
Abstract
Congenital anomalies of the kidneys or lower urinary tract (CAKUT) encompass a spectrum of anomalies that result from aberrations in spatio-temporal regulation of genetic, epigenetic, environmental, and molecular signals at key stages of urinary tract development. The Rearranged in Transfection (RET) tyrosine kinase signaling system is a major pathway required for normal development of the kidneys, ureters, peripheral and enteric nervous systems. In the kidneys, RET is activated by interaction with the ligand glial cell line-derived neurotrophic factor (GDNF) and coreceptor GFRα1. This activated complex regulates a number of downstream signaling cascades (PLCγ, MAPK, and PI3K) that control proliferation, migration, renewal, and apoptosis. Disruption of these events is thought to underlie diseases arising from aberrant RET signaling. RET mutations are found in 5–30 % of CAKUT patients and a number of Ret mouse mutants show a spectrum of kidney and lower urinary tract defects reminiscent of CAKUT in humans. The remarkable similarities between mouse and human kidney development and in defects due to RET mutations has led to using RET signaling as a paradigm for determining the fundamental principles in patterning of the upper and lower urinary tract and for understanding CAKUT pathogenesis. In this review, we provide an overview of studies in vivo that delineate expression and the functional importance of RET signaling complex during different stages of development of the upper and lower urinary tracts. We discuss how RET signaling balances activating and inhibitory signals emanating from its docking tyrosines and its interaction with upstream and downstream regulators to precisely modulate different aspects of Wolffian duct patterning and branching morphogenesis. We outline the diversity of cellular mechanisms regulated by RET, disruption of which causes malformations ranging from renal agenesis to multicystic dysplastic kidneys in the upper tract and vesicoureteral reflux or ureteropelvic junction obstruction in the lower tract.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mutations in Rearranged in Transfection (RET) are associated with several diseases including congenital anomalies of the kidneys or urinary tract (CAKUT), Hirschsprung's disease, and multiple endocrine neoplasia type 2 (MEN2) syndromes. Loss-of-function germline mutations in RET are associated with Hirschsprung's disease in 30–70 % of patients [1]. Germline gain-of-function RET mutations are associated with cancer syndromes: MEN2A, MEN2B, and familial medullary thyroid cancer [2]. Somatic RET mutations have been linked to pheochromocytoma and sporadic medullary thyroid cancer. RET gene rearrangements are a cause in a subset of papillary thyroid carcinomas and lung cancers [3]. It is reported that approximately 5 % of living patients with CAKUT harbor mutations in the RET pathway [4], and RET mutations are present in 7 % [5] of fetuses with CAKUT, and in 30 % of fetuses with unilateral or bilateral renal agenesis [6]. The pleiotropic roles of RET in several organ systems in humans has generated considerable interest in understanding the molecular mechanisms of RET function within and across different systems.
Almost two decades ago, RET was found to be required for kidney development. Mice with absent Ret signaling did not develop kidneys and therefore died at birth [7]. Since then the urinary tract of the mouse has been a model system to study RET’s physiological roles during development and disease. New genetic tools are providing key insights into (1) the basis for redundancy and specificity during urinary tract development, (2) determining interactions between RET and other genes important in kidney development, and (3) determining the roles of RET in renal homeostasis throughout embryonic development as well as postnatally. The insights from recent and ongoing studies will potentially lead to the discovery of biomarkers and therapeutic targets for congenital renal disease, acute kidney injury, and cancer treatment. Here, we will discuss the current knowledge of RET signaling in kidney development and CAKUT with a focus on insights derived from studies in vivo using mice with aberrant Ret signaling.
RET structure
RET was first discovered as a gene rearrangement in fibroblast cell culture [8]. The RET gene is located on chromosome 10q11.2 in humans. It consists of 20 exons and encodes a single-pass transmembrane protein that exists as two main isoforms with a molecular mass of 150 and 170 kDa, due to alternative splicing in the carboxy-terminus (Fig. 1a). The long isoform (RET51) harbors 1115 aa and the short isoform (RET9) harbors 1072aa [9]. RET is a member of the receptor tyrosine kinase (RTK) family [10]. The extracellular region consists of four cadherin-like domains and a cysteine-rich domain that mediates co-receptor and ligand binding. The cytoplasmic portion has two kinase domains with specific tyrosine residue phosphorylation sites which upon activation trigger downstream signaling [11].

RET structure and signaling system. a Illustration shows RET gene structure with 20 exons (solid boxes) and the putative two major RET isoforms: short RET 9 (1072 aa) or long RET 51 (1114 aa). The different isoforms are generated by alternative splicing and differ only in the number of aa residues in the cytosolic tail. RET 9 has nine amino acids at the carboxyl terminus that are different than the 51 terminal residues in RET 51. The extracellular domain consists of four cadherin motifs. The key tyrosine (Y) residues in the cytoplasmic domain are labeled. Note that the RET 51 isoform has an additional tyrosine residue Y1096. b Schematic shows RET signaling components using RET 51 as an example. GFRα1, the essential co-receptor of RET, is a glycophosphatidylinositol linked protein present on cell surface in the epithelia or in the mesenchyme. Two molecules of GDNF, ligand for RET signaling, bind to homodimers of Gfrα1 which then recruit two RET molecules in the cell membrane. Formation of this hexameric complex induces autophosphorylation of specific tyrosine residues in the intracellular kinase domain that serve as docking sites for the intracellular adaptors and activates the indicated downstream signaling cascades. Asterisks on SHC denote that other adaptors including SHC, IRS1-2, FRS2, DOK 1–6, and ENIGMA can also bind to the multi-docking Y1062 site. Activated SHC can either trigger the PI3K-AKT pathway or the ERK-MAPK pathway depending on the adaptor complex recruited. RET 51 has the additional residue Y1096 which can also activate the PI3K-AKT pathway. (aa amino acids; UB ureteric bud; PLCγ phospholipase Cγ; PKC protein kinase C)
RET signaling components
RET is activated by its interaction with one of the four members of the glial cell line-derived neurotrophic factor (GDNF) family ligands (GFLs) and one of the four GFL coreceptors (GFRα 1–4). The four GFLs are GDNF, Neurturin, Artemin, and Persephin. Each GFL preferentially binds one of the GFRα coreceptors to activate RET, although some cross-reactivity exists. Since GDNF and GFRα1 are the physiologically relevant partners of RET in the urinary tract we will focus on GDNF-GFRα1-RET signaling. GDNF is a secreted glycoprotein related to the transforming growth factor β superfamily and is primarily expressed in the mesenchymal compartment of the urinary system. In contrast, RET is expressed only in the urinary tract epithelium [12]. RET is the only known RTK that does not bind directly to its ligand. It requires a co-receptor for activation. Interestingly, the co-receptor, GFRα1, is expressed in both the mesenchyme and the urinary tract epithelium [13–16].
The current view is that the activated GDNF-GFRα1-RET complex is a hexameric structure consisting of homodimers of GDNF, GFRα1 and RET [17]. Formation of this multimeric complex results in phosphorylation of specific tyrosine residues in the cytoplasmic domain (Fig. 1b). The major tyrosine (Y) residues are Y981, Y1015 and Y1062 and they serve as interacting sites for intracellular adaptor proteins that activate downstream signaling cascades [9, 17]. The residue Y1015 is a docking site for phospholipase C-γ (PLCγ) and activates the protein kinase C (PKC) pathway; Y981 binds the adaptor SRC and activates MAPK cascade, and Y1062 can bind multiple docking proteins including SHC, IGF, FRS1 and FRS2 (please note in Fig. 1b only SHC is illustrated for simplicity). Activated SHC recruits either the adaptor complex GRB2-GAB to activate the PI3K-AKT pathway or the GRB2-SOS complex to activate the ERK-MAPK pathway. The RET51 isoform harbors an additional docking site, Y1096, for binding GRB2, which can also activate the PI3K cascade. Depending on tissue and cell type, these docking sites have redundant or unique biological roles (see later).
Overview of kidney development
In mammals, early kidney development occurs in an anterior-posterior (cephalad-caudal) direction. Three pairs of kidneys are initiated from the intermediate mesoderm (IM) in the following order: pronephros, mesonephros, and metanephros [18, 19]. The metanephros ultimately becomes the permanent kidney. A critical event in urinary tract development is the formation of the Wolffian duct (WD), a simple epithelial tube, from a collection of cells that undergo mesenchyme-to-epithelial transition in the IM. A subset of cells in the IM also gives rise to the mesenchymal compartment adjacent to the developing WD. These structures initially constitute the first embryonic kidney, the pronephros. The pronephros is a nonfunctional lateral extension from the WD that develops in humans at about 3 weeks of gestation (embryonic day E8.5 in mice). The pronephros degenerates by 4 weeks of gestation (E9 in mice) and the next kidney, the mesonephros begins to form. The mesonephros consists of linearly arranged mesonephric tubules connected to the WD. Although functional, the mesonephros is also a transient structure and regresses in a caudal to rostral direction except the most rostral mesonephric tubules will become part of the epididymis in males. The permanent kidney, the metanephros, appears at about 4.5 weeks of gestation in humans (E10.5 in mice) as an outgrowth, called the ureteric bud (UB), from the caudal aspect of the WD that by now has reached the cloaca (future bladder). The formation of this out-pouching from the WD is believed to occur because of inductive signals from the surrounding metanephric mesenchyme (MetM) and is termed induction of the UB. The UB invades the MetM, forms an initial branch to create a T-shaped structure with two tips and a stalk. Each tip subsequently undergoes branching morphogenesis. This is a reiterative process that is dependent on reciprocal interactions between the mesenchyme and the UB. The kidney grows in a centripetal pattern with the greatest cell proliferation at the outer most edge called the nephrogenic zone (NZ). There is a high concentration of RET-expressing cells within UB tips. Nephrogenesis and branching morphogenesis occur concurrently and influences each other’s development through a process called reciprocal interactions. Nephrogenesis is completed before birth in humans but it continues for about a week after birth in mice.
To glean insights into the function of RET signaling components, we and others have reported expression of Gdnf, Gfrα1, and Ret during different stages of kidney development in mice (Fig. 2). Ret expression is detected as early as E8.5d in a subset of cells in the anterior WD during the pronephros and mesonephros stages. Cells that express both Ret and Gfrα1 are the major cell types that populate the growing WD. Gfrα1 and Gdnf are expressed in the adjacent mesonephric mesenchyme (MesM) at E10–10.5 and in the MetM at E11.5 [13, 20, 21]. Ret-Gfrα1-expressing cells undergo cellular rearrangement in the WD and migrate towards the putative UB budding site at E10.5 in the mouse [22]. During UB induction, the greatest Ret-Gfrα1 expression is in the growing UB with weaker expression in the UB stalk. During branching morphogenesis Ret-Gfrα1 expression continues to be highest in the distal UB tips. The condensing mesenchyme harbors Gdnf and Gfrα1-expressing cells (albeit Gfrα1 expression is much weaker than that in the UB) [13]. Ret-Gfrα1 expression in the UB stalks, ureter, and the WD becomes weaker to undetectable except in the common nephric duct (CND, the distal most portion of the WD after UB induction) [20, 23]. Using Ret and Gfrα1-reporter mice, we have observed Ret in a subset of cortical and medullary collecting ducts and Gfrα1 in a subset of collecting ducts and proximal tubules postnatally (unpublished data).

RET, GDNF, and GFRα1 expression during murine kidney development. Schematic illustrates Ret, Gdnf, and Gfrα1 expression at the indicated time points during kidney development in the mouse. Ret and Gfrα1 proteins are expressed along the entire WD whereas Gdnf is expressed in the adjacent MesM at E9.5 (corresponds to approximately 3 weeks in humans). At E10.5, the site of UB induction shows high Gfrα1 and Ret expression compared to that in the anterior WD. Note that Gfrα1 is also expressed in the MesM and MesT. The Wolffian duct attaches to the cloaca by E10.5 and the MesM begins to degenerate. At E11.5, and during subsequent branching high expression of Ret and Gfrα1 is present in the UB tips. Gfrα1 and Gdnf are also expressed in the MetM and activate Ret signaling in the UB. The common nephric duct (CND) has high Ret and Gfrα1 expression and undergoes apoptosis to ensure proper ureter migration and insertion into the bladder. WD Wolffian duct; MesM mesonephric mesenchyme; UB ureteric bud; MesT mesonephric tubules; MetM metanephric mesenchyme
Physiological roles of Gdnf, Gfrα1 and Ret in the kidney
Biological roles of RET signaling components in the kidney and urinary tract have been revealed by three types of experimental model systems in vivo: (1) knockout mice with complete loss of protein from beginning of gestation, (2) tissue-specific transgenic mice or mice that express mutant forms of Ret knocked-in the Ret locus, and (3) mice that enable conditional or tissue-specific deletion of Ret. Work of several laboratories has shown that Ret, Gdnf, and Gfrα1-null mice die at birth due to bilateral renal aplasia or agenesis [7, 24–28]. Mice lacking GFLs other than Gdnf or lacking Gfrα2–4 have normal kidneys, indicating that Gdnf-Gfrα1-Ret is the relevant Ret signaling complex in the developing kidney [29–32]. Several studies show that mice with aberrant Ret expression cause a spectrum of renal anomalies, suggesting the importance of precise regulation of Ret during development. For example, when Ret was expressed throughout the collecting system (normally localized only to the UB tips) using the HoxB7 promoter, it resulted in multicystic dysplastic kidneys [33]. Mice expressing a dominant negative Ret mutation that severely diminishes ERK activity exhibited a variety of anomalies, including unilateral agenesis, bilateral hypoplasia, cysts, and bilateral agenesis [34]. More recently, knock-in mice expressing Ret isoforms or specific mutations in key docking tyrosines have provided evidence for much broader roles of Ret pathways in renal development and CAKUT [35–37]. Studies of mice expressing either wild-type Ret9 or Ret51 or Ret mutations with abrogation of adaptor sites for PLCγ or Src or Shc reveal novel roles of Ret signaling and identify mechanisms of specificity and redundancy in kidney development (Fig. 3) [35, 37, 38]. Here, we will summarize the spectrum of renal anomalies in Ret mutant mice and then discuss the pathogenetic mechanisms that lead to these defects.

Role of specific Ret tyrosine residues and downstream signaling cascades in kidney development. Schematic showing Ret9 intracellular structure with three major tyrosine phosphorylation docking sites and their downstream targets. Loss of Y1015 signaling in either isoform of Ret prevents recruitment of PLCγ causing severe renal dysplasia with multiplexed kidneys, UVJ obstruction, megaureter, VUR, and gonadal dysgenesis. Y1062 is a multiadaptor docking site (asterisk, only SHC shown, see text for other binding proteins). Loss of Y1062 in Ret9 abrogates MAPK and PI3K signaling and causes bilateral renal agenesis. The loss of signaling through the Y981 docking site affects recruitment of SRC resulting in reduced MAPK activity and causes an incompletely penetrant megaureter. Note that in the Ret51 isoform (not shown) the additional Y1096 site provides redundancy and normal kidney formation occurs when only Y981 or Y1062 are mutated. UVJ ureterovesical junction; VUR vesicoureteral reflux
In order to study the difference between signaling by the two Ret isoforms, human cDNAs harboring individual docking tyrosine mutations of each isoform were introduced in the Ret locus using knockout-knock-in approach [35]. Ret docking tyrosine mutations (Y981F, Y1015F, or Y1062F) in the context of the Ret9 isoform (also lacks the Grb2 Y1096 adaptor site) in general had more severe defects than docking tyrosine mutations in the Ret51 isoform, suggesting that the C-terminus Grb2-PI3K signaling provides redundancy in kidney development [35, 38]. Mice lacking only the Src adaptor site (Ret51Y981F) had normal kidneys but a subset of those lacking the Src and Grb2 site (Ret9Y981F) displayed unilateral or bilateral kidney and ureter defects (Fig. 3). Abrogating RetY1015 activity (Y1015) results in loss of PLCγ recruitment and bilateral megaureters, dysplastic multiplexed kidneys, and failure of the gonads to descend in both males and females revealing a novel role of Ret in genitourinary development and CAKUT (Fig. 3) [35, 39]. A similar phenotype has been reported in chimeric mice deficient for PLC-γ1 [40]. Abrogation of the multidocking adaptor site Y1062 (Ret51Y1062F) alone did not significantly affect kidney development. However, abrogating Y1062 and Grb2 site (Ret9Y1062F) results in bilateral agenesis or severe aplasia, a phenotype similar to that seen in Ret knockout mice (Fig. 3). Mice that express both mutant Ret9Y1062F and Ret51Y1062F have a milder phenotype: hypoplastic kidneys with reduced branching. This finding suggests that the extra Y1096 in Ret51 mice provides redundancy [35, 37, 38]. Mice with deletion of Frs2α from the ureteric epithelium show mild renal hypoplasia with decreased Ret expression, suggesting that the Frs2α adaptor may be one of the physiologically relevant adaptor proteins docking at RetY1062 [41]. Finally, a recent study also showed that Ret activity can be negatively regulated by protein kinase (PKA) via a mutation of the serine phosphorylation site Ser697. Abrogation of this PKA site results in renal hypoplasia [42]. These analyses provide evidence that some of the phenotypic diversity seen in CAKUT can occur due to differential regulation of signaling cascades regulated by the same gene RET. In contrast to above, relative to the different isoforms of RET, it should be mentioned that a separate study using a chimeric protein with human RET in the intracellular domain and mouse Ret in the extracellular domain reported that Ret51 did not support normal kidney development [36]. The reason for the different results, one study finding that either isoform alone, RET9 or RET51, is sufficient for normal kidney development [35] versus a second study that showed mutations in RET51 resulted in loss of normal kidney development [36] remains unknown, but likely due to the different properties of human RET51 and chimeric murine Ret51.
One of the challenges in the study of Ret has been to determine the role of Ret signaling after UB budding in vivo. Based on continued Gfrα1 and Ret expression during branching morphogenesis, RET signaling has been hypothesized to play a critical role during branching morphogenesis [43]. While most of the above models investigate the role of Gdnf-Gfrα1-Ret loss from the onset of emergence of renal primordia, little is known if these proteins have a role after UB induction in vivo. Experiments in vitro using kidney explant cultures exposed to Gdnf show a 1.5–2-fold increase in branching [44, 45]. Meanwhile, branching is inhibited when renal primordia are grown in the presence of Gdnf antibodies [46, 47]. To directly examine the role of the Ret signaling pathway after UB induction, we created Gfrα1 and Ret conditional alleles that allow Cre-mediated cell-type-specific or tamoxifen-inducible deletion of these genes after UB induction in mice [35, 48]. Interestingly, we found that the requirement of Gfrα1 depends on the stage of kidney formation. In the preUB induction stage, deletion of Gfrα1 from the WD directed by the HoxB7-Cre transgene resulted in renal agenesis or aplasia. This finding demonstrates that Gfrα1 in trans from the MetM is not sufficient to compensate for its loss in the WD and that Gfrα1 in the WD is required for kidney formation [13]. Surprisingly, Gfrα1 deletion in the UB after induction with a tamoxifen-inducible Cre-lox system in vivo and in explant cultures had only a modest effect on overall kidney size (a 20–25 % reduction) and branching morphogenesis. This suggests that post-UB induction branching may also depend on signals other than Gfrα1 in the UB epithelium. Since Gfrα1 was not completely deleted from the MetM using the tamoxifen-inducible model, it is also possible that Gfrα1 from the MetM partially compensates by activating Ret in trans during branching. While Gfrα1 expression in the MetM is dispensable for normal kidney development [14], it remains to be seen if conditional deletion of Gfrα1 from both the UB and MetM has a more severe effect on branching. Nevertheless, two important conclusions are reached from this study: (1) Gfrα1 is more critical in preUB urinary epithelium and less so post-UB induction, and (2) collecting duct progenitors do not require Gfrα1 to colonize the UB tips after UB induction. The studies with Ret and Gdnf conditional mice are in progress and will be highly informative in definitively establishing the physiological roles of the Ret signaling components at different stages of kidney development.
The role of Ret in the lower urinary tract
The lower urinary tract consists of the ureter, bladder, and urethra. The ureter arises from the WD while the bladder and urethra arise from the cloaca and urogenital sinus. Specific cellular events are required for proper connection of the ureter to the bladder. These events have been described in several reviews [49–52]. Briefly, these include growth of the WD toward the cloaca, proper location of the UB induction site on the distal WD and degeneration of the CND. This is accompanied by a series of ureter maturation events including apoptosis of the CND, migration and tunneling into the bladder trigone forming the ureterovesical junction, and separation of the WD from the ureter. In males, the WD becomes the vas deferens and connects to the seminal vesicle and prostate whereas in females, it becomes a rudiment. Disruption of these processes can lead to a spectrum of urinary tract anomalies such as hydronephrosis, ureterovesical junction (UVJ) obstruction, megaureter, ureterocele, and vesicoureteral reflux (VUR) [53].
Ret is critical for ureter formation and maturation because Ret-null mice harbor a number of ureter defects including no ureters, small ureters, and abnormally connected ureters to the gonadal ducts [23, 53]. Analysis of Ret-null mice show that Ret is required for proper insertion of the WD into the primitive bladder prior to UB formation [53]. The WDs of Ret-null mice either do not reach the cloaca or show a delayed fusion with the cloaca causing abnormal insertion into the bladder [53]. Mice that overexpress Ret also develop VUR [54], indicating that both loss and overexpression of Ret can cause lower tract anomalies. We recently analyzed mice with a mutation in the Ret-PLCγ docking site (Y1015) and found that the CNDs do not undergo timely degeneration and fail to separate from the WD, resulting in obstructive nephropathy [20]. The ureter defect in RetY1015F mice is different than that in Ret-null mice as it occurs during the ureter maturation stage after the WD has reached cloaca. It remains to be determined if the Ret-null phenotype is due to collective abrogation of all Ret-activated pathways or if there is a specific signal that ensures that WD reaches the cloaca.
Ret and other genes in the urinary system
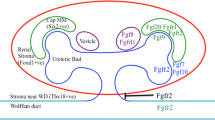
Several genes known to be important during kidney development are upstream regulators of Gdnf-Ret signaling or are regulated by this pathway downstream (Fig. 4). Here we report a few examples of genes that affect, or are affected by, the Ret signaling complex.

Gene and pathways associated with the RET signaling complex. The schematic depicts the major direct and indirect regulators of RET-GFRα1- GDNF signaling (upstream) and those that are affected by RET-GFRα1- GDNF signaling (downstream). In the upstream panel, red indicates inhibitors and green indicates stimulators or activators of RET signaling. In the downstream panel, proteins in red inhibit GDNF-RET activity and those in green are activated or are positively regulated by GDNF-RET either at transcription or posttranslational level
Upstream regulators of Gdnf-Ret signaling in vivo
Characterization of a number of mice with mutations of key kidney development genes show similar phenotypes to those seen in mice with Gdnf-Gfrα1-Ret mutations. Functional analysis of these genetic mutations often reveals perturbations in cellular pathways that are upstream or downstream of the Ret signaling pathway. For instance, conditional deletion of Rar results in hypoplastic kidneys or agenesis due to transcriptional downregulation of Ret [55]. Rar and Gata3 signaling modulate Ret expression and thereby regulate the joining of the WD to cloaca [53]. Bmp4-null mice show increased Gdnf expression and extra UBs indicating that Bmp4 is a negative regulator of Gdnf expression and Ret signaling [56]. Pax2-null mice show severe reduction in mesonephros development and reduction in Gdnf expression [57]. Slit2 and Robo2-null mice show expansion of the Gdnf-expression domain, suggesting that these proteins negatively regulate Gdnf expression and inhibit Ret [58].
Downstream proteins interacting with Gdnf-Ret signaling in vivo
Activity of many genes is modulated by Ret signaling. Ret regulates the expression of Wnt11, Etv4, and Etv5 [44, 59]. Further, Wnt11-null and Etv4-Etv5 double-null mice show renal hypoplasia or agenesis, phenotypes similar to those seen in a subset of Ret-mutant mice. Mice that are null for Shp2, a tyrosine phosphatase, also have hypoplastic kidneys [45]. These mice exhibit down-regulation of Wnt11, Etv4, and Etv5, indicating that Shp2 is a downstream modulator of RET signaling [45]. A phenotype similar to Y1015F ureter defects was described in mice deficient for leukocyte antigen-related (Lar) family protein tyrosine phosphatase, receptor type S and F (Ptprs and Ptprf). Increased activation of Ret-docking tyrosines in these mice suggests an interaction between Lar and Ret signaling pathways [60].
Gdnf-Ret and Spry1 interactions
Sprouty1 protein (Spry1) inhibits RTK signaling by suppressing RAS-MAPK activity (Figs. 3 and 4) [61]. Therefore, a Spry1-null mutation results in hyperactivation of the RAS-MAPK cascade [62]. Spry1-null mice show multiplexed dysplastic kidneys with megaureters and supernumerary UBs [62]. This is in contrast to the renal agenesis phenotype in the Ret-null or Ret-Y1062F mutant mice where UB induction is inhibited due to severe reduction in RAS-MAPK signaling. These observations suggest Spry1 is a negative regulator of UB development. Spry1-nulls and Ret-nulls represent extreme forms of CAKUT that emerge from dysregulation of MAPK signaling. Several laboratories performed experiments to test genetic interactions between these proteins in vivo. Double homozygous mutant mice that lack Ret-PI3K-MAPK signaling and Spry1 (Spry1 –/− : Ret Y1062F/Y1062F) surprisingly showed morphological correction of the disparate renal anomalies seen in these individual null mice [63]. This remarkable correction of two different phenotypes has potential implications in modulating these pathways in CAKUT therapy. The mechanism for normalization of the renal defects was postulated to be through an increase in RAS-MAPK signaling via a pathway other than Ret. A likely candidate for this was FGFR signaling as in the absence of Gdnf/Ret signaling FGF7 can induce UBs [64]. Further, support for alternative tyrosine kinase activity comes from compound homozygous mice lacking Gdnf or Ret, and Spry1. Mice lacking Gdnf and Spry1 (Spry1 −/− : Gdnf −/−) or Ret and Spry1 (Spry1 −/− : Ret −/−) surprisingly did not show the renal agenesis or complex CAKUT seen in Ret-null or Spry1-null mice, respectively. These double mutant mice develop mildly hypoplastic kidneys (∼70 % of size of controls) almost 90 % of the time. This suggests that if basal MAPK suppression is removed (no Spry1), UB budding and kidneys can be induced without Ret. Removing Fgf10 from Ret-Spry1 double nulls results in no UBs, suggesting that Fgf10 presumably through activation of the receptor Fgfr2 was the alternative RTK signal for UB induction [65]. The ampullae of the Ret-Spry1 or Gdnf-Spry1 double mutant mice are dilated, suggesting that Ret may be still required to regulate some aspects of branching. Because the kidneys are essentially of normal size in Spry1 −/− : Ret Y1062F/Y1062F mice compared to double-null Spry1 −/− : Gdnf −/−or Spry1 −/− : Ret −/− mice, it is possible that Ret signals other than those from Y1062 may also have a role in UB branching.
Developmental and molecular mechanisms of kidney and urinary tract regulation by Ret
The phenotypes observed in different Gdnf, Gfrα1, and Ret mutants represent an allelic series that are reminiscent of CAKUT in humans. Early developmental analysis of Ret-tyrosine docking site mutants and null mice reveal that Ret signaling regulates WD morphogenesis and kidney development at four stages: (1) PreUB induction, (2) UB induction, (3) branching morphogenesis, and (4) ureter maturation (Table 1). Further, Ret has dual roles in the urinary tract. Normal Ret signaling maintains a balance between inhibitory signals mediated through Ret-Y1015 and activating signals through RetY1062. In the anterior WD, RetY1015 signaling suppresses ectopic UBs in the mesonephros by ensuring timely degradation of the surrounding mesonephric mesenchyme by apoptosis before primary UB budding and branching morphogenesis takes place [20]. In RetY1015F mutant mice, the MesM degeneration is delayed, leading to sustained exposure of the anterior WD to Gdnf that promotes supernumerary buds. These buds merge with the growing metanephric kidney and result in multiplexed, dysplastic kidneys. During UB induction, we also found that Ret, through Y1015, keeps a distinct boundary between the MetM and the WD. Disruption of Y1015 signals results in MetM abutting the WD in an extended UB budding domain, which provides a further stimulus for multiple UBs. In Ret-null mice, the UB either fails to initiate or grows into the MetM. This defect results in agenesis, hypoplasia, or aplasia. Thus, proper UB induction is under precise regulation by activating and inhibitory signals. Ret-null mice also show failure of the WD to make contact with the cloaca by E10.5 due to slow or arrested distal WD growth. In contrast, in RetY1015F mutant mice, the WD reaches the cloaca but does not undergo timely CND apoptosis between E12.5 and E14.5. This results in ureters that are ectopically inserted or that have a blind ending and fail to separate from the WD, leading to megaureters [20]. The mechanism of this is sustained ERK activity, which promotes proliferation and inhibits apoptosis. This suggests that Ret has a dual role in distal WD growth and maintains a balance between activating and inhibiting signals to prevent ureter abnormalities. In elegant experiments where the WD consists of chimeric Ret-null and wild-type cells, it was recently shown that Ret signaling is critical in controlling cell movements in the primary UB budding site [22]. Ret-deficient cells have delayed migration towards the primary UB budding and this finding explains the UB induction defects that result in hypoplasia or agenesis. The mechanism of hypoplasia when Gfrα1 is conditionally deleted from the UB epithelium after UB induction (E11.5–12.5) is decreased branching due to a reduction in UB tip cell proliferation and ERK activity [13]. These studies also show that the adjacent wild-type UB tip cells have a remarkable capability to compensate for Gfrα1 loss.
GDNF, GFRα1, and RET mutations in patients with CAKUT
We recently reported a study of 122 living patients, encompassing various CAKUT, and found GDNF or RET variations in about 5 % of the patients [4]. These patients are living, therefore these mutations were not deleterious enough to cause lethality, but occur in a context that affects RET signaling in combination with other interacting genes. For example, we detected rare and novel variants in GDNF and RET that coexist with a common polymorphism. Biochemical characterization showed that the presence of a common SNP RETG691S and a mutation RET982 in the same allele together caused a severe reduction in MAPK signaling. Targeted capture and whole-exome sequencing identified a novel GFRα1 mutation, in addition to the RET-G691S and RET982, in a child with complex renal anomalies including megaureters, dysplasia, and cryptorchidism. This suggests that the net burden of common and rare/novel deleterious variants in the same gene or its interacting partners determines the output from key signaling cascades and ultimately determines if anomalous renal development will occur.
Conclusions
About 5–30 % of CAKUT patients (from analysis of about 250 patients from three studies) harbor GDNF, GFRα1, or RET mutations, making this pathway highly relevant to renal malformations. The analyses of Gdnf, Gfrα1, and Ret allelic series of mice reveal the importance of this pathway in regulating various aspects of early kidney and urinary tract development. CAKUT are genetically heterogeneous disorders—many genes interacting together are responsible for these defects. This means that co-expression of mutant forms of genes in the same pathway can exacerbate CAKUT or result in apparently normal kidneys. Several of these studies show that kidney and urinary tract development is sensitive to precise timing and net output of MAPK activity. This can be regulated by altering the dosage of the Ret signaling complex, or activity of key docking tyrosine of Ret, or by interactions with negative regulators such as Spry1. While significant advances have been made in RET biology, many exciting challenges and new questions have emerged.
The RET pathway is a key regulator of kidney size and nephron endowment [66], and decreased nephron number at birth is related to hypertension and CKD [67]. An unanswered question is if variations in kidney development genes such as RET, that are not lethal, are risk factors or predictive of CKD progression, essential hypertension, or ESRD. It is also time to explore the contribution of structures outside the urinary tract that may be regulated by RET and affect the kidney. For example, RET shows strong expression in the autonomic and sensory nervous systems, but we do not know how RET activity in these systems influences kidney and urinary tract pathophysiology. Recently, GFLs were reported to be highly expressed in immunosuppressed transplant patients, suggesting a possible role in the immune system [68]. Genes important in development may also play a role in repair and regeneration after injury. Whether mutations in the RET pathway render one more susceptible to chemical, hypoxic, toxic, or obstructive insults in the kidney needs to be investigated. There are exciting times ahead in RET translational and basic research that will continue to advance our understanding of congenital, acute, and chronic kidney diseases. With interdisciplinary efforts and collaborations between basic scientists, geneticists, and clinicians and commitment of funding agencies, the field of RET signaling will reach new heights in the coming years.
References
Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, Brooks AS, Antinolo G, de Pontual L, Clement-Ziza M, Munnich A, Kashuk C, West K, Wong KK, Lyonnet S, Chakravarti A, Tam PK, Ceccherini I, Hofstra RM, Fernandez R, Hirschsprung Disease Consortium (2008) Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet 45:1–14
Margraf RL, Crockett DK, Krautscheid PM, Seamons R, Calderon FR, Wittwer CT, Mao R (2009) Multiple endocrine neoplasia type 2 RET protooncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum Mutat 30:548–556
Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, Iwakawa R, Ogiwara H, Oike T, Enari M, Schetter AJ, Okayama H, Haugen A, Skaug V, Chiku S, Yamanaka I, Arai Y, Watanabe S, Sekine I, Ogawa S, Harris CC, Tsuda H, Yoshida T, Yokota J, Shibata T (2012) KIF5B-RET fusions in lung adenocarcinoma. Nat Med 18:375–377
Chatterjee R, Ramos E, Hoffman M, VanWinkle J, Martin DR, Davis TK, Hoshi M, Hmiel SP, Beck A, Hruska K, Coplen D, Liapis H, Mitra R, Druley T, Austin P, Jain S (2012) Traditional and targeted exome sequencing reveals common, rare and novel functional deleterious variants in RET-signaling complex in a cohort of living US patients with urinary tract malformations. Hum Genet 131:1725–1738
Jeanpierre C, Mace G, Parisot M, Moriniere V, Pawtowsky A, Benabou M, Martinovic J, Amiel J, Attie-Bitach T, Delezoide AL, Loget P, Blanchet P, Gaillard D, Gonzales M, Carpentier W, Nitschke P, Tores F, Heidet L, Antignac C, Salomon R, Societe Francaise de F (2011) RET and GDNF mutations are rare in fetuses with renal agenesis or other severe kidney development defects. J Med Genet 48:497–504
Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ (2008) Renal aplasia in humans is associated with RET mutations. Am J Hum Genet 82:344–351
Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V (1994) Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367:380–383
Takahashi M, Ritz J, Cooper GM (1985) Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 42:581–588
Jain S (2009) The many faces of RET dysfunction in kidney. Organogenesis 5:177–190
Song R, El-Dahr SS, Yosypiv IV (2011) Receptor tyrosine kinases in kidney development. J Signal Transduct 2011:869281
Parkash V, Goldman A (2009) Comparison of GFL-GFRalpha complexes: further evidence relating GFL bend angle to RET signalling. Acta Crystallogr Sect F Struct Biol Cryst Commun 65:551–558
Sariola H, Saarma M (2003) Novel functions and signalling pathways for GDNF. J Cell Sci 116:3855–3862
Keefe Davis T, Hoshi M, Jain S (2013) Stage specific requirement of Gfrα1 in the ureteric epithelium during kidney development. Mech Dev 130:506–518
Enomoto H, Hughes I, Golden J, Baloh RH, Yonemura S, Heuckeroth RO, Johnson EM Jr, Milbrandt J (2004) GFRalpha1 expression in cells lacking RET is dispensable for organogenesis and nerve regeneration. Neuron 44:623–636
Golden JP, DeMaro JA, Osborne PA, Milbrandt J, Johnson EM Jr (1999) Expression of neurturin, GDNF, and GDNF family-receptor mRNA in the developing and mature mouse. Exp Neurol 158:504–528
Towers PR, Woolf AS, Hardman P (1998) Glial cell line-derived neurotrophic factor stimulates ureteric bud outgrowth and enhances survival of ureteric bud cells in vitro. Exp Nephrol 6:337–351
Ibanez CF (2013) Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a009134
Dressler GR (2009) Advances in early kidney specification, development and patterning. Development 136:3863–3874
Costantini F, Kopan R (2010) Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev Cell 18:698–712
Hoshi M, Batourina E, Mendelsohn C, Jain S (2012) Novel mechanisms of early upper and lower urinary tract patterning regulated by RetY1015 docking tyrosine in mice. Development 139:2405–2415
Kobayashi H, Kawakami K, Asashima M, Nishinakamura R (2007) Six1 and Six4 are essential for Gdnf expression in the metanephric mesenchyme and ureteric bud formation, while Six1 deficiency alone causes mesonephric-tubule defects. Mech Dev 124:290–303
Chi X, Michos O, Shakya R, Riccio P, Enomoto H, Licht JD, Asai N, Takahashi M, Ohgami N, Kato M, Mendelsohn C, Costantini F (2009) Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev Cell 17:199–209
Batourina E, Choi C, Paragas N, Bello N, Hensle T, Costantini FD, Schuchardt A, Bacallao RL, Mendelsohn CL (2002) Distal ureter morphogenesis depends on epithelial cell remodeling mediated by vitamin A and Ret. Nat Genet 32:109–115
Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H (1996) Defects in enteric innervation and kidney development in mice lacking GDNF. Nature 382:73–76
Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EM Jr, Milbrandt J (1998) GFR alpha1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron 21:317–324
Cacalano G, Farinas I, Wang LC, Hagler K, Forgie A, Moore M, Armanini M, Phillips H, Ryan AM, Reichardt LF, Hynes M, Davies A, Rosenthal A (1998) GFRalpha1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron 21:53–62
Gould TW, Yonemura S, Oppenheim RW, Ohmori S, Enomoto H (2008) The neurotrophic effects of glial cell line-derived neurotrophic factor on spinal motoneurons are restricted to fusimotor subtypes. J Neurosci 28:2131–2146
Jain S, Golden JP, Wozniak D, Pehek E, Johnson EM Jr, Milbrandt J (2006) RET is dispensable for maintenance of midbrain dopaminergic neurons in adult mice. J Neurosci 26:11230–11238
Airaksinen MS, Saarma M (2002) The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3:383–394
Airaksinen MS, Titievsky A, Saarma M (1999) GDNF family neurotrophic factor signaling: four masters, one servant? Mol Cell Neurosci 13:313–325
Nishino J, Mochida K, Ohfuji Y, Shimazaki T, Meno C, Ohishi S, Matsuda Y, Fujii H, Saijoh Y, Hamada H (1999) GFR alpha3, a component of the artemin receptor, is required for migration and survival of the superior cervical ganglion. Neuron 23:725–736
Rossi J, Luukko K, Poteryaev D, Laurikainen A, Sun YF, Laakso T, Eerikainen S, Tuominen R, Lakso M, Rauvala H, Arumae U, Pasternack M, Saarma M, Airaksinen MS (1999) Retarded growth and deficits in the enteric and parasympathetic nervous system in mice lacking GFR alpha2, a functional neurturin receptor. Neuron 22:243–252
Srinivas S, Wu Z, Chen CM, D’Agati V, Costantini F (1999) Dominant effects of RET receptor misexpression and ligand-independent RET signaling on ureteric bud development. Development 126:1375–1386
Jain S, Naughton CK, Yang M, Strickland A, Vij K, Encinas M, Golden J, Gupta A, Heuckeroth R, Johnson EM Jr, Milbrandt J (2004) Mice expressing a dominant-negative Ret mutation phenocopy human Hirschsprung disease and delineate a direct role of Ret in spermatogenesis. Development 131:5503–5513
Jain S, Encinas M, Johnson EM Jr, Milbrandt J (2006) Critical and distinct roles for key RET tyrosine docking sites in renal development. Genes Dev 20:321–333
de Graaff E, Srinivas S, Kilkenny C, D’Agati V, Mankoo BS, Costantini F, Pachnis V (2001) Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev 15:2433–2444
Jijiwa M, Fukuda T, Kawai K, Nakamura A, Kurokawa K, Murakumo Y, Ichihara M, Takahashi M (2004) A targeting mutation of tyrosine 1062 in Ret causes a marked decrease of enteric neurons and renal hypoplasia. Mol Cell Biol 24:8026–8036
Wong A, Bogni S, Kotka P, de Graaff E, D’Agati V, Costantini F, Pachnis V (2005) Phosphotyrosine 1062 is critical for the in vivo activity of the Ret9 receptor tyrosine kinase isoform. Mol Cell Biol 25:9661–9673
Jain S, Knoten A, Hoshi M, Wang H, Vohra B, Heuckeroth RO, Milbrandt J (2010) Organotypic specificity of key RET adaptor-docking sites in the pathogenesis of neurocristopathies and renal malformations in mice. J Clin Invest 120:778–790
Shirane M, Sawa H, Kobayashi Y, Nakano T, Kitajima K, Shinkai Y, Nagashima K, Negishi I (2001) Deficiency of phospholipase C-gamma1 impairs renal development and hematopoiesis. Development 128:5173–5180
Sims-Lucas S, Cullen-McEwen L, Eswarakumar VP, Hains D, Kish K, Becknell B, Zhang J, Bertram JF, Wang F, Bates CM (2009) Deletion of Frs2alpha from the ureteric epithelium causes renal hypoplasia. Am J Physiol Renal Physiol 297:F1208–F1219
Asai N, Fukuda T, Wu Z, Enomoto A, Pachnis V, Takahashi M, Costantini F (2006) Targeted mutation of serine 697 in the Ret tyrosine kinase causes migration defect of enteric neural crest cells. Development 133:4507–4516
Shakya R, Watanabe T, Costantini F (2005) The role of GDNF/Ret signaling in ureteric bud cell fate and branching morphogenesis. Dev Cell 8:65–74
Pepicelli CV, Kispert A, Rowitch DH, McMahon AP (1997) GDNF induces branching and increased cell proliferation in the ureter of the mouse. Dev Biol 192:193–198
Willecke R, Heuberger J, Grossmann K, Michos O, Schmidt-Ott K, Walentin K, Costantini F, Birchmeier W (2011) The tyrosine phosphatase Shp2 acts downstream of GDNF/Ret in branching morphogenesis of the developing mouse kidney. Dev Biol 360:310–317
Qiao J, Sakurai H, Nigam SK (1999) Branching morphogenesis independent of mesenchymal–epithelial contact in the developing kidney. Proc Natl Acad Sci USA 96:7330–7335
Vega QC, Worby CA, Lechner MS, Dixon JE, Dressler GR (1996) Glial cell line-derived neurotrophic factor activates the receptor tyrosine kinase RET and promotes kidney morphogenesis. Proc Natl Acad Sci USA 93:10657–10661
Uesaka T, Jain S, Yonemura S, Uchiyama Y, Milbrandt J, Enomoto H (2007) Conditional ablation of GFRalpha1 in postmigratory enteric neurons triggers unconventional neuronal death in the colon and causes a Hirschsprung’s disease phenotype. Development 134:2171–2181
Woolf AS, Davies JA (2013) Cell biology of ureter development. J Am Soc Nephrol 24:19–25
Mendelsohn C (2009) Using mouse models to understand normal and abnormal urogenital tract development. Organogenesis 5:306–314
Stewart K, Bouchard M (2011) Kidney and urinary tract development: an apoptotic balancing act. Pediatr Nephrol 26:1419–1425
Stahl DA, Koul HK, Chacko JK, Mingin GC (2006) Congenital anomalies of the kidney and urinary tract (CAKUT): a current review of cell signaling processes in ureteral development. J Pediatr Urol 2:2–9
Chia I, Grote D, Marcotte M, Batourina E, Mendelsohn C, Bouchard M (2011) Nephric duct insertion is a crucial step in urinary tract maturation that is regulated by a Gata3-Raldh2-Ret molecular network in mice. Development 138:2089–2097
Yu OH, Murawski IJ, Myburgh DB, Gupta IR (2004) Overexpression of RET leads to vesicoureteric reflux in mice. Am J Physiol Renal Physiol 287:F1123–F1130
Rosselot C, Spraggon L, Chia I, Batourina E, Riccio P, Lu B, Niederreither K, Dolle P, Duester G, Chambon P, Costantini F, Gilbert T, Molotkov A, Mendelsohn C (2010) Non-cell-autonomous retinoid signaling is crucial for renal development. Development 137:283–292
Raatikainen-Ahokas A, Hytonen M, Tenhunen A, Sainio K, Sariola H (2000) BMP-4 affects the differentiation of metanephric mesenchyme and reveals an early anterior-posterior axis of the embryonic kidney. Dev Dyn 217:146–158
Brophy PD, Ostrom L, Lang KM, Dressler GR (2001) Regulation of ureteric bud outgrowth by Pax2-dependent activation of the glial derived neurotrophic factor gene. Development 128:4747–4756
Grieshammer U, Le M, Plump AS, Wang F, Tessier-Lavigne M, Martin GR (2004) SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev Cell 6:709–717
Lu BC, Cebrian C, Chi X, Kuure S, Kuo R, Bates CM, Arber S, Hassell J, MacNeil L, Hoshi M, Jain S, Asai N, Takahashi M, Schmidt-Ott KM, Barasch J, D’Agati V, Costantini F (2009) Etv4 and Etv5 are required downstream of GDNF and Ret for kidney branching morphogenesis. Nat Genet 41:1295–1302
Uetani N, Bertozzi K, Chagnon MJ, Hendriks W, Tremblay ML, Bouchard M (2009) Maturation of ureter-bladder connection in mice is controlled by LAR family receptor protein tyrosine phosphatases. J Clin Invest 119:924–935
Hanafusa H, Torii S, Yasunaga T, Nishida E (2002) Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol 4:850–858
Basson MA, Akbulut S, Watson-Johnson J, Simon R, Carroll TJ, Shakya R, Gross I, Martin GR, Lufkin T, McMahon AP, Wilson PD, Costantini FD, Mason IJ, Licht JD (2005) Sprouty1 is a critical regulator of GDNF/RET-mediated kidney induction. Dev Cell 8:229–239
Rozen EJ, Schmidt H, Dolcet X, Basson MA, Jain S, Encinas M (2009) Loss of Sprouty1 rescues renal agenesis caused by Ret mutation. J Am Soc Nephrol 20:255–259
Maeshima A, Sakurai H, Choi Y, Kitamura S, Vaughn DA, Tee JB, Nigam SK (2007) Glial cell-derived neurotrophic factor independent ureteric bud outgrowth from the Wolffian duct. J Am Soc Nephrol 18:3147–3155
Michos O, Cebrian C, Hyink D, Grieshammer U, Williams L, D’Agati V, Licht JD, Martin GR, Costantini F (2010) Kidney development in the absence of Gdnf and Spry1 requires Fgf10. PLoS Genet 6:e1000809
Zhang Z, Quinlan J, Hoy W, Hughson MD, Lemire M, Hudson T, Hueber PA, Benjamin A, Roy A, Pascuet E, Goodyer M, Raju C, Houghton F, Bertram J, Goodyer P (2008) A common RET variant is associated with reduced newborn kidney size and function. J Am Soc Nephrol 19:2027–2034
Vikse BE, Irgens LM, Leivestad T, Hallan S, Iversen BM (2008) Low birth weight increases risk for end-stage renal disease. J Am Soc Nephrol 19:151–157
Sigdel TK, Li L, Tran TQ, Khatri P, Naesens M, Sansanwal P, Dai H, Hsieh SC, Sarwal MM (2012) Non-HLA antibodies to immunogenic epitopes predict the evolution of chronic renal allograft injury. J Am Soc Nephrol 23:750–763
Acknowledgments
We thank Dr. Feng Chen for many useful comments and discussions in preparation of this manuscript. We apologize to all colleagues if we overlooked to cite their work. Work reported in this review was partly supported by the National Institutes of Health grants DK081644 and DK082531 (S.J.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Davis, T.K., Hoshi, M. & Jain, S. To bud or not to bud: the RET perspective in CAKUT. Pediatr Nephrol 29, 597–608 (2014). https://doi.org/10.1007/s00467-013-2606-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-013-2606-5