/dL, and estimated glomerular filtration rate of 21.2 mL/min/1.73 m2. The total serum protein level was 4.9 g/dL with a serum albumin level of 3.0 g/dL. The total serum cholesterol was 229 mg/dL. Liver dysfunction and carbohydrate metabolism disorder were not observed. Immunological studies showed a serum IgG concentration of 491 mg/dL, IgA concentration of 83 mg/L, and IgM concentration of 56 mg/dL. The serum complement C3 level was 97.8 mg/dL (normal range 86–160 mg/dL); the C4 level was 40.6 mg/dL (normal range 17–45 mg/dL); and the total complement activity (CH50) level was 63.2 U/mL (normal range 30–45 U/mL). The findings of the tests for hepatitis B and C, anti-nuclear antibody, anti-DNA antibody, and cryoglobulin were all negative. As a result of the additional examinations, the serum and urine immunoelectrophoresis showed no monoclonal bands. Furthermore, the entity of monoclonal protein was not identified on immunofixation electrophoresis. The serum-free kappa light chain level was 47.6 mg/L; the lambda light chain level was 36.3 mg/L; and the kappa lambda ratio was 1.311 (normal range 0.248–1.804). Percutaneous kidney biopsy of the allograft was performed to establish an accurate diagnosis. The findings of transplantation biopsy are described below.
Transplantation biopsy
First (1 h after transplantation)
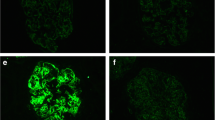
The sample contained 13 glomeruli, none of which showed global sclerosis. LM revealed a segmental mesangial hypercellularity in one glomerulus. Interstitial fibrosis and tubular atrophy were slight. No findings of acute rejection were observed (Fig. 1). IF staining showed no significant positive findings (Fig. 2). EM presented no electron-dense deposits (EDD) in the tissue (Fig. 3).

Light microscopic findings of transplantation biopsy. a, e, i First transplantation biopsy. The presented glomerulus is almost intact. Proliferative lesions are not observed. Minor interstitial fibrosis and tubular atrophy are detected. Arteriosclerosis corresponding to age is confirmed in the interlobular artery. b, f, j Second transplantation biopsy. No significant pathological findings are observed in the glomeruli. There is no observation of rejection reactions. Interstitial findings, such as fibrosis and tubular atrophy, remain mild. c, g, k Third transplantation biopsy. The representative glomerulus shows a mesangial matrix expansion and mesangial cell proliferation. The chronic change in the interstitial fibrosis and tubular atrophy is progressing. d, h, l Fourth transplantation biopsy. The glomeruli become lobulated. In addition to the mesangial proliferation, double contours of the glomerular basement membranes, extracapillary proliferation, and epithelial cell hyperplasia are observed. The tubulointerstitial injury is further exacerbated. a–d Periodic acid–Schiff staining, original magnification 400×. e–h Periodic acid–methenamine silver staining, original magnification 400 ×. i–l Masson’s trichrome staining, original magnification 100×

Immunofluorescence findings of transplantation biopsy. First transplantation biopsy. Immunofluorescence staining shows no significant positive findings for IgG (a), C3 (e), and C1q (i) in the glomeruli. b, f, j Second transplantation biopsy. The findings for IgG (b), C3 (f), and C1q (j) are positive in the mesangial area. c, g, k Third transplantation biopsy. Immunofluorescence microscopy reveals granular staining in the mesangium and along the capillary walls for IgG (c), C3 (g), and C1q (k). d, h, l Fourth transplantation biopsy. Although similar findings are found as in the third biopsy, the positive region of the heavy chain and complement becomes more prominent in the periphery. a–l Original magnification 400 ×

Electron microscopic findings of transplantation biopsy. a First transplantation biopsy. Electron microscopy (EM) shows no electron dense deposits (EDD). b Second transplantation biopsy. On EM, segmental granular mesangial EDD are observed. c EM reveals granular EDD in the subendothelial areas. Mesangial interposition is also confirmed. d A large amount of EDD are detected linearly in the subendothelial areas. The EDD have no organized structures
Second (1 month after transplantation)
On LM, 17 glomeruli were observed, none of which showed global sclerosis. There was no mesangial proliferation, endocapillary hypercellularity, and extracapillary proliferation. Mild interstitial fibrosis and tubular atrophy were observed. Pathological findings suggestive of an acute rejection were not confirmed (Fig. 1). IF microscopy revealed a moderate intensity (2 +) for IgG and C3 and mild intensity (1 +) for C4 and C1q in the mesangium (Fig. 2). EM showed EDD in the mesangial regions (Fig. 3).
Third (3 years and 4 months after transplantation)
Nine glomeruli were microscopically examined, one of which showed global sclerosis. The other glomeruli exhibited diffuse mesangial matrix expansion and mesangial cell proliferation. Adhesion to the Bowman’s capsule was also observed. Interstitial fibrosis and tubular atrophy have developed. Focal cellular infiltration was observed in the interstitium; however, tubulitis was not detected (Fig. 1). On IF, granular staining in the mesangium and along the capillary walls for IgG (2 +), C3 (1 +), and C1q (1 +) was observed (Fig. 2). EM revealed mesangial and subendothelial deposits. Duplication of the glomerular basement membranes with a mesangial interposition was observed (Fig. 3).
Fourth (4 years and 10 months after transplantation)
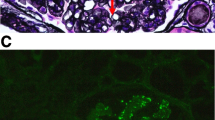
The specimen for LM included three glomeruli, one of which was globally sclerotic. The nonsclerosed glomeruli showed predominantly a finding of MPGN mainly based on the duplication of the basement membranes and mesangial proliferation. The glomeruli were lobulated, and the tissue revealed a crescentic formation, epithelial cell hyperplasia, and segmental sclerosis. The interstitial architecture showed an advanced interstitial fibrosis and tubular atrophy without a significant interstitial inflammation or tubulitis. Severe hyalinosis of the arterioles with a lumen occlusion was observed (Fig. 1). IF microscopy examination demonstrated peripheral and mesangial granular staining for IgG (2 +), C3 (2 +), and C1q (1 +) (Fig. 2). The staining for the IgG subclasses revealed a strong IgG3 signal; however, the findings for the IgG1, IgG2, and IgG4 were entirely negative. In the staining for light chains, the finding for kappa was positive in the mesangium and along the capillary walls; however, the finding for lambda was negative (Fig. 4). On EM, a subendothelial EDD was detected, and a large amount of deposits were observed in the form of strips (Fig. 3). The result of immunostaining for detection of amyloid deposits was negative.

Immunofluorescent staining of IgG subclasses and light chains. Immunofluorescence staining for the IgG subclasses and light chains was performed on the fourth transplantation biopsy. Only IgG3 (c) is intensely positive in the mesangium and along the glomerular basement membranes. The findings of the staining for IgG1 (a), IgG2 (b), and IgG4 (d) are entirely negative in the same specimen. The glomerulus shows a strong staining for the kappa light chain (e) with a negative staining for the lambda light chain (f). a–f Original magnification 400 ×
Discussion
We report a case of PGNMID in a renal allograft diagnosed with clinicopathological features. The clinicopathological findings, at the time of each renal transplantation biopsy, are summarized in Table 1.
PGNMID is a newly established disease concept and a rare disease. PGNMID was first described in 2004 as a novel glomerulonephritis related to monoclonal IgG deposition by Nasr et al. [3]. Subsequently, Nasr et al. reported a clinical research including 37 cases in 2009 [4]. Clinical findings of PGNMID were characterized by proteinuria and progressive renal dysfunction, and approximately half of the patients had nephrotic syndrome. Only 30% of the cases had identified monoclonal proteins in the serum or urine. There were usually no hematologic diseases. The pathological findings on LM were mostly MPGN. In addition, endocapillary proliferative glomerulonephritis and membranous nephropathy pattern were described. IF microscopy revealed granular deposits for a single heavy chain subclass and a single light chain isotype in the mesangium and along the capillary walls. Most cases were positive for IgG3 kappa. Complement deposits, including C3 and C1q, were often detected in a similar region to that of the heavy and light chain. On EM, granular EDD were observed mainly in the subendothelial area and mesangium. Subepithelial deposits were less frequent.
Recently, recurrent PGNMID after kidney transplantation has been reported [6, 7]. Nasr et al. described the clinicopathological findings of recurrent PGNMID [6]. Renal function deterioration and proteinuria were present in all cases. None of the patients had monoclonal proteins. There were cases in which the pathological findings of LM were different between the first incidence and recurrence. Conversely, the IF study findings were consistent with the native kidney. The subclass of the deposited IgG was IgG3 in all cases, and the light chain was kappa in most cases. The mean time from the transplantation to the recurrence was 3.8 months.
This case was diagnosed as PGNMID in a renal allograft. In a previous report [3], Nasr et al. described the following diagnostic criteria: (1) presence of glomerular monoclonal IgG deposits restricted to a single IgG subclass and a single light chain isotype, associated with endocapillary proliferative, membranoproliferative, or membranous features; (2) presence of granular (“immune complex type”) deposits on EM; and (3) absence of clinical and laboratory evidence of cryoglobulinemia. This case fulfilled these criteria, and the clinical manifestations coincided with the previously reported clinical features. As a differential diagnosis, type I cryoglobulinemia, Randall-type light and heavy chain deposition disease (LHCDD), light and heavy chain amyloidosis, immunotactoid glomerulonephritis, primary MPGN, and infection-related glomerulonephritis (IRGN) can be considered. In this case, the findings of cryoglobulinemia were not observed. There was no deposition of IgG3 kappa in the tubular basement membranes, vessel walls, and interstitium. LHCDD is characterized by the deposition on the glomerular and tubular basement membranes, as opposed to PGNMID. No substructures and microtubules suggestive of cryoglobulinemia, immunotactoid glomerulopathy and amyloidosis were confirmed based on the EM findings. Immunostaining for amyloid was negative. The results of the IgG subclass and light chain staining made it possible to distinguish from primary MPGN and IRGN.
In our case, monoclonal protein in the serum and urine was not detected, and the serum-free light chain assay showed a normal kappa and lambda ratio. It may be explained that monoclonal IgG has a high affinity to the glomeruli, and even if it remains in the serum, it does not reach the measurable concentration in an extremely small amount.
Glomerulonephritis may recur after kidney transplantation; however, it may develop as a de novo glomerular disease. Recurrent PGNMID has been reported to develop early after transplantation [6]. Albawardi et al. described a de novo PGNMID case in a kidney allograft [7]. The concern is whether the presented PGNMID case was recurrent or de novo. The disease recurrence is considered to be caused by persistent circulating factors in the recipient. The finding for IgG was already positive, and EDD was observed in the mesangial area only 1 month after transplantation. These findings indicate that this case is recurrent nephritis. To our knowledge, the pathological features of this presented case suggested the earlier recurrence among the recurrent PGNMID in kidney allografts reported up to date. Unfortunately, owing to the lack of specimens, the IgG subclasses and light chains could not be stained.
This case took a long time to be diagnosed. Recurrent PGNMID is possibly an underrecognized disease and is often misled to respective diseases, such as MPGN. Initially, this case did not lead to the diagnosis of PGNMID because the analysis of the IgG subclasses and light chains was not performed on IF. Indeed, the findings for IgG3 kappa were positive upon staining the third biopsy specimen. To diagnose earlier without overlooking PGNMID, it is essential to perform routine immunostaining for light chains including kappa and lambda [8]. This case demonstrates the change in the pathological findings of recurrent PGNMID over time, which is very valuable. Positive findings on IF studies and EDD on EM were confirmed at the stage when the clinical symptoms, such as proteinuria, and histological changes on LM were not observed. On LM, mesangial proliferation first occurred and then progressed to MPGN-like features. EDD confirmed by EM was found in the mesangial region; thereafter, subendothelial deposits became apparent. Although the characteristic histopathology of PGNMID is a MPGN-like change, it may be pathologically diagnosed with mesangial proliferative glomerulonephritis at an early stage of recurrence.
The renal survival of PGNMID in the native kidneys is poor [4]. Recurrent cases in the allografts seem to have a better prognosis than the native diseases [6]. However, effective treatment for PGNMID has not been established yet to date. Transplantation patients receive continuous immunosuppressive therapy; however, PGNMID still relapses. As such, immunosuppressive regimen after kidney transplantation is considered insufficient as a prevention of PGNMID. As the etiology of PGNMID, a hypothesis that clonal B lymphocytes or plasma cells induce super-secretion of monoclonal IgG with a strong affinity to the kidney has been assumed. In that respect, use of rituximab may be reasonable [9]. However, this case recurred immediately; nevertheless, rituximab was administered before transplantation. According to the review of MGRS in recent years, bortezomib, cyclophosphamide, and dexamethasone are proposed as treatment options of IgG or IgA type PGNMID [10]. In our patient, the deterioration of his renal function was rapid, and finally, maintenance hemodialysis was reintroduced 6 months after the fourth renal biopsy.
In summary, we described a case of PGNMID that developed in the allograft, suggesting the possibility of a relapse at an early stage after transplantation. Rituximab and immunosuppressive treatment after transplantation were also ineffective in our patient. The awareness of this disease and accurate pathological diagnosis are necessary for the determination of aptitude in kidney transplantations or early treatment interventions at recurrence. Further investigations are needed to clarify the pathophysiology of PGNMID and establish therapeutic methods.
References
O’Shaughnessy MM, Liu S, Montez-Rath ME, Lenihan CR, Lafayette RA, Winkelmayer WC. Kidney transplantation outcomes across GN subtypes in the United States. J Am Soc Nephrol. 2017;28:632–44.
Ponticelli C. Recurrence of focal segmental glomerular sclerosis (FSGS) after renal transplantation. Nephrol Dial Transplant. 2010;25:25–31.
Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, Appel GB, Aucouturier P, D’Agati VD. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65:85–96.
Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, Stokes MB, Nadasdy T, D’Agati VD. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20:2055–64.
Bridoux F, Leung N, Hutchison CA, Touchard G, Sethi S, Fermand JP, Picken MM, Herrera GA, Kastritis E, Merlini G, Roussel M, Fervenza FC, Dispenzieri A, Kyle RA, Nasr SH. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. 2015;87:698–711.
Nasr SH, Sethi S, Cornell LD, Fidler ME, Boelkins M, Fervenza FC, Cosio FG, D’Agati VD. Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol. 2011;6:122–32.
Albawardi A, Satoskar A, Von Visger J, Brodsky S, Nadasdy G, Nadasdy T. Proliferative glomerulonephritis with monoclonal IgG deposits recurs or may develop de novo in kidney allografts. Am J Kidney Dis. 2011;58:276–81.
Gowda KK, Nada R, Ramachandran R, Joshi K, Tewari R, Kohli HS, Jha V, Gupta KL. Proliferative glomerulonephritis with monoclonal immunoglobulin deposition disease: the utility of routine staining with immunoglobulin light chains. Indian J Nephrol. 2015;25:344–8.
Merhi B, Patel N, Bayliss G, Henriksen KJ, Gohh R. Proliferative glomerulonephritis with monoclonal IgG deposits in two kidney allografts successfully treated with rituximab. Clin Kidney J. 2017;10:405–10.
Caravaca-Fontán F, Gutiérrez E, Delgado Lillo R, Praga M. Monoclonal gammopathies of renal significance. Nefrologia. 2017;37:465–77.
Acknowledgements
The authors appreciate the excellent technical assistance of N. Asano, Y. Sawa, and N. Suzuki. The authors also acknowledge Editage for providing editorial and publication support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors have declared no competing interest.
Human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from the participant included in the article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Katsuno, T., Kato, M., Fujita, T. et al. Chronological change of renal pathological findings in the proliferative glomerulonephritis with monoclonal IgG deposits considered to have recurred early after kidney transplantation. CEN Case Rep 8, 151–158 (2019). https://doi.org/10.1007/s13730-019-00384-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-019-00384-6