
Abstract
Background
Deficiency of complement factor H-related (CFHR) proteins and CFH autoantibody-positive hemolytic uremic syndrome (DEAP-HUS) represents a unique subgroup of complement-mediated atypical HUS (aHUS). Autoantibodies to the C-terminus of CFH block CFH surface recognition and mimic mutations found in the genetic form of (CFH-mediated) aHUS. CFH autoantibodies are found in 10–15 % of aHUS patients and occur—so far unexplained—almost exclusively in the background of CFHR1 or CFHR3/CFHR1 deletions.
Methods
As a well-defined role for eculizumab in the treatment of complement-mediated aHUS is becoming established, its role in DEAP-HUS is less conspicuous, where a B-cell-depleting and immunosuppressive treatment strategy is being proposed in the literature.
Results
We here show eculizumab to be safe and effective in maintaining a disease-free state, without recurrence, in a previously plasma-therapy-dependent DEAP-HUS patient, and in another patient in whom, although showing a good clinical response to plasma therapy, the therapy was hampered by allergic reactions to fresh frozen plasma and contend there is a rationale for the use of eculizumab in concert with an immunosuppressive strategy in the treatment of DEAP-HUS. Considering the high rate of early relapse, the possible coexistence and contribution of both known and unknown complement-gene mutations, the probable pathogenic role of CFHR1 as a complement alternative pathway (CAP) regulator, the experimental nature of measuring and using anti-CFH autoantibodies to guide management, and until the positive reports of immunosuppression in addition to plasma therapy are confirmed in prospective studies, we feel that a complement-directed therapy should not be neglected in DEAP-HUS. Serial CFH autoantibody titer testing may become a valuable tool to monitor treatment response, and weaning patients off eculizumab may become an option once CFH autoantibody levels are depleted.
Conclusions
A prospective study of eculizumab treatment in a larger cohort of DEAP-HUS patients is required to validate the applicability of our positive experience.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hemolytic uremic syndrome (HUS) describes the triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. In 80 % of cases, HUS is initiated by Shiga toxin (Stx)-producing Escherichia coli (STEC) [1]. HUS in association with pregnancy, drugs such as calcineurin inhibitors, defective cobalamin metabolism, and genetic defects of complement regulation make up the remainder of cases, collectively termed atypical HUS (aHUS) [2].
STEC-HUS was traditionally not recognized as complement-mediated disease, although there is emerging evidence that complement plays a role in its pathology [3, 4]. In aHUS, however, 40–60 % of cases are linked to mutations in components of the complement alternative pathway (CAP) [5, 6], where the disease-linked regulators are the plasma proteins complement factors H (CFH) and I (CFI), and the cell-surface-associated membrane cofactor protein (MCP/CD46) and thrombomodulin (THBD/CD141). These proteins interfere with the assembly, activity, and stability of the CAP C3 convertase complex (C3bBbP) both in plasma and on cell surfaces. Gain of function mutations in the C3 convertase components C3 and complement factor B (CFB) and loss-of-function mutations in regulators (above; primarily CFH) have been linked to CAP hyperactivity and aHUS [5]. A substantial subgroup of patients with complement-mediated aHUS (10–15 % of cases) [7–9] have also been found to have autoantibodies to CFH. More than 90 % of these cases are associated with homozygous deletions within the complement factor H-related (CFHR) gene cluster on chromosome 1q32, resulting in the loss of genes coding for CFH-related proteins 3 (CFHR3) and 1 (CFHR1). This condition has a typical age of onset ranging from 4 to 17 years and is termed deficiency of CFHR proteins and CFH autoantibody-positive HUS (DEAP-HUS) [8].
Approximately 50 % of DEAP-HUS cases present with a diarrheal prodrome similar to STEC-HUS [10], and the disease has a similar clinical course to non-CFH mutation associated aHUS in terms of chronic kidney disease (CKD), progression to end-stage kidney disease (ESKD), and death (occurring in about 40 %, 30 %, and 10 % of patients, respectively). There is a high rate of disease relapse (58 %) due to accumulation of autoantibodies [10], and with each recurrence, the risk of CKD and ESKD increases [11]. In the largest series of patients with DEAP-HUS (44 patients), almost 27 % had ESKD and almost 40 % had evidence of CKD [10].
Specific clinical practice guidelines for DEAP-HUS are lacking; strategies have been adopted from genetically determined forms of aHUS or other autoimmune diseases [12, 13]. The available literature supports therapeutic plasma exchange (TPE) initially, as in the other forms of aHUS, but with the addition of an antibody-suppressing or depleting treatment, such as azathioprine, mycophenolate mofetil (MMF), cyclophosphamide, or rituximab [10, 14, 15]. Expanding the scope of treatment options for DEAP-HUS, we report here the safe and successful use of the complement C5 blocker eculizumab (Soliris®) in two DEAP-HUS patients. One patient was in a more acute phase of the disease and responding to plasma exchange but developed increasingly severe allergic reactions to fresh frozen plasma (FFP); the other was dependent on biweekly plasma infusions as maintenance therapy. We also discuss current treatment strategies for DEAP-HUS and highlight the distinctive characteristics of this aHUS subgroup that support the use of a complement-directed therapy, at least initially.
Patients, methods, and results
Patient 1
An 11-year-old girl with no past medical history of note travelled to Syria where she became unwell with nausea, vomiting, fever, and hematuria. There was evidence of acute kidney injury (creatinine 180 μmol/L), hyperuricemia [506 μmol/L (normal 110–310 μmol/L)] anemia (hemoglobin 89 g/l), and thrombocytopenia (68 × 109/L). She maintained good urine output and avoided dialysis. Initial management consisted of blood and platelet transfusions and pulsed methylprednisolone followed by prednisone and allopurinol. She underwent a bone marrow biopsy, which showed a hypocellular marrow with normal maturation. Her creatinine peaked at 660 μmol/L. At the time of discharge from the hospital, her creatinine was 447 μmol/L and platelets were still low (68 × 109/L). Upon return to Canada, she presented to our institution a month post discharge from the hospital in Syria and 7 weeks after initial presentation. On admission, she was cushingoid, edematous, and hypertensive (blood pressure 134/100 mmHg). Initial hematological investigations were remarkable for anemia (hemoglobin 109 g/L), reticulocytosis [651 × 109/L (normal 10–100 × 109/L)], low haptoglobin [<0.07 g/L (normal 0.32–1.98)], and thrombocytopenia (platelets 48 × 109/L). There were schistocytes on blood film. This was accompanied by significant renal impairment (creatinine 403 μmol/L, urea 26.7 mmol/L), hypokalemia (potassium 2.9 mmol/L), hypocalcemia (ionized calcium 1.01 mmol/L), hyperuricemia [510 μmol/L (normal 120–360)], and hyperphosphatemia (phosphate 3.7 mmol/L). Serum bilirubin was normal but aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) were elevated (LDH 4,510 U/L). Parathyroid hormone (PTH) at presentation was also significantly raised [301 ng/L (normal 10–65)]. Urine analysis showed >3 g/L protein and large blood, there were no casts on microscopy, and a 24-h assessment had 5.54 g of protein (protein/creatinine ratio 1,183 mg/mmol). Regarding complement studies, her C3 was normal [0.79 g/L (normal 0.77–1.43)], as was her C4 [0.25 g/L (normal 0.07–0.4 g/L)]. Her a disintegrin and metalloprotease with thrombospondin motifs (ADAMTS-13) protease activity was normal (52 %, normal range 30–120 %). A full vasculitic and microbiological screen was negative; renal biopsy showed a thrombotic microangiopathy (TMA) with acute and chronic features, with both the glomerular capillaries and smaller-caliber interstitial vessels being affected.
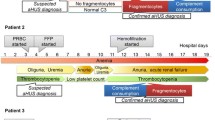
Total plasma exchange (TPE) was commenced on day 12 of admission, with FFP at 1.5 × plasma volume. She received a total of nine TPE sessions over the first 2 weeks, with a slow increase in platelet count and no real improvement in creatinine (Fig. 1a). She was switched to FFP infusions daily for a week, then three times weekly for 2 weeks, then twice weekly for 2 months. The steroids (60 mg/m2) initiated at first presentation were weaned off over the course of 2 months (Fig. 1a). Autoantibodies to CFH were confirmed. Complete absence of CFHR1 and CFHR3 was found on Western blot analysis and a homozygous deletion of CFHR3/CFHR1 on chromosome 1q32 subsequently confirmed by multiplex ligation-probe amplification (MLPA) analysis. Genetic analysis for mutations in CFH, CFI, Factor B, MCP/CD46, CFHR5, and C3 genes was negative.

a Clinical course of patient 1 during initial presentation and first major relapse. b Clinical course of patient 1 during maintenance plasma infusion (PI) and chronic eculizumab therapy. FFP, fresh frozen plasma; TPE, therapeutic plasma exchange
Two weeks after discontinuation of steroids and while on twice-weekly FFP infusions, the patient developed a recurrence, as evidenced by an increase in creatinine (398 μmol/L), and drop in platelets (116 × 109/L), hemoglobin (85 g/L), and C3 (0.71 g/L). She began daily FFP infusions and was recommenced on high-dose prednisone, which was continued for 4 months (Fig. 1a). The FFP infusions were continued biweekly for 38 months with no further relapse. On two separate occasions, with an intercurrent illness, biochemical changes (LDH, platelets) were suggestive of a possible impending relapse; therefore, a decision in favor of chronic plasma therapy was made. Her creatinine slowly improved and was 119 μmol/L 1 year later.
Four years from initial presentation, she was entered into an open-label, multicenter, controlled clinical trial of eculizumab for adolescent patients with plasma-therapy-sensitive aHUS (C08-003 ClinicalTrials.gov numbers, NCT00844428 [adolescent)]. This consisted of weekly eculizumab for 4 weeks (induction phase) and thereafter biweekly eculizumab (maintenance phase) for 2.5 years. Plasma therapy was successfully discontinued (Fig. 1b). CFH autoantibodies remained positive on a low level, which was in keeping with ongoing signs for complement activation [e.g., soluble terminal complement complex (SC5b-9)], but there was no further evidence of disease activity (Table 1) [16]. Creatinine remained stable at 100 μmol/L, with an estimated glomerular filtration rate (eGFR) of 80 ml/min/1.73 m2. A plan to attempt an antibody-depleting or immunosuppressive strategy with a careful wean of eculizumab is now underway.
Patient 2
An 8-year-old boy presented with a 3-day history of vomiting, abdominal pain, pallor, lethargy, and decreased fluid intake. He had passed one loose, non-bloody stool, and although his urine was described as being dark in color, there was no decline in urine output noted. On examination, he was pale with icteric sclerae and a few petechiae. Blood pressure was elevated at 119/67 mmHg, and he was clinically hypovolemic. The rest of his examination was unremarkable. Urine dipstick showed protein >3 g/L and 3+ blood, and microscopy showed >3 red blood cells/high power field (hpf), occasional white blood cells, and coarse granular casts. Complete blood count revealed a low hemoglobin (67 g/l), thrombocytopenia (22 × 109/L), neutrophil leukocytosis (neutrophils 9.4 × 109/L, WBC 13.8 × 109/L), and spherocytes and fragments on blood film consistent with a microangiopathic process. Further evidence of hemolysis included a reduced haptoglobin [<0.01 (normal 0.32–1.98 g/L)], as well as elevated reticulocyte count [329 (normal 10–100 × 109/L)], bilirubin (38 μmol/L), AST (176 U/L), and LDH (6,450 U/L). There was evidence of significant acute kidney injury (urea 57.2 mmol/L, creatinine 472 μmol/L), mild hyponatremia (sodium 133 mmol/L), borderline hyperkalemia (5 mmol/L), and hyperphosphatemia (3.94 mmol/L).
Initial management was supportive and consisted of fluid resuscitation with normal saline, a packed red blood cell transfusion, and monitoring of his fluid status. Complement studies subsequently revealed a low C3 [0.5 (normal 0.77–1.43 g/L)] and normal C4. Antineutrophil antibodies (ANA) and antineutrophil cytoplasmic antibodies (ANCA) were negative. Antistreptolysin O (ASO) titer was normal (200 IU/L), and throat swab was negative for beta hemolytic group A Streptococci. Verotoxin (Shiga toxin) polymerase chain reaction (PCR) examination of his stool was negative, and no enterohemorrhagic bacteria were detected on stool culture. A spot urine protein/creatinine ratio was elevated at 910 mg/mmol. An abdominal ultrasound showed enlarged and echogenic kidneys.
The patient maintained a good urine output and avoided dialysis. Although his creatinine improved over the first week of admission, his platelets were showing no sign of recovery, his LDH was still markedly elevated (15,764 U/L), C3 remained low (0.47 g/L), and he required a total of three red blood cell transfusions for anemia. A central line was inserted through his right internal jugular vein (RIJ) on day 9 of admission, and daily (1.5×) plasma volume exchanges commenced with FFP. There was a good response to TPE in terms of blood parameters, his C3 transiently normalized (0.73 g/L) by day 3, and his creatinine dropped to 91 μmol/L after 5 days of TPE. After the first four exchanges, he developed increasing allergic reactions to FFP (one requiring cessation of treatment). He received three further exchanges with predominantly 5 % albumin, but after a total of seven TPEs, a decision was made to transfer him to eculizumab therapy (Fig. 2). He received his first dose of eculizumab (600 mg) on day 16 of admission. He received meningococcal C, Prevnar 13 pneumococcal, and Haemophilus influenzae B vaccinations prior to eculizumab and was additionally placed on penicillin prophylaxis for 14 days. His C3 was low, at 0.69 g/L, platelets 146 × 109/L, and creatinine 86 μmol/L on day 16. Four days after eculizumab initiation, his C3 (0.87 g/L), creatinine (67 μmol/L), and platelets (169 × 109/L) had normalized. He received weekly eculizumab for 3 weeks (induction phase) prior to converting to biweekly infusions (maintenance phase) (Fig. 2). Further complement studies were performed both pre- and post-eculizumab infusions on two separate occasions (at 5 and 6 months) during biweekly maintenance therapy. High pre-eculizumab soluble terminal complement complex (SC5b-9) levels, possibly indicating emerging complement activation toward the end of the treatment cycle, were significantly lowered upon eculizumab reinfusion (Table 2) [16]. Whereas in-depth complement workup detected no mutations in CFH, CFI, CFB, MCP/CD46, THBD/CD141, CFHR5, or C3 genes, and ADAMTS13 activity was within normal range, autoantibodies to CFH were positive (Table 2), and a homozygous deletion of CFHR3/CFHR1 was confirmed by MLPA. Interestingly, we observed a positive impact of eculizumab on anti-CFH titers. However, C3 levels remained low, most probably due to continuous CAP dysregulation via the anti-CFH autoantibodies and/or a transiently positive C3 nephritic factor (C3NeF) (Table 2).

Clinical course of patient 2. TPE, therapeutic plasma exchange
Discussion
We describe the safe and effective use of the complement C5 inhibitor, eculizumab, in maintaining a disease-free state in the presence of ongoing anti-CFH autoantibodies in a previously plasma-therapy (infusion)-dependent DEAP-HUS patient. In another patient, plasma therapy (exchange)—although clinically successful—was hampered by allergic reactions to FFP and had to be replaced by eculizumab, which maintained this patient in remission despite ongoing presence of antibodies and evidence of complement activation.
DEAP-HUS
Autoantibodies [immunoglobulin G (IgG)1 and 3 subtypes] to CFH were first described in association with aHUS in three patients [7]. The CFH autoantibodies bind to the short consensus repeats 19–20 (SCR 19–20) in the C-terminal-recognition region of CFH, a known “hot spot” for aHUS-associated mutations in the CFH gene, containing—among others—the binding sites for C3b, heparin, and endothelial cells. Thus, antibodies—similar to CFH mutations—result in failure of regulation of the CAP C3 convertase (C3bBb) on the endothelial surface [17]. Recognizing a correlation between the presence of CFH autoantibodies and the homozygous deletion of genes for CFH-related proteins CFHR3 and CFHR1 (CFHR3/CFHR1) caused by nonallelic homologous recombination on chromosome 1q32—a phenomenon initially described as an independent susceptibility factor for aHUS [18]—a novel subgroup of aHUS was defined as deficiency of CFHR proteins and CFH autoantibody-positive HUS, or DEAP-HUS [8].
Whereas genetic predisposition is not fully understood, CFHR3/CFHR1 deficiency was found in up to 93 % of patients with autoimmune aHUS, suggesting a causal role for the development of these antibodies [19]. Furthermore, a regulatory function of CFHR1 on the C5 convertase has recently been recognized via which CFHR1—when deficient—may also contribute to aHUS pathogenesis [20]. Similarly, CFHR3 has regulatory effects on the C3 convertase of complement [21].
CFHR3 and CFHR1 lie in contiguous regions on chromosome 1q32, downstream of CFH, and sequence homologies render this region at risk of chromosomal recombination, often affecting both CFHR3 and CFHR1 together. However, it has recently been suggested that deficiency of CFHR1 rather than CFHR3 seems to be a precursor to the development of CFH autoantibodies [9], and so far, only five patients with autoantibody-mediated aHUS but not lacking CFHR3/CFHR1 have been described [9, 22]. In addition, mutations in CFH, CFI, MCP/CD46, and C3 have been detected in five of 13 patients with CFHR3/CFHR1 deletion and autoantibody-mediated aHUS, suggesting a “multiple-hit” mechanism to the etiology of autoimmune aHUS in at least some of these patients [22]. Further corroboration of a genetic multiple-hit hypothesis comes from the recent publication of the European Working Party on Complement Genetics in Renal Diseases, in which certain risk haplotypes, especially in CFH and MCP/CD46, screened for in 795 patients across multiple cohorts predicted a worse prognosis [6].
More recent studies on the pathobiology of the autoantibodies have shed more light on their pathogenicity [23, 24]. The autoantibodies affect CFH binding to pentraxin 3, a protein family, including C-reactive protein (CRP) that normally aids CFH binding to cell surfaces and enhances endothelial cell protection against complement-mediated cytotoxicity [23]. It is now known that the antibodies do not just affect CFH binding to C3b on the cell surface but have many effects on CFH function, both in the fluid phase and on the surface, including: (i) reduced CFH binding to C3(H20) in the fluid phase, (ii) reduced binding of CFH to C3b, (iii) reduced CFH dissociation of the C3bBb complex (decay activity), and (iv) altered cofactor activity [24].
Treatment concepts
The principal treatment concept for complement-mediated aHUS hinges on restoring proper CAP regulation by removing circulating factors or replacing deficient or defective regulatory proteins, and to date, it centers on the prompt initiation of TPE using FFP or virus-inactivated, pooled plasma (SD-plasma; Octaplas®), as outlined in evidence-based consensus guidelines [25, 26]. Ongoing management can be achieved with prophylactic plasma infusions to maintain remission [27], but complications are multiple and include primary or secondary treatment failure, allergic reactions, and access-related infections or thrombosis [28]. There are no consensus guidelines for managing DEAP-HUS, but treatment concepts can be developed following those for other autoimmune diseases. TPE will remove CFH autoantibodies and simultaneously—with plasma as replacement fluid—enhance the circulating CFH pool, which contributes to CAP regulation and reduces the antibody load via specific binding. In fact, Strobel et al. proposed using the CFH autoantibody-binding capacity of the CFHR1 C-terminus, which is almost identical to the CFH C-terminus, as a potential treatment tool in DEAP-HUS patients [29]. However, as none of these strategies are capable of blocking ongoing antibody production, a treatment strategy limiting antibody synthesis would be useful, especially after the initial acute management, to prevent recurrence.
In keeping with this concept several immunosuppressive agents have been employed and allowed termination of TPE or plasma infusions, including prednisone, intravenous immunoglobulin (IVIG), azathioprine, MMF, cyclophosphamide, and rituximab (Table 3) [7, 9, 11, 14, 15, 22, 30–32]. Cyclophosphamide was used in three DEAP-HUS patients at a dosage of 1 g/1.73 m2 for five pulses in one and 0.5 g/1.73 m2 for two pulses in the other two patients and resulted in a sustained reduction in CFH autoantibodies without recurrence of aHUS or the ongoing need for TPE [15]. In the large series reported by Dragon-Durey et al., plasma exchange and immunosuppression, when used in combination from disease onset, had a favorable outcome in eight of nine patients [10].
The successful use of rituximab as an antibody-depleting strategy in a plasmatherapy-dependent patient with DEAP-HUS has been reported. Dosage employed was 375 mg/m2 weekly for 4 weeks, and this effected a sustained reduction in CFH IgG autoantibodies and normal C3 for a 4-month period [14]. Rituximab was also used in another child, but there was a recurrence 3 months later despite complete depletion of CD19- and CD20-positive cells [15]. A similar experience was reported by Skerka et al. in which rituximab actually failed to lower CFH autoantibody titers and to prevent recurrence despite depleted CD19 and CD20 cells [32]. CFH antibodies disappeared in one patient but were unaffected in another two DEAP-HUS patients, as reported by Dragon-Durey et al. [10]. Why there is a variable effect on the antibody titers despite confirmed CD19 and CD20 B-cell depletion is uncertain, and close monitoring of titers is warranted. It must also be noted that an exclusive immunosuppressive strategy fails to take into account the potential contribution of a lack of the C5 convertase inhibitor CFHR1 [18, 20] or the possibility of other mutations in complement regulatory proteins that might be contributing to the pathogenesis of aHUS in some patients, as suggested by the findings of Moore et al. [22].
Eculizumab
There is strong evidence from murine models for the pivotal role of C5 in the pathogenesis of aHUS [33]. Eculizumab is a humanized murine monoclonal IgG that binds with high affinity to complement C5, thus preventing its cleavage and the formation of terminal complement components, including the membrane attack complex (MAC), SC5b-9 [34]. Although originally developed for treating rheumatoid arthritis and various nephritides [35], reports of its success in paroxysmal nocturnal hemoglobinuria (PNH) [36] and subsequently aHUS defined its current role [37]. Since the original report by Nuernberger [37], eculizumab has successfully been used as rescue therapy in plasma-unresponsive aHUS [38–40] and in the peritransplant period, when it can actually negate the need for plasmatherapy and prevent or rescue posttransplant recurrence [41–44].
We observed that eculizumab works in maintaining remission in a plasmatherapy-dependent DEAP-HUS patient over the longer term (patient 1) and, more acutely, in a patient already showing a response to plasma exchange but who had allergic reactions to FFP. Moving forward, we envisage that the role of eculizumab in DEAP-HUS may be fourfold, namely: (i) in the acute phase, (ii) with relapses, (iii) as maintenance therapy, and (iv) in the peritransplant setting. Eculizumab can effectively block the terminal complement cascade and arrest the damage associated with the presence of anti-CFH autoantibodies, as it does in the other forms of genetic aHUS. In DEAP-HUS, reducing the antibody load with an immunosuppressive agent likely also plays an important role. However, until a full screen for all known mutations in complement regulatory proteins associated with aHUS other than the CFHR3/CFHR1 deletion is performed, a treatment concept of blocking complement via eculizumab rather than focusing on CFH autoantibodies alone is likely safest. Also, because relapses are frequent, occurring in up to 58 % of patients and predominantly within the first 6 months after first presentation [10], continuing eculizumab therapy at least for this time period seems rational. Measuring antibody titers has been suggested as having a possible role in monitoring and long-term management of these patients [10, 45]. Where immunosuppressive agents fail to reduce antibody titers or while awaiting depletion in anti-CFH autoantibodies, eculizumab could also be employed as maintenance therapy. A careful wean off eculizumab could be considered once antibody titers are depleted, with close monitoring for relapses using hematological (hemoglobin and platelets) and biochemical (LDH and haptoglobin) parameters of hemolysis and measures of complement activation (C3 and SC5b-9), especially during intercurrent illnesses.
Abandoning a treatment concept of definite complement control must be carefully considered, in light of the risk imparted by the presence of additional—known or as yet undiscovered—aHUS causing mutations that may coexist with the CFHR3/CFHR1 deletion. As these mutations could trigger aHUS via a “multiple-hit” phenomenon, continued eculizumab as a definitive treatment strategy could also be justified, and discontinuing therapy in these patients could expose them to a relapse with its inherent consequences.
Monitoring treatment outcome
Measuring CFH autoantibody titers is still experimental, there is no standardized assay as yet, and the level at which these antibodies turn pathogenic in an individual are not known [46]. Issues pertaining to antibody measurement, as well as utilization of antibody titers for diagnosis, prognosis, and management, has been a longstanding problem in other autoimmune diseases, such as in systemic lupus erythematosus (SLE) [47–49], ANCA-positive vasculitis [50], and antiglomerular basement membrane (anti-GBM) antibody-mediated disease [50]. On the other hand, antiphospholipase A2 receptor antibodies (anti-PLA2R) in membranous nephropathy are not only useful in establishing the diagnosis but also in monitoring treatment response [51, 52]. Whereas CFH autoantibody titers have been suggested in recent practice guidelines to aid risk stratification for kidney transplantation in aHUS patients [53], such guidelines are still lacking for the primary disease. Monitoring response to therapy by measuring ongoing complement activity, such as CH50, APH50, SC5b9, and the C3 split product C3d, as performed in our patients (see Tables 1 and 2), remains experimental and needs to be prospectively studied and validated. Finally, a recent study suggests that measuring the circulating autoantibody–CFH immune complexes may correlate better with disease activity in DEAP-HUS [24].
Conclusion
DEAP-HUS represents a unique subgroup of complement-mediated aHUS. As the role for eculizumab in the treatment of aHUS (and STEC-HUS) becomes more clearly defined, its use in managing DEAP-HUS needs to be considered. Given the high rate of early relapse, the possible coexistence of both known and unknown complement-gene mutations, the possible pathogenic role of CFHR1 as a CAP regulator, and the experimental nature of measuring and using CFH autoantibodies to guide management, we feel that a complement-directed therapy is crucial in DEAP-HUS. We propose that eculizumab be used acutely to arrest the complement-mediated damage and that it be continued for at least 6 months considering the high rate of recurrence in DEAP-HUS during this time frame—and certainly until a full complement abnormality screen has been completed. Immunosuppressive agents targeted at B cells and ongoing antibody production can also be added. However, results of using agents such as rituximab have not been as encouraging as might have been hoped. Although limitations to measuring CFH autoantibody titers exist, antibody testing may become a valuable tool for monitoring treatment response. Thus, it may be possible to wean patients off maintenance therapy with eculizumab once antibody levels are depleted and perhaps if other markers of complement activation, such as SC5b-9, remain normal. It must be appreciated that there is currently not enough evidence to define a final treatment algorithm for DEAP-HUS, and the optimal combination of immunosuppression, antibody depletion, and complement control needs to be prospectively studied in a larger cohort of DEAP-HUS patients [54].
References
Tarr PI, Gordon CA, Chandler WL (2005) Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365:1073–1086
Noris M, Remuzzi G (2009) Atypical hemolytic-uremic syndrome. N Engl J Med 361:1676–1687
Orth D, Khan AB, Naim A, Grif K, Brockmeyer J, Karch H, Joannidis M, Clark SJ, Day AJ, Fidanzi S, Stoiber H, Dierich MP, Zimmerhackl LB, Wurzner R (2009) Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol 182:6394–6400
Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, Pagani C, Noris M, Gobbi M, Stravalaci M, Rottoli D, Tedesco F, Remuzzi G, Zoja C (2011) Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol 187:172–180
Loirat C, Noris M, Fremeaux-Bacchi V (2008) Complement and the atypical hemolytic uremic syndrome in children. Pediatr Nephrol 23:1957–1972
Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, Pinto S, Goodship TH, Alberti M, Ribes D, Valoti E, Remuzzi G, Noris M (2013) Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol 24:475–486
Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, Weiss L, Fridman WH, Fremeaux-Bacchi V (2005) Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol 16:555–563
Jozsi M, Licht C, Strobel S, Zipfel SL, Richter H, Heinen S, Zipfel PF, Skerka C (2008) Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 111:1512–1514
Abarrategui-Garrido C, Martinez-Barricarte R, Lopez-Trascasa M, de Cordoba SR, Sanchez-Corral P (2009) Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood 114:4261–4271
Dragon-Durey MA, Sethi SK, Bagga A, Blanc C, Blouin J, Ranchin B, Andre JL, Takagi N, Cheong HI, Hari P, Le Quintrec M, Niaudet P, Loirat C, Fridman WH, Fremeaux-Bacchi V (2010) Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol 21:2180–2187
Le Quintrec M, Zuber J, Noel LH, Thervet E, Fremeaux-Bacchi V, Niaudet P, Fridman WH, Legendre C, Dragon-Durey MA (2009) Anti-Factor H autoantibodies in a fifth renal transplant recipient with atypical hemolytic and uremic syndrome. Am J Transplant 9:1223–1229
Elliott MA, Heit JA, Pruthi RK, Gastineau DA, Winters JL, Hook CC (2009) Rituximab for refractory and or relapsing thrombotic thrombocytopenic purpura related to immune-mediated severe ADAMTS13-deficiency: a report of four cases and a systematic review of the literature. Eur J Haematol 83:365–372
Harambat J, Lamireau D, Delmas Y, Ryman A, Llanas B, Brissaud O (2011) Successful treatment with rituximab for acute refractory thrombotic thrombocytopenic purpura related to acquired ADAMTS13 deficiency: a pediatric report and literature review. Pediatr Crit Care Med 12:e90–e93
Kwon T, Dragon-Durey MA, Macher MA, Baudouin V, Maisin A, Peuchmaur M, Fremeaux-Bacchi V, Loirat C (2008) Successful pre-transplant management of a patient with anti-factor H autoantibodies-associated haemolytic uraemic syndrome. Nephrol Dial Transplant 23:2088–2090
Boyer O, Balzamo E, Charbit M, Biebuyck-Gouge N, Salomon R, Dragon-Durey MA, Fremeaux-Bacchi V, Niaudet P (2010) Pulse cyclophosphamide therapy and clinical remission in atypical hemolytic uremic syndrome with anti-complement factor H autoantibodies. Am J Kidney Dis 55:923–927
Prufer F, Scheiring J, Sautter S, Jensen DB, Treichl R, Wurzner R, Zimmerhackl LB (2006) Terminal complement complex (C5b-9) in children with recurrent hemolytic uremic syndrome. Semin Thromb Hemost 32:121–127
Jozsi M, Strobel S, Dahse HM, Liu WS, Hoyer PF, Oppermann M, Skerka C, Zipfel PF (2007) Anti factor H autoantibodies block C-terminal recognition function of factor H in hemolytic uremic syndrome. Blood 110:1516–1518
Zipfel PF, Edey M, Heinen S, Jozsi M, Richter H, Misselwitz J, Hoppe B, Routledge D, Strain L, Hughes AE, Goodship JA, Licht C, Goodship TH, Skerka C (2007) Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet 3:e41
Dragon-Durey MA, Blanc C, Marliot F, Loirat C, Blouin J, Sautes-Fridman C, Fridman WH, Fremeaux-Bacchi V (2009) The high frequency of complement factor H related CFHR1 gene deletion is restricted to specific subgroups of patients with atypical haemolytic uraemic syndrome. J Med Genet 46:447–450
Heinen S, Hartmann A, Lauer N, Wiehl U, Dahse HM, Schirmer S, Gropp K, Enghardt T, Wallich R, Halbich S, Mihlan M, Schlotzer-Schrehardt U, Zipfel PF, Skerka C (2009) Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood 114:2439–2447
Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, Pandey MK, Kohl J, Zipfel PF, Weber BH, Skerka C (2010) An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum Mol Genet 19:4694–4704
Moore I, Strain L, Pappworth I, Kavanagh D, Barlow PN, Herbert AP, Schmidt CQ, Staniforth SJ, Holmes LV, Ward R, Morgan L, Goodship TH, Marchbank KJ (2010) Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood 115:379–387
Kopp A, Strobel S, Tortajada A, Rodriguez de Cordoba S, Sanchez-Corral P, Prohaszka Z, Lopez-Trascasa M, Jozsi M (2012) Atypical hemolytic uremic syndrome-associated variants and autoantibodies impair binding of factor h and factor h-related protein 1 to pentraxin 3. J Immunol 189:1858–1867
Blanc C, Roumenina LT, Ashraf Y, Hyvarinen S, Sethi SK, Ranchin B, Niaudet P, Loirat C, Gulati A, Bagga A, Fridman WH, Sautes-Fridman C, Jokiranta TS, Fremeaux-Bacchi V, Dragon-Durey MA (2012) Overall neutralization of complement factor H by autoantibodies in the acute phase of the autoimmune form of atypical hemolytic uremic syndrome. J Immunol 189:3528–3537
Ariceta G, Besbas N, Johnson S, Karpman D, Landau D, Licht C, Loirat C, Pecoraro C, Taylor CM, Van de Kar N, Vandewalle J, Zimmerhackl LB (2009) Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol 24:687–696
Taylor CM, Machin S, Wigmore SJ, Goodship TH (2010) Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 148:37–47
Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T (2010) Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 36:673–681
Madore F, Lazarus JM, Brady HR (1996) Therapeutic plasma exchange in renal diseases. J Am Soc Nephrol 7:367–386
Strobel S, Abarrategui-Garrido C, Fariza-Requejo E, Seeberger H, Sanchez-Corral P, Jozsi M (2011) Factor H-related protein 1 neutralizes anti-factor H autoantibodies in autoimmune hemolytic uremic syndrome. Kidney Int 80:397–404
Lee BH, Kwak SH, Shin JI, Lee SH, Choi HJ, Kang HG, Ha IS, Lee JS, Dragon-Durey MA, Choi Y, Cheong HI (2009) Atypical hemolytic uremic syndrome associated with complement factor H autoantibodies and CFHR1/CFHR3 deficiency. Pediatr Res 66:336–340
Strobel S, Hoyer PF, Mache CJ, Sulyok E, Liu WS, Richter H, Oppermann M, Zipfel PF, Jozsi M (2010) Functional analyses indicate a pathogenic role of factor H autoantibodies in atypical haemolytic uraemic syndrome. Nephrol Dial Transplant 25:136–144
Skerka C, Zipfel PF, Muller D, Micklisch S, Riedl M, Zimmerhackl LB, Hofer J (2010) The autoimmune disease DEAP-hemolytic uremic syndrome. Semin Thromb Hemost 36:625–632
de Jorge EG, Macor P, Paixao-Cavalcante D, Rose KL, Tedesco F, Cook HT, Botto M, Pickering MC (2011) The development of atypical hemolytic uremic syndrome depends on complement C5. J Am Soc Nephrol 22:137–145
Dubois EA, Cohen AF (2009) Eculizumab. Br J Clin Pharmacol 68:318–319
Kaplan M (2002) Eculizumab (Alexion). Curr Opin Investig Drugs 3:1017–1023
Hillmen P, Hall C, Marsh JC, Elebute M, Bombara MP, Petro BE, Cullen MJ, Richards SJ, Rollins SA, Mojcik CF, Rother RP (2004) Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med 350:552–559
Nurnberger J, Philipp T, Witzke O, Opazo Saez A, Vester U, Baba HA, Kribben A, Zimmerhackl LB, Janecke AR, Nagel M, Kirschfink M (2009) Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med 360:542–544
Gruppo RA, Rother RP (2009) Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med 360:544–546
Lapeyraque AL, Fremeaux-Bacchi V, Robitaille P (2011) Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 26:621–624
Kim JJ, Waller SC, Reid CJ (2012) Eculizumab in atypical haemolytic-uraemic syndrome allows cessation of plasma exchange and dialysis. Clin Kidney. doi:10.1093/ckj/sfr174
Krid S, Roumenina L, Beury D, Charbit M, Boyer O, Fremeaux-Bacchi V, Niaudet P (2012) Renal transplantation under prophylactic eculizumab in atypical hemolytic uremic syndrome with CFH/CFHR1 hybrid protein. Am J Transplant 12:1938–1944
Al-Akash SI, Almond PS, Savell VH Jr, Gharaybeh SI, Hogue C (2011) Eculizumab induces long-term remission in recurrent post-transplant HUS associated with C3 gene mutation. Pediatr Nephrol 26:613–619
Larrea CF, Cofan F, Oppenheimer F, Campistol JM, Escolar G, Lozano M (2010) Efficacy of eculizumab in the treatment of recurrent atypical hemolytic-uremic syndrome after renal transplantation. Transplantation 89:903–904
Davin JC, Gracchi V, Bouts A, Groothoff J, Strain L, Goodship T (2010) Maintenance of kidney function following treatment with eculizumab and discontinuation of plasma exchange after a third kidney transplant for atypical hemolytic uremic syndrome associated with a CFH mutation. Am J Kidney Dis 55:708–711
Dragon-Durey MA, Blanc C, Garnier A, Hofer J, Sethi SK, Zimmerhackl LB (2010) Anti-factor H autoantibody-associated hemolytic uremic syndrome: review of literature of the autoimmune form of HUS. Semin Thromb Hemost 36:633–640
Zipfel PF, Mache C, Muller D, Licht C, Wigger M, Skerka C (2010) DEAP-HUS: deficiency of CFHR plasma proteins and autoantibody-positive form of hemolytic uremic syndrome. Pediatr Nephrol 25:2009–2019
Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB (2003) Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med 349:1526–1533
Tomer Y, Buskila D, Shoenfeld Y (1993) Pathogenic significance and diagnostic value of lupus autoantibodies. Int Arch Allergy Immunol 100:293–306
Hanly JG, Su L, Farewell V, Fritzler MJ (2010) Comparison between multiplex assays for autoantibody detection in systemic lupus erythematosus. J Immunol Methods 358:75–80
Sinclair D, Stevens JM (2007) Role of antineutrophil cytoplasmic antibodies and glomerular basement membrane antibodies in the diagnosis and monitoring of systemic vasculitides. Ann Clin Biochem 44:432–442
Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ (2009) M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 361:11–21
Hofstra JM, Beck LH Jr, Beck DM, Wetzels JF, Salant DJ (2011) Anti-phospholipase A(2) receptor antibodies correlate with clinical status in idiopathic membranous nephropathy. Clin J Am Soc Nephrol 6:1286–1291
Zuber J, Fakhouri F, Roumenina LT, Loirat C, Fremeaux-Bacchi V (2012) Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol 8:643–657
Rees L (2013) Atypical HUS: time to take stock of current guidelines and outcome measures? Pediatr Nephrol 28:675–677
Acknowledgments
DN, AW, FGP, DFG, MK: none
PFZ received speaking honoraria form Alexion Pharmaceuticals, Inc.
CL has a financial relationship with Alexion Pharmaceuticals, Inc. via consultancy, paid speaking, and unrestricted research grants
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Noone, D., Waters, A., Pluthero, F.G. et al. Successful treatment of DEAP-HUS with eculizumab. Pediatr Nephrol 29, 841–851 (2014). https://doi.org/10.1007/s00467-013-2654-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-013-2654-x