
Abstract
Preeclampsia (PE) development is often associated with placental immune and inflammatory dysregulation, as well as endoplasmic reticulum (ER) stress. However, the mechanisms linking ER stress and inflammatory dysregulation to PE have not been elucidated. It has been reported that thioredoxin-interacting protein (TXNIP), which can bind with and activate the NLR family pyrin domain containing 3 (NLRP3) inflammasome, is a key point in immune regulation. Recent experimental evidence suggests that activated NLRP3 inflammasomes can activate interleukin-1β (IL-1β) production in the placenta of patients with PE. The objective of the current study was to explore if TXNIP plays a critical signaling role linking ER stress with NLRP3 inflammasome activation in PE. We hypothesized that ER stress would induce TXNIP production, which would bind with NLRP3 inflammasomes to activate IL-1β production. These cells showed a higher protein level of NLRP3 and IL-1β, as well as a higher enzymatic activity of caspase-1, indicating enhanced inflammatory dysregulation and ER stress. Cells transfected with TXNIP siRNA showed reduced NLRP3 inflammasome activation. Cells treated with 4-phenylbutyric acid, an inhibitor of ER stress, showed a similar result. Outgrowth of the explant with TXNIP lentivirus in H/R or tunicamycin (inducers of ER stress) was also measured to verify our hypothesis. These findings demonstrated that TXNIP could influence inflammatory dysregulation by mediating ER stress and NLRP3 inflammasome activation in PE. This novel mechanism may further explain the inflammation observed at the maternal-fetal interface, which leads to placental dysfunction in a patient with PE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Preeclampsia (PE) is a pregnancy-specific disorder which is one of the important causes of morbidity and mortality in mother and baby (Steegers et al. 2010). Pregnancy is a state of controlled mild maternal systemic inflammation: imbalances in this inflammation at the maternal-fetal interface (placenta) may be actively involved in the development and/or progression of pregnancy disorders, such as PE (Christopher W.G. Redman 1999). Cytokines and chemokines are known to be the most important inflammatory mediators contributing to maternal inflammation. In PE, trophoblast cells express inflammatory cytokines including interleukins (ILs), such as IL-1β (Amash et al. 2012; Siljee et al. 2013), which have been shown to mediate maternal endothelial dysfunction and regulate cytotrophoblast invasion in vitro (Librach et al. 1994; Rusterholz et al. 2007). It has been reported that NLR family pyrin domain containing 3 (NLRP3) inflammasomes can elevate the production of IL-1β by via caspase-1 (Ting et al. 2008; Schroder and Tschopp 2010), and thus, an increase in NLRP3 will lead to the increase of IL-1β, an important inflammatory marker of PE.
NLRP3 is also known to be involved in the downstream cascade of endoplasmic reticulum (ER) stress (Lerner et al. 2012; Guo et al. 2018). ER stress is a rescue response when the cellular secretory pathway is overwhelmed by large secretory loads and results in the accumulation of unfolded/misfolded proteins within the ER lumen (Wang et al. 1999). This cellular stress aims to restore normal cellular function by slowing protein translation, breaking down misfolded proteins, and increasing the production of molecular chaperones (Oyadomari et al. 2002; Merksamer and Papa 2010). Under ER stress, thioredoxin-interacting protein (TXNIP), a binding protein of thioredoxin that modulates its antioxidant functions, can bind and activate NLRP3 inflammasomes to slice procaspase-1 to its active form, thus triggering maturation and secretion of the IL-1β, which is a kind of inflammatory cytokine (Ting et al. 2008; Lerner et al. 2012; Guo et al. 2018). If ER stress persists, abnormal cellular inflammation and apoptosis will occur (Burton et al. 2009).
Although it has been reported that the role of ER stress in the pathogenesis of PE, research into the interaction between inflammation and ER stress in PE is limited. In this study, we hypothesise that TXNIP acts as the link between inflammation and ER stress, by mediating ER stress-activated NLRP3 inflammasomes in trophoblast cells of women who develop PE.
Materials and methods
This study was ratified by the Ethics Committee of The First Affiliated Hospital of Chongqing Medical University, China. All the tissue samples and data from patient were obtained with written informed consent.
Study population
Our study was conducted in two parts. Firstly, we enrolled 30 independent pregnant women into this study from the Department of Obstetrics and Gynecology at The First Affiliated Hospital of Chongqing Medical University between 1 December 2013 and 1 October, 2014. Among the pregnant women included in the study, 15 were diagnosed with PE and the other 15 were normotensive pregnancies without essential hypertension, cardiovascular disease, diabetes mellitus, metabolic diseases, or pre-existing renal disease. The recruitment of PE patients for this study was based on the diagnostic criteria outlined by the American College of Obstetricians and Gynecologists (ACOG) (Bulletins—Obstetrics 2002). PE was defined as a maternal systolic blood pressure ≧ 140 mmHg and/or a diastolic blood pressure ≧ 90 mmHg, measured on two occasions separated by at least 6 hours; and proteinuria of qualitatively > 1. Placental samples from PE and control participants were obtained during elective non-labored cesarean deliveries. Three small pieces of tissue in separate lobules were collected from each placenta (avoiding the calcification region) and rinsed three times with phosphate-buffered saline (PBS). Samples were snap-frozen in liquid nitrogen and then stored at − 80 °C until further analysis.
The second part of the study included the enrolment of 10 independent pregnant women without any complications, who had chosen to terminate their pregnancy between 6 and 10 weeks of pregnancy. We collected villus from those women. Once acquired, the villus was rinsed three times with PBS. The samples were stored in icy PBS and transported to our lab within 1 h, for subsequent processing.
Reagents
Tunicamycin (TM) was purchased from Shanghai Yuanye Bio-Technology Co., Ltd. (S17119). 4-Phenylbutyric acid (4-PBA) was bought from Sigma-Aldrich (P21005).
Cell cultures and treatment
HTR8/SVneo is a mammalian transformed primary extravillous trophoblast cell line and it is so kind that the cell line was provided by Dr. C. H. Graham (Queen’s University, Kingston, ON, Canada). HTR8/SVneo cells were cultured in RPMI1640 (Gibco, USA) containing 10% fetal bovine serum (Gibco, USA). Cells were treated in hypoxic conditions (1% O2) for 6 h followed by 6 h of reoxygenation (20% O2) to mimic the hypoxia-reoxygenation in the placenta of PE (Hung 2002; Leach et al. 2008). Some cells were treated with TM (5 μg/ml) for 8 h or 4-PBA(5 mmol/ml) for 4 h, in normal conditions. Cells were then harvested for further analysis.
Extravillous explant culture and treatments
The explant culture was performed as described previously (Genbacev et al. 1993; Chen et al. 2015). In brief, placental villous (about 2 mm) were dissected from first trimester (6–10 weeks’ gestation) human placenta and cultured in a 48-well plate, which was pre-coated with 100 μl of diluted matrigel (BD, USA). The explants were cultured for 4 h in DMEM/F12 with 10% FBS and 100 IU/ml penicillin. Once the explants were anchored on the matrigel, we administer different treatments and explants were incubated in a 5% CO2 incubator. After 24 h, the EVT cell migration distance was assessed by an invert stereomicroscope (Life Technologies, USA).
Immunohistochemistry
Firstly, the paraffin section was deparaffinized and then rehydrated. The paraffin section was further quenched in 3% hydrogen peroxide for 20 min and then incubated in goat serum for 30 min. The slides were incubated at 4 °C overnight with polyclonal rabbit anti-ASC antibody (Santa Cruz, USA), monoclonal rabbit anti-TXNIP antibody (Abcam, USA), or monoclonal rabbit anti-NLRP3 antibody (Cell Signaling Technology, USA). The slides were rinsed with PBS and then incubated with goat anti-rabbit IgG for 30 min at 37 °C. DAB (ZSGB-BIO; China) was used as the chromogen and hematoxylin (Sigma, USA) was used for nuclear counterstain. The primary antibodies were omitted as the negative controls. Experiments were repeated at least three times. Images were acquired using an EVOS FL Auto Imaging System (Life Technologies, USA).
Immunofluorescence
After treatment, the cells were fixed with formaldehyde (4%) and blocked with goat serum (Sigma, USA), then incubated with anti-IL-1β antibody for 24 h (Abcam, USA). A fluorescein second antibody (Santa Cruz Biotechnology, USA) was then incubated for 1 h at 37 °C. Vectashield Mounting Medium with DAPI (VECTOR, USA) was used for nuclear staining. Images were acquired using a fluorescence microscope (Life Technologies, USA).
siRNA transfection
To specifically decrease TXNIP expression in HTR8/SVneo cells, the cell line was transfected with siRNA against TXNIP (ACAGACUUCGGAGUACCUG). Scrambled siRNA (UUCUCCGAA CGUGUCACGUTT) was used as a negative control. Cells were incubated in the transfection medium with 100 pmol of scrambled siRNA or TXNIP siRNA for 8 h, according to the manufacturer’s instructions, then left to recover in complete medium for 48 h. TXNIP expression was then assessed by western blotting.
Lentivirus transfection
To decrease TXNIP expression in the placental explant, the explant was transfected with lentivirus against TXNIP (GGATGTCATTCCTGAAGAT), using the lentivirus (TTCTCCGAACGTGTCACGT) as a negative control. The explants were incubated in the transfection medium with 80 titers of TXNIP lentivirus or the negative control lentivirus for 72 h, according to the manufacturer’s instructions. TXNIP expression was then assessed by western blotting. The invasiveness of the explant was assessed using the transwell invasion assay.
Western blotting
Treated HTR-8/SVneo cells were lysed with RIPA buffer (Beyotime, China). According to the manufacturer’s instructions, protein concentration was determined using a Bicinchoninic Acid (BCA) Protein Assay Kit (Beyotime, China). Protein samples (20 μg) were loaded on 10% SDS-polyacrylamide gels, resolved by electrophoresis, and transferred to polyvinylidene difluoride membranes (Millipore, USA). Immunoblotting was performed using polyclonal rabbit anti-BIP antibody (Abcam, USA), monoclonal rabbit anti-TXNIP antibody (Abcam, USA), monoclonal rabbit anti-NLRP3 antibody (Cell Signaling Technology, USA), polyclonal rabbit anti-ASC antibody (Santa Cruz, USA), or polyclonal rabbit anti-caspase-1 (p10) antibody (Santa Cruz, USA) and β-actin (ZSGB-BIO, China). Following incubation with the secondary antibodies (Beyotime, China), the bands of specific proteins on the membranes were developed with Immobilon Western Chemiluminescent HRP Substrate (Millipore, USA). The levels of proteins were quantified by a ChemiDoc™ XRS+ (Bio-Rad, USA). β-actin was used as the loading control.
Immunoprecipitation
Cells were lysed in a cell lysis buffer for western blotting and immunoprecipitation (Beyotime, China). Cell lysates were centrifuged at 14,000g for 10 min at 4 °C. After removing the sediment, the protein concentration of the supernatant was determined using a BCA Protein Assay Kit (Beyotime, China), which switched the concentration to 1 μg/μl. A 200-μl suspension was incubated with a 1:200 dilution of rabbit anti-NLRP3 antibody overnight, at 4 °C. Then, 40 μl Protein A/G Agarose (Beyotime, China) was added and incubated for an additional 4 h in a shaker, at 4 °C. After washing with PBS five times, the immune complexes were boiled in the sample buffer. These samples were then immunoblotted with rabbit primary antibody, as previously described.
HTR8/SVneo transwell invasion assay
Twenty-four-well plates were used as the outer chambers of the transwell chamber (Millipore, USA). The transwell chamber was coated with 60 μl of matrigel (BD, USA). 1.0 × 105 HTR8/SVneo cells in 200 μl serum-free media were seeded into each of the transwell chambers and 600 μl of medium with 10% FBS was added to the 24-well plates. After 48 h of incubation at 37 °C, the cells on the upper surface of the transwell chamber were removed with a cotton swab and the invasive cells were fixed with formaldehyde (4%) for 10 min and then stained with 0.5% crystal violet. A light microscope (Life Technologies, USA) was used to count the number of migrated cells, and the experiment was performed in triplicate.
Real-time invasion assay
A real-time invasion assay was performed as previously described (Keogh 2010; Liu et al. 2012). HTR8/SVneo cells were analyzed using the technique of the xCELLigence system from Roche Applied Sciences, which is composed of a specialized transwell apparatus, cell invasion migration (CIM) proliferation plate, and the Real-Time Cell Analyzer-Dual Plate (RTCA-DP) software. The transwell membrane coated with gold microelectrodes can monitor impedance changes when cells invade through and reach the membrane. The upper chamber of the CIM plate was covered with 10 μl of diluted matrigel (BD, USA); 165 μl of complete medium was added to the lower chamber; and 30 μl DMEM was added into the upper chamber. After assembling the two chambers, background impedance of the media was detected. One hundred ten microliter of the HTR8/SVneo cell (5.0 × 105) suspension was seeded into the upper chamber. After cells were standing for 30 min, the CIM plate was placed in the RTCA-DP analyzer to record the electrical impedance every 15 min for 48 h. The invasion data was analyzed using the RTCA software.
Proliferation assay
HTR8/SVneo cells were seeded in cell culture E-plates at a cell density of 4000 cells per well and incubated overnight in culture medium at 37 °C and 5% CO2. Cell count was performed using TC20TM Automated Cell Counter after cells were treated with TM, 4-PBA, or TXNIP siRNA. The cell growth curves were automatically recorded on the xCELLigence system in real-time. The cell index was followed for 48 h.
Caspase-1 activity assay
After treatment, HTR8/SVneo cells in six wells were collected and lysed. The caspase-1 activity assay was performed using a commercial Caspase-1 Activity Assay Kit (Beyotime, China), according to the manufacturer’s manual.
ELISA
The IL-1β levels in the supernatants of the PE and control groups were measured by enzyme-linked immunosorbent assay (ELISA), using a Human IL-1β/IL-1F2 Quantikine ELISA Kit (R&D, Minneapolis, MN, USA), according to the manufacturer’s instructions.
Statistical analysis
All data was expressed as mean ± standard deviation (SD). T tests were performed to test for differences between the control and treatment groups. Statistical analyses were performed using GraphPad Prism software (version 5.0). P < 0.05 was considered statistically significant.
Results
Participant characteristics
Pregnant females experiencing normal (n = 15) and PE (n = 15) pregnancies were recruited for this study. Clinical characteristics for all pregnant women are displayed in Table 1. PE patients exhibited a significantly higher systolic blood pressure, diastolic blood pressure, and proteinuria than the normal pregnancies. Normal and PE pregnancies were matched for age, BMI, gestational age at delivery, and parity (p value > 0.05).
The expression of inflammatory and ER stress biomarkers were elevated in PE placenta
To investigate whether inflammation was raised in the placenta of women with PE, the expression of apoptosis-associated speck-like protein (ASC), TXNIP, and NLRP3 was assessed using immunohistochemistry in normal and PE placenta. The results in Fig. 1(a–c) visually demonstrate that the protein expressions were increased in the placenta of PE, when compared with the normal placenta. Furthermore, we used western blotting to show the specific levels of NLRP 3, caspase-1 (p10), TXNIP, and ASC (Fig. 1(d, d’) ). BIP, a biomarker of ER stress, was observed in higher levels in PE-complicated placenta, implying increased levels of ER stress. TXNIP is known as an ER stress downstream effector. Taken together, increased levels of TXNIP and BIP suggest that ER stress and NLRP3 inflammasome activation are associated with PE.

The expression of inflammatory and endoplasmic reticulum (ER) stress biomarkers in both preeclampsia (PE) and normal (N) placentae. (a–b”) Immunohistochemistry images show the percentage of cells that expressed thioredoxin-interacting protein (TXNIP), NLR family pyrin domain containing 3 (NLRP3), and apoptosis-associated speck-like protein (ASC) in placenta from healthy pregnant women and women with PE. (c) The right bar graph reveals the semi-quantitative percentages of the cells expressing ASC, TXNIP, and NLRP3 proteins (*p < 0.05; **p < 0.005). Magnification of images is × 400 and the white bar length represents 200 μm. (d, d’) Western blotting analysis of relative expression levels of NLRP3, caspase-1 (p10), heavy-chain–binding protein (BIP), TXNIP, and ASC in the placenta from PE and normal pregnancies (**p < 0.05). Caspase-1 (p10) represents the cleaved caspase-1 form, which indicates phosphorylated caspase-1 as the active counterpart
The relationship between ER stress and inflammation in HTR8/SVneo cells
To identify the link between ER stress and inflammation in trophoblasts, tunicamycin (TM), an inducer of ER stress, was administered to HTR8/SVneo cells. The data showed that TM treatment resulted in the upregulation of NLRP3 expression (Fig. 2a) and caspase-1 activity (Fig. 2b). On the contrary, the addition of an inhibitor of ER stress, 4-phenylbutyric acid (4-PBA), suppressed NLRP3 expression and caspase-1 activity in HTR8/SVneo cells. Similarly, HTR8/SVneo cells subjected to 6 h of hypoxia and 6 h of reoxygenation (H/R), an in vitro cellular PE model, enhanced NLRP3 and BIP expressions. Moreover, the activity of caspase-1 was promoted by H/R. The effect of H/R was partially moderated by the addition of 4-PBA (Fig. 2a).

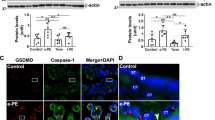
The expression of inflammatory proteins (BIP, TXNIP, NLRP3) in the HTR8/SVneo cells following treatments associated with ER stress and PE (dimethyl sulfoxide (DMSO), hypoxia/reoxygenation (H/R), tunicamycin (TM), and 4-phenylbutyric acid (4-PBA)), compared with normal cells. (a) Western blotting analysis demonstrates the relative protein expression levels of BIP, TXNIP, and NLRP3 in the HTR8/SVneo cells with DMSO, H/R, TM, and 4-PBA treatments, compared with normal cells (N). (b) The activity of caspase-1 in cells with H/R, DMSO, TM, and 4-PBA treatments compared with normal cells (N) (*p < 0.05)
ER stress enhances TXNIP activation and NLRP3 inflammasome formation in HTR8/SVneo cells
To investigate whether ER stress induced TXNIP activation and NLRP3 inflammasome formation in trophoblast cells, HTR8/SVneo cells were treated with TM. The treatment resulted in an enhancement of the interaction between ASC, NLRP3, and TNXIP. Similarly, H/R also induced TXNIP-ASC-NLRP3 co-formation in HTR8/SVneo cells (Fig. 3a, a’, c, c’. To further explore the role of TXNIP in PE-associated ER stress, TXNIP expression was knocked down in HTR8/SVneo cells by siRNA (Fig. 3b, b’), and then, the cells were treated with TM or H/R. The results demonstrated that TXNIP siRNA partially inhibited both NLRP3 inflammasome and ASC activation in the presence of TM or H/R (Fig. 3c, c’). Lastly, levels of NLRP3’s downstream target IL-1β were shown by ELISA and immunofluorescence (IF) to be elevated in the extracellular excretion from the medium supernatants (Fig. 4a) and increased expression was observed in HTR8/SVneo cells following either H/R or TM treatment (Fig. 4b–e”), when compared with the control group.

The effect of TXNIP and NLRP3 inflammasomes on ER stress in HTR8/SVneo cells. (a, a’) Immunoprecipitation analysis demonstrates the interaction between NLRP3 and TXNIP in HTR8/SVneo cells treated with TM or subjected to H/R, compared with normal (N). (b, b’) Western blotting analysis of TXNIP shows the efficiency of TXNIP siRNA. (c, c’) Western blotting analysis demonstrates the relative expression levels of NLRP3 and ASC in HTR8/SVneo cells treated with or without TXNIP siRNA

The extracellular and intracellular level of IL-1β under various treatments. (a) The ELISA analysis shows the excretion level of IL-1β in conditioned medium supernatants: *p < 0.05 compared with normal (N) group; #ap < 0.05 compared with H/R (hypoxia and reoxygenation) group; #bp < 0.05 compared with TM (tunicamycin) group. (b–e”) Immunofluorescence image analysis shows the percentage of cells expressing IL-1β in HTR8/SVneo cells. DAPI-labeled nucleus is indicated by blue fluorescence. IL-1β is labeled by green fluorescence. Magnification of images is × 400 and the white bar length represents 200 μm
TXNIP impacts the invasion and proliferation of HTR8/SVneo cells
To determine whether increased ER stress and the microenvironment of H/R influence the invasiveness of the trophoblast, HTR8/SVneo cells were analyzed using a transwell assay and real-time cell analyzer. The data showed that the number of HTR8/SVneo cells that had migrated was significantly reduced in the presence of TM. However, trophoblast invasiveness was partially restored by siRNA transfection (Fig. 5a, a’). Furthermore, we found that proliferation of HTR8/SVneo cells was dramatically compromised by TM, but significantly improved by silencing TXNIP. However, siRNA TXNIP failed to TM-induced inhibition of proliferation in HTR8/SVneo cells (Fig. 5b, b’). To validate the above findings in a PE model, the traditional transwell assay was performed. The results demonstrated that both H/R and TM significantly impaired invasion of HTR8/SVneo cells, while siRNA TXNIP reversed the H/R and TM-induced inhibition (Fig. 5c, d”). These findings are consistent with the data gained from the real-time cell analyzer. Taken together, the data strongly suggests that TXNIP is critical for ER stress–induced inhibition of trophoblast invasion.

The influence of TXNIP on the invasion and proliferation of HTR8/SVneo cells. (a, a’) The real-time cell analyzer (RTCA) invasion assay of HTR8/SVneo cells in vitro. (b, b’) The proliferation assay of HTR8/SVneo cells in vitro. Means for four replicates ± standard deviations are shown for both the RTCA invasion assay and proliferation assay (*p < 0.05). (d’, c) Trophoblast cell invasion was reduced in the presence of TM (5 μg/ml), compared with the normal group (N). (c”, d”, d) Trophoblast cell invasion was partially inhibited by transfecting with TXNIP SiRNA, compared with the negative control (NC) group. (d’, d”) In the presence of TM, decreased levels of TXNIP partially reversed the inhibition of cell invasion. Results were taken after 40 h of cell incubation. The black bar length represents 400 μm
BIP,TXNIP,and NLRP3 expressions in placental explants
To validate the findings from our cell experiments, the changes of BIP, TXNIP, and NLRP3 were further examined in first trimester placental explants. Both H/R and TM induced the overexpression of BIP, TXNIP, and NLRP3 (Fig. 6a, a’). However, only lentivirus TXNIP reversed the overexpression of TXNIP by H/R and TM, while lentivirus NLRP3 had little effect on NLRP3 overexpression induced by H/R and TM (Fig. 6b, 6b’).

The expression of inflammatory (TXNIP and NLRP3) and ER stress (BIP) biomarkers in placental explants with different treatments. (a, a’) Western blotting analysis demonstrates the relative expression levels of BIP, TXNIP, and NLRP3 in placental explants with H/R and TM treatments. (b, b’) Western blotting shows the expression level of TXNIP and NLRP3 in placental explants treated with H/R and TM, and with the addition of lentivirus
H/R-reduced outgrowth in placental extravillous explants is mediated by TXNIP
Outgrowth experiments were also performed on the first trimester placental explants. As shown in Fig. 7a, b, d, and e, both H/R and TM treatments compromised explant migration distance. The inhibited placental explant outgrowth by TM treatment was restored following lentivirus TXNIP treatment. However, this result did not occur in H/R treatment (Fig. 7b, c, e, f). This result confirmed our previous findings in the HTR8/SVneo cell transwell experiments, indicating that ER stress inhibits trophoblast invasion through TXNIP, suggesting a potential causative relationship with PE development.

The outgrowth of the placental extravillous explants in normal and H/R conditions. Extravillous explants were cultured on matrigel and incubated with different treatments. Images were taken using a light microscope. The black bar length represents 100 mm. (a) CON.; (b) H/R treatment; (c) H/R + TXNIP lentivirus treatment; (d) negative control using lentivirus; (e) TM treatment; (f) TM + TXNIP lentivirus treatment
Discussion
In the pathophysiology of PE, the role of ER stress has been reported previously (Fu et al. 2015). In response to the accumulation of abnormal unfolded and misfolded proteins, ER stress is induced and results in the upregulation of several intracellular signaling pathways involved in trophoblastic apoptosis and inflammation, which is thought to be an important feature in the placental pathogenesis of PE (Cindrova-Davies 2014; Zou et al. 2014).
PE is commonly perceived to be associated with inflammatory dysregulation, and thus signaling pathways related to IL-1β production, which have been proposed to be linked to PE development (Liu et al. 2018). In this study, we have shown that BIP, a biomarker of ER stress, as well as the upstream markers of the signaling pathways of IL-1β (e.g., ASC, NLPR3, caspase-1) were increased in PE placenta when compared with normal placenta. These findings demonstrated that ER stress and inflammatory dysregulation were elevated in PE. Despite two seemingly independent mechanisms, consistent changes in ER stress and inflammation were observed in the PE placenta. When we investigated ER stress and signaling pathways associated with IL-1β, TXNIP was found to act as an intersection between ER stress and inflammation, and similar findings have been documented in other diseases (Tavakkol Afshari et al. 2016). Previous studies have demonstrated that ER stress induces TXNIP production, which is a thioredoxin (Trx)-binding protein involved in the modulation of cellular redox status by binding and inhibiting Trx (Luo et al. 2017). TXNIP is known to be a critical component of the cells’ antioxidant system and evidence suggests that it is important for activating NLRP3 inflammasomes under oxidative stress (Stodle et al. 2018). Based on these findings, we proposed ER stress and inflammatory dysregulation may be linked by TXNIP.
The inflammatory mediators released by trophoblast cells are the key participants in the placental pathogenesis of PE. Therefore, we used HTR-8/SVneo cells as a cell model to study the potential role of TXNIP under ER stress in vitro. Our HTR-8/SVneo cell study demonstrated that the expression of TXNIP, NLRP3, active caspase-1, and IL-1β was all increased in H/R treatment (PE model) and TM treatment (ER stress–induced model). In contrast, the expression of all the proteins investigated was decreased when 4-PBA (ER stress–suppressed model) was added. On the other hand, when TXNIP siRNA was applied, the expression of TXNIP was reduced and its downstream proteins such as NLRP3, active caspase-1, and IL-1β were also decreased in both the PE model and ER stress–induced model. To account for the biological differences between cells and tissues, the trophoblastic extravillous explant was studied using a similar experimental design as performed on the HTR-8/SVneo cells. The outcomes were consistent in the extravillous explants, further strengthening our findings. These results verified that ER stress may modulate TXNIP to activate NLRP3 and, as a consequence, result in the inflammatory dysregulation observed in PE. Furthermore, our study indicates that TXNIP is involved in compromised trophoblast invasion, a feature well-recognized as being involved in the pathogenesis of PE (Daneva et al. 2016; C Weel et al. 2017; Kumar and Mittal 2017). Both the HTR-8/SVneo cells and extravillous explants exhibited reduced invasiveness under H/R and TM treatments, while reduced invasiveness was partially recovered by reversing TXNIP via siRNA and lentivirus.
One of the limitations in this study is that the IL-1β ELISA in conditioned medium supernatants of explant cultures should be performed in order to prove that IL-1β secretion occurs as a functional result of ER stress–associated TXNIP involved in NLRP3 inflammasome activation. Last, the immunohistochemistry should be given in a serial sections to show the co-localizations of ASC, TXNIP, and NLRP3 proteins in the same placental villi.
In conclusion, this study demonstrates the role of TXNIP in ER stress–induced NLRP3 inflammasome activation associated with PE. Moreover, we found that TXNIP was involved in regulating trophoblast invasion. Our research confers that TXNIP is a nexus linking ER stress and inflammation in PE. Future studies should consider validating these results in a larger sample size and using different models of PE. This protein may be a potential interventional target for PE prevention and therapeutic management, warranting further investigation.
Change history
07 December 2019
The authors apologize that in our published paper entitled “Endoplasmic reticulum stress may activate NLRP3 inflammasomes via TXNIP in preeclampsia” <Emphasis Type="Italic">Cell and Tissue Research</Emphasis> (Published online: 22 October 2019)
07 December 2019
The authors apologize that in our published paper entitled ���Endoplasmic reticulum stress may activate NLRP3 inflammasomes via TXNIP in preeclampsia��� Cell and Tissue Research (Published online: 22 October 2019)
References
Amash A, Holcberg G, Sapir O, Huleihel M (2012) Placental secretion of interleukin-1 and interleukin-1 receptor antagonist in preeclampsia: effect of magnesium sulfate. J Interf Cytokine Res 32(9):432–441
Bulletins--Obstetrics, A. C. o. P (2002) ACOG practice bulletin. Diagnosis and management of preeclampsia and eclampsia. Number 33, January 2002. Obstet Gynecol 99(1):159–167
Burton, G. J., H. W. Yung, T. Cindrova-Davies and D. S. Charnock-Jones (2009). Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 30 Suppl A: S43–48
C Weel I, Romao-Veiga M, Matias ML, Fioratti EG, Peracoli JC, Borges VT, Araujo JP Jr, Peracoli MT (2017) Increased expression of NLRP3 inflammasome in placentas from pregnant women with severe preeclampsia. J Reprod Immunol 123:40–47
Chen H, Sun M, Liu J, Tong C, Meng T (2015) Silencing of paternally expressed gene 10 inhibits trophoblast proliferation and invasion. PLoS One 10(12):e0144845
Cindrova-Davies T (2014) The therapeutic potential of antioxidants, ER chaperones, NO and H2S donors, and statins for treatment of preeclampsia. Front Pharmacol 5:119
Daneva AM, Hadzi-Lega M, Stefanovic M (2016) Correlation of the system of cytokines in moderate and severe preeclampsia. Clin Exp Obstet Gynecol 43(2):220–224
Fu J, Zhao L, Wang L, Zhu X (2015) Expression of markers of endoplasmic reticulum stress-induced apoptosis in the placenta of women with early and late onset severe pre-eclampsia. Taiwan J Obstet Gynecol 54(1):19–23
Genbacev O, Jensen KD, Powlin SS, Miller RK (1993) In vitro differentiation and ultrastructure of human extravillous trophoblast (EVT) cells. Placenta 14(4):463–475
Guo M, Wang X, Zhao Y, Yang Q, Ding H, Dong Q, Chen X, Cui M (2018) Ketogenic diet improves brain ischemic tolerance and inhibits NLRP3 inflammasome activation by preventing Drp1-mediated mitochondrial fission and endoplasmic reticulum stress. Front Mol Neurosci 11:86
Hung TH (2002) Hypoxia-reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ Res 90(12):1274–1281
Keogh RJ (2010) New technology for investigating trophoblast function. Placenta 31(4):347–350
Kumar A, Mittal R (2017) Mapping Txnip: key connexions in progression of diabetic nephropathy. Pharmacol Rep 70(3):614–622
Leach RE, Kilburn BA, Petkova A, Romero R, Armant DR (2008) Diminished survival of human cytotrophoblast cells exposed to hypoxia/reoxygenation injury and associated reduction of heparin-binding epidermal growth factor-like growth factor. Am J Obstet Gynecol 198(4):471 e471–471 e477 discussion 471 e477–478
Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR (2012) IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16(2):250–264
Librach CL, Feigenbaum SL, Bass KE, Cui TY, Verastas N, Sadovsky Y, Quigley JP, French DL, Fisher SJ (1994) Interleukin-1 beta regulates human cytotrophoblast metalloproteinase activity and invasion in vitro. J Biol Chem 269(25):17125–17131
Liu NC, Hsieh PF, Hsieh MK, Zeng ZM, Cheng HL, Liao JW, Chueh PJ (2012) Capsaicin-mediated tNOX (ENOX2) up-regulation enhances cell proliferation and migration in vitro and in vivo. J Agric Food Chem 60(10):2758–2765
Liu L, Zhang Y, Wang Y, Peng W, Zhang N, Ye Y (2018) Progesterone inhibited endoplasmic reticulum stress associated apoptosis induced by interleukin-1beta via the GRP78/PERK/CHOP pathway in BeWo cells. J Obstet Gynaecol Res 44(3):463–473
Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H, Zeng Z, Huang W, He Y (2017) NLRP3 inflammasome as a molecular marker in diabetic cardiomyopathy. Front Physiol 8:519
Merksamer PI, Papa FR (2010) The UPR and cell fate at a glance. J Cell Sci 123(Pt 7):1003–1006
Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M (2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 109(4):525–532
Christopher W.G. Redman, M., Gavin P. Sacks, MD, and Ian L. Sargent, PhD (1999). "Preeclampsia_ an excessive maternal inflammatory response to pregnancy." Am J Obstet Gynecol: 499–506
Rusterholz C, Hahn S, Holzgreve W (2007) Role of placentally produced inflammatory and regulatory cytokines in pregnancy and the etiology of preeclampsia. Semin Immunopathol 29(2):151–162
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140(6):821–832
Siljee JE, Wortelboer EJ, Koster MP, Imholz S, Rodenburg W, Visser GH, de Vries A, Schielen PC, Pennings JL (2013) Identification of interleukin-1 beta, but no other inflammatory proteins, as an early onset pre-eclampsia biomarker in first trimester serum by bead-based multiplexed immunoassays. Prenat Diagn 33(12):1183–1188
Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R (2010) Pre-eclampsia. Lancet 376:631–644
Stodle, G. S., G. B. Silva, L. H. Tangeras, L. M. Gierman, I. Nervik, U. E. Dahlberg, C. Sun, M. H. Aune, L. C. V. Thomsen, L. Bjorge and A. C. Iversen (2018). "Placental inflammation in pre-eclampsia by nod-like receptor protein (NLRP)3 inflammasome activation in trophoblasts." Clin Exp Immunol
Tavakkol Afshari Z, Rahimi HR, Ehteshamfar SM, Ganjali R, Tara F, Shapouri Moghadam A (2016) Tumor necrosis factor-alpha and interleukin-1-beta polymorphisms in pre-eclampsia. Iran J Immunol 13(4):309–316
Ting JP, Willingham SB, Bergstralh DT (2008) NLRs at the intersection of cell death and immunity. Nat Rev Immunol 8(5):372–379
Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, Takata K, Koizumi A, Izumi T (1999) A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest 103(1):27–37
Zou Y, Jiang Z, Yu X, Zhang Y, Sun M, Wang W, Ge Z, De W, Sun L (2014) MiR-101 regulates apoptosis of trophoblast HTR-8/SVneo cells by targeting endoplasmic reticulum (ER) protein 44 during preeclampsia. J Hum Hypertens 28(10):610–616
Funding
This work was supported by the National Natural Science Foundation of China (No.81571453, 81771607, 81871185, 81701477), The 111 Project (Yuwaizhuan (2016)32), The National Key Research and Development Program of Reproductive Health & Major Birth Defects Control and Prevention (2016YFC1000407), Chongqing Health Commission (2017ZDXM008,2018ZDXM024), and Chongqing Science & Technology Commission (cstc2017jcyjBX0062).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, Y., Li, J., Han, TL. et al. Endoplasmic reticulum stress may activate NLRP3 inflammasomes via TXNIP in preeclampsia. Cell Tissue Res 379, 589–599 (2020). https://doi.org/10.1007/s00441-019-03104-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-019-03104-9