
Abstract
Human pregnancy is a metabolic and immune challenge for the mother who has to accommodate in her womb a semi-allogeneic fetus whose energy needs increase tremendously with gestation. Recent compelling research has suggested that proper inflammatory changes and oxidative balance are a requisite for successful pregnancy. The placenta is an integral component of this inflammatory response as it actively produces a variety of cytokines and immunomodulatory hormones. In preeclampsia, a life-threatening disorder of pregnancy that is characterized by widespread damage and dysfunction of the maternal endothelium, placental oxidative stress and aberrant cytokine expression induces an exaggerated maternal systemic inflammatory response to pregnancy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Human pregnancy imposes a massive stress to the maternal body, which has to accommodate the increasing energy needs of the developing fetus at the expense of its own needs. Therefore, several physiologic and metabolic changes take place in the maternal body to adapt to such a challenge. The most striking physiologic alteration during pregnancy, apart from the evident weight gain as a consequence of fat and protein deposition in maternal stores, is the increase in blood volume, which is needed for extra blood flow to the uterus and increased perfusion of other organs, especially the kidneys. The pregnant uterus undergoes important tissue and vascular remodeling, the most remarkable of which is the transformation of the uterine spiral arteries into low-resistance flow vessels that enable large volumes of blood to gain access to the placental intervillous space [1]. Additional metabolic changes, such as increased red blood cell counts, insulin resistance and elevated cardiac output further guaranty that the placental villous tree bathes in a nutrient- and oxygen-rich milieu.
The success of pregnancy requires an appropriate immunological interplay between the mother and the developing fetus, as the latter, which expresses paternal antigens, is considered to be a semi-allograft to the maternal immune system [2]. Thus, mechanisms have developed to allow the fetal entity to escape from maternal immune attack, and above all, avoid rejection. To begin with, the fetus is physically secluded from the maternal immune system by the protective shell of embryonic trophectoderm-derived trophoblast cells, which make up the placenta and the chorionic membrane. Most interestingly, the placenta has been identified as a site of immune privilege. This tissue is the source of production of many immunomodulatory hormones and cytokines, and one prevalent hypothesis is that several of these factors released at the feto–maternal interface or into the maternal blood stream contribute to the regulation of the local and systemic immune changes required for a successful pregnancy. By contrast, aberrant placental production of these factors has been shown to be associated with pregnancy-related disorders or even failed pregnancies. The action of several cytokines in the success of pregnancy and in the development of preeclampsia will be reviewed here.
The immunology of pregnancy
The placenta, which is in direct contact with maternal cells in the uterine wall and with maternal blood, is an important immunological barrier between maternal and fetal antigens. This tissue lacks expression of the conventional polymorphic major histocompatibility molecules (MHC) class I, human leukocyte antigen (HLA)-A and HLA-B, and class II [3], and thus, is protected from cytotoxic T lymphocyte (CTL)-mediated destruction. To avoid killing by natural killer (NK) cells, which are programmed to recognize HLA-null cells, trophoblast cells express instead a combination of the nonclassical MHC molecules HLA-G [4], HLA-E, and HLA-F [5]. Whereas the role of HLA-F in pregnancy still awaits elucidation, accumulating research has provided convincing evidence that HLA-G and HLA-E possess immunosuppressive properties, especially towards NK cells and subsets of T lymphocytes, and the idea that placental HLAs facilitate pregnancy, in part, by inhibiting maternal immune cytotoxic responses towards placental cells is now well established [6, 7]. However, a different regulatory mechanism must apply to decidual NK cells, which constitute 50 to 70% of all maternal immune cells present in the pregnant uterus, as these cells do not appear to possess lytic activity [8]. Instead, they are thought to positively regulate pregnancy through secretion of cytokines and angiogenic factors, which have crucial actions on the vascular and decidual transformations occurring in the uterine wall during the early weeks of pregnancy [9–11]. Nevertheless, their regulation might also be mediated by HLA-G and by HLA-C, also present in high amounts on extravillous trophoblasts invading the pregnant uterus [12, 13].
In addition to strategies conferring a relative immune “invisibility” to the placenta, adjustments in the maternal immune system have been suggested to help in sustaining a successful pregnancy. The concept of a maternal CD4-positive T helper cell (Th) 2-biased immunity in pregnant women has been appreciated for over a decade [14]. Th2 immunity is characterized by the dominance of humoral immune responses over cell-mediated, more destructive responses, more likely to be detrimental to the fetal allograft. CD4-positive Th2 lymphocytes develop from naïve T helper cells in the presence of interleukin (IL)-4 and IL-10, whereas Th1 cells arise when IL-2 and interferon (IFN)-γ are present. In turn, Th1 and Th2 lymphocytes express a panel of membrane-bound or soluble immunomodulatory mediators, which influence the activity of other cells of the adaptive immune system, therefore controlling the immune response. A type 2 bias seems to take place both in the intrauterine milieu and in the systemic maternal circulation during pregnancy. It has been reproducibly demonstrated that the placental bed encloses a high incidence of the Th2 factors IL-4 and IL-10 [15–19]. Several isoforms of the immunosuppressive transforming growth factor (TGF)β have also been localized in the placenta, adding to the immune privilege of this tissue [20–22]. Trophoblast cells themselves contribute to the generation of this cytokine milieu as they spontaneously secrete these immunoregulatory molecules, locally, but also into the intervillous space where these may likely participate in the constitution of the peripheral type 2 response. By contrast, type 1 cytokines appear to be marginally expressed or completely absent from the feto–maternal interface [23, 24]. Moreover, the profile of cytokine synthesized by maternal peripheral blood lymphocytes during pregnancy further sustains the development of type 2 immune responses [25]. Conversely, failure to generate a Th2 cytokine milieu, or alternatively, a Th1 cytokine environment has been associated with poor pregnancy outcome [26–28]. However, this long-standing paradigm has been recently disputed [29], as the networks of cytokines targets and actions have become increasingly intricate.
Normal pregnancy and inflammation
A series of intriguing findings have lead to the suggestion that pregnancy is, in fact, a condition of controlled mild maternal systemic inflammation, during which cells of the innate immune system display activated phenotypes [30–32] and circulating levels of particular pro-inflammatory cytokines, such as tumor necrosis factor (TNF)α, IL-6, and IL-1 are raised compared to nonpregnant women [33, 34]. By some aspects, the inflammatory changes in peripheral blood leukocytes were found to be similar to those occurring in patients with sepsis [30]. In addition, monocytes from normal pregnant women were shown to be primed to produce the potent Th1 cytokine IL-12 [35], fueling the debate on the strict dependence on a type 1 vs type 2 adaptive immunity in pregnancy. What is more confounding is the fact that trophoblast cells as well appear to be the source of production of these pro-inflammatory cytokines [36–41]. In vitro, in a dually perfused placental cotyledon, most of placental TNFα was released to the maternal side [42], confirming the importance of placentally produced inflammatory mediators in the induction of the maternal systemic changes. Yet, generalized maternal inflammation is not associated with illness in pregnancy, but has been suggested to be part of the mother’s adaptation to pregnancy and might be crucial for successful pregnancy by not only potentially compensating for a suppressed maternal adaptive immune system and therefore help protect against infection, but also by promoting the physiologic and metabolic changes taking place in the mother’s body [43].
Preeclampsia
Preeclampsia is a serious complication of the second half of human pregnancy, which can have harmful effects on the immediate and long-term health of the mother and the baby [44, 45]. This disease is characterized by multiple maternal disturbances, among which the more prominent symptoms are de novo hypertension, proteinuria, and edema. Additional metabolic dysfunctions may be present, such as activation of the clotting system, impaired liver function, renal failure or pulmonary edema, in particular, in cases of severe, early onset disease [46]. In the absence of intervention, preeclampsia can progress in generalized convulsions or eclampsia. The symptoms resolve only once the placenta is removed, and thus, preeclampsia remains one of the most common reasons for induced preterm delivery.
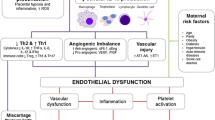
While the etiology of the disorder is still elusive, it is quite clear that it requires a placenta to develop. Risk factors are known and include primiparity, multiple pregnancies, a previous history of preeclampsia, and chronic medical conditions such as obesity, hypertension, vascular disease, or diabetes [47]. However, there is no definitive predictive factor and no preventive treatment available so far. There may undoubtedly be a genetic component at the basis of some cases of preeclampsia, at least in those with a familial history [48, 49]. However, such a genetic cause has not been convincingly demonstrated until now, most likely because polymorphisms in not only one but in several genes are likely to predispose to the development of this complex multifactorial disease. On the other hand, recent work clearly reveals that immune maladaptation and overt activation of the maternal innate immune system are involved in preeclampsia [50, 51]. Remarkably, although the maternal symptoms of preeclampsia appear very heterogeneous at first sight, they can all be ascribed to a generalized endothelium dysfunction [52], which is undeniably part of this exaggerated systemic inflammatory response to pregnancy [53].
Two-stage model of the disease
The symptomatic phase of the disease is, in reality, the second step of a two-stage pathological process, which has its origins in the placenta and occurs during the first weeks of pregnancy [54, 55]. There is evidence of inadequate immunological interactions at the feto–maternal interface already very early in pregnancy. It has been described that HLA-G is under-expressed in preeclamptic placentas [56–58], whereas certain combinations of placental HLA-C and their receptors on maternal NK cells appear to be selectively linked to the risk of developing the disease [13, 59]. Moreover, the number and distribution of macrophages in placental beds are significantly altered in preeclampsia in comparison to normal pregnancy [60–62]. Furthermore, it was also shown that activated macrophages have the potential to induce apoptosis of extravillous trophoblasts in vitro [63]. Increased trophoblast cell death is believed to be central to the impaired placentation that is observed in most cases of preeclampsia. These cells detach from the tip of the primitive trophoblastic outgrowths of the embryo, the anchoring villi, soon after embryo implantation, and actively invade the uterine wall around and into the spiral arteries, to initiate vessel remodeling and establish the uteroplacental circulation. In normal pregnancy, extravillous trophoblasts migrate deep into the decidual part of the arteries and ultimately adopt an endothelial cell-like phenotype and replace their musculo-endothelial lining. The arteries are thus transformed into low-resistance, high-flow channels that provide the appropriate blood flow to the fetus. Preeclampsia is associated with widespread apoptosis of cytotrophoblasts that invade the uterus [64]. Correspondingly, extravillous trophoblasts invasion is abnormally shallow, and the remodeling and enlargement of the spiral arteries is restricted to their placental–proximal part [65, 66].
It has been hypothesized that as a consequence of failed remodeling, blood supply to the placenta is greatly reduced, and this may trigger placental hypoxia [67, 68]. An alternative interpretation of the histopathologic findings has proposed that after incomplete modification of the spiral arteries by the extravillous cytotrophoblasts, these retain a certain capacity to contract in their myometrial part. Phases of contraction followed by relaxation would then lead to cycles of hypoxia/reoxygenation within the placenta [69]. Whatever mechanism at play, the end result would be placental oxidative stress and dysfunction.
Oxidative stress occurs when the cellular levels of reactive oxygen species (ROS) exceed the cell antioxidant capacities. It has been reproducibly observed that ROS are increased, and the levels of several detoxifying enzymes are reduced in preeclamptic placentas, leading to damage of cellular lipids and various other cellular components [70–74]. Moreover, in vitro triggered oxidative stress of a trophoblast cell line reproduced the placental abnormalities of preeclampsia [75]. Deficient oxygenation and increased oxidative stress most probably occur as early as week 10–12 of gestation when maternal blood first gains access to the placenta; however, the antioxidant capacity of placental cells might counteract or contain the insult for some time, until the levels of ROS, by far, surpass the placental redox capacity. Subsequently, placental damage and dysfunction becomes disproportionate. Hence, preeclamptic placentas are often aberrantly structured, with histological evidence of vasculitis, thrombosis, and areas of ischemic or necrotic tissue. These placentas also exhibit increased trophoblast apoptosis [76, 77], a feature that can be reproduced in vitro in models of placental hypoxia or hypoxia/reoxygenation [78–80]. It has been proposed that unusual loads of placental debris and toxic factors, among which pro-inflammatory cytokines, are released in the intervillous space, and in turn, interact with maternal endothelium and cells of the innate immunity, causing the vast array of maternal symptoms that distinguish the disease [81]. We and others have shown that the amount of placental debris, such as cell-free apoptotic DNA [82, 83] and syncytial microparticles, is indeed elevated in peripheral blood of women with preeclampsia [84, 85].
Cytokines in preeclampsia
It is generally agreed that preeclampsia is associated with both local and systemic changes in type 1/type 2 cytokine balance compared to normal pregnancy. Decidual lymphocytes and peripheral blood mononuclear cells from patients with preeclampsia are generally primed to synthesize high levels of the Th1 cytokines, IL-2, IL-12, and IFN-γ [86–89]. On the other hand, they exhibit low spontaneous or phytohemaglutinin-induced expression of the Th2 cytokines IL-10 and IL-5 [90–92]. Whereas these findings initially lead to the conclusion that a maternal T lymphocyte-mediated cytotoxic reaction against the fetal allograft was possibly associated with, and maybe the cause of preeclampsia, it is now believed that such a cytokine environment rather reflects the state of exaggerated inflammation that characterizes the disease. Monocytes and granulocytes present an activated pattern of leukocyte adhesion molecules on their surface and show an increased incidence of basal or induced oxidative stress response compared to their counterparts from normal pregnancy [30, 93]. Spontaneous monocytic cytokine expression is higher in preeclampsia in comparison with normal pregnancy [94]. This is mirrored in the maternal plasma cytokine environment. The circulating levels of TNFα and IL-6, which are already more elevated in healthy pregnant women compared to nonpregnant controls, are further raised in patients with preeclampsia [95–98]. There are also numerous reports of increased serum or plasma levels of several other pro-inflammatory cytokines and of their modulators, such as, IL-2, IL-8, IL-12, IL-15, IL-18, IL-1 receptor antagonist, soluble IL-4 receptor, and soluble TNF receptor (Table 1) [96, 99–104]. The increased levels of soluble cytokine receptors found in patients with preeclampsia may represent a protective response to increased cytokine activity and be a marker for overt inflammation. However, these findings remain very controversial as others did not detect alterations in the levels of pro-inflammatory molecules between healthy pregnant controls and preeclamptic patients [104–106]. Of course, plasma cytokine milieu does not reflect the strict contribution of the placenta, as cytokine production by the dysfunctional maternal endothelium and peripheral blood mononuclear cells is obvious. However, it is undoubted that placental contribution is likely to be significant, as cytokine imbalance and elevated expression of pro-inflammatory molecules is also evident in preeclamptic placentas. Increased production of TNFα, IL-1, and IFN-γ has been documented in these placentas [107–110].
Oxidative stress and placental production of pro- and anti-inflammatory cytokines
A recent set of data has demonstrated that placental expression of many cytokines and soluble mediators of inflammation is tightly regulated by the oxygen tension and cellular oxidative stress that are imposed on the tissue. They provide, therefore, a direct link between the aberrant placental tissue oxygenation and altered cytokine patterns observed in preeclamptic placentas. It was shown that explants of placental villous tissue, derived from normal term placentas, significantly enhance their production of TNFα, IL-1α, and IL-1β when they are incubated in hypoxia, in comparison with the levels produced upon incubation in normoxia [111, 112]. The expression levels of these cytokines are similarly raised under alternative culture conditions which reduced oxygen availability, such as in the presence of an iron chelator or cobalt chloride. It is interesting to note that late first trimester villous tissue also responds to hypoxia by increasing pro-inflammatory cytokine secretion, whereas early first trimester villous tissue is relatively insensitive to the changes in oxygen tension [111]. Furthermore, villous explants derived from preeclamptic placentas were found to be equally sensitive to the effect of hypoxia on TNFα and IL-1 expression [113].
Hypoxia/reoxygenation also increases TNFα synthesis by placental villous tissue in vitro [114]. Notably, in this particular experimental setting, hypoxia/reoxygenation was shown to be a more potent inducer of cytokine secretion than hypoxia alone. Finally, chemicals which trigger placental oxidative stress also stimulates elevated TNFα and IL-1 production by villous explants [112]. In this context, it is interesting to note that in the presence of the antioxidant vitamin C, the secretion of TNFα by term placental villous explants is significantly reduced by 20% (Fig. 1). In contrast, IL-1β secretion did not show such a regulation. This would suggest that therapeutic interventions with antioxidant agents could possibly be developed, even though clinical trials with vitamin supplementation have so far failed to prevent disease occurrence in women at increased risk [115, 116].

Placental villous explants were cultured for 24 h in the absence (−) or in the presence (+) of vitamin C and levels of TNFα (left panel) and IL-1β (right panel) secreted in the culture supernatant were measured by enzyme linked immunosorbent assay and related to explant wet weight. Paired Student t test was used. *p < 0.05, N = 10
Whether IL-6 expression is also controlled by oxygen levels is still debatable. No hypoxia-driven changes in IL-6 synthesis by placental explants were documented. In contrast, isolated cytotrophoblast cells derived from preeclamptic placentas were shown to express higher levels of this molecule than their normal counterparts and to respond to hypoxia by further increasing IL-6 secretion [117]. These latter data seem to contradict the finding that preeclampsia is associated with decreased placental IL-6 production [113, 118].
It is interesting to note that placental expression of immunosuppressor cytokines also seems to be modulated by oxygen tension. Cytotrophoblasts isolated from preeclamptic placentas show a hypoxia-induced reduction in IL-10 synthesis [117]. This feature is in agreement with the description of deficient placental IL-10 production in preeclampsia [119, 120]. Trophoblast expression of TGFβ3 is also regulated by oxygen tensions, in particular, during the switch from early to late first trimester. This gestational age-dependent dichotomy in cytokine production might be explained by the fact that early placental development takes place in the total absence of oxygen, as at that time, spiral arteries remodeling is not fully complete, and the intervillous space is largely devoid of blood flow. But by the end of the first trimester, maternal blood reaches the placenta, and TGFβ3 production is down-regulated [121]. In contrast, TGFβ3 remains overexpressed in preeclamptic placentas [122], a finding that may reflect the state of tissue oxidative stress, but could also participate in the development of later placental dysfunctions. Recently, it was shown that IFN-γ synthesis by trophoblast cells similarly failed to switch from high to low expression as placental environment switches from early hypoxic to late normoxic condition [110].
Cytokines and placentation
During early pregnancy, the interstitial and endovascular trophoblast cells, which invade the decidua and the lumen of the uterine spiral arteries, respectively, differentiate from a fraction of highly proliferative extravillous trophoblast stem cells located in the anchoring villi. These differentiated cells express a new repertoire of adhesion molecules and secrete numerous proteinases, which enables their migration through the uterine stoma and along the endothelial lining of the vessels. In preeclampsia, invading trophoblast cells exhibit an altered protease expression profile and fail to acquire a vascular adhesion phenotype [123, 124]. Remarkably, the proliferation, invasion, and apoptosis of extravillous trophoblasts are orchestrated by several growth factors and almost all cytokines present in the intrauterine wall.
The switch in phenotype from a proliferative to an invasive state appears to be principally controlled by epidermal growth factor and hepatocyte growth factor [125, 126]. This process is also very sensitive to oxygen tension [127], possibly because of the overproduction of cytokines and other mediators of angiogenesis by the extravillous trophoblasts themselves. For instance, IL-1α, IL-1β, and IL-6 have been described as positive regulators of trophoblast differentiation along the invasive pathway as these cytokines can stimulate matrix metalloproteinase (MMP) 9 and MMP2 expression and activity [128, 129]. Specifically, IL-1β expression was shown to parallel the invasive potential of cytotrophoblasts in vitro [130]. On the contrary, IL-10 and TGFβ inhibit trophoblast production of proteases and trophoblast invasion [131, 132]. Failure to down-regulate TGFβ3 expression in proliferative cytotrophoblasts has been linked to shallow placentation and predisposes the pregnancy to preeclampsia [122]. TNFα has also been implicated in the control of trophoblast cell fate. Notably, TNFα, in combination with IFN-γ, stimulates apoptosis of cultured cytotrophoblasts and syncytiotrophoblasts [133, 134]. This suggests a mechanism for deficient placentation in preeclampsia, where local concentrations of both cytokines are high. In contrast, conflicting results exist relative to the role of TNFα in modulating extravillous trophoblast migration potential [135, 136]. Nevertheless, the sum of these in vitro data suggests that the unbalanced placental expression of several cytokines early in pregnancies destined to develop preeclampsia likely plays a role in the etiology of the disease.
TNFα also negatively affects the integrity of the syncytiotrophoblast layer by stimulating increased apoptosis in vitro [137]. Under normoxia, the underlying cytotrophoblasts were shown to reconstitute the syncytiotrophoblast. In contrast, when the placental environment was hypoxic, the syncytiotrophoblast failed to regenerate. TNFα has also been reported to impair the syncytialization process [138]. Thus, in preeclampsia, increased production of TNFα by trophoblast cells in response to deficient uteroplacental blood flow could, in an autocrine manner, lead to continuous syncytiotrophoblast damage and dysfunction. In addition, placental inflammation might be triggered locally, via the release of TNFα after adhesion of activated maternal monocytes to the syncytiotrophoblast [139].
The placental expression of several gestational hormones and mediators of metabolic pathways is also determined by the local cytokine milieu [140]. This area of research has been extensively studied in pregnancies affected with obesity or diabetes mellitus, but less in pregnancies complicated with preeclampsia. It is, nevertheless, likely that the dysregulated expression of these molecules is also occurring in preeclampsia as a result of the aberrant placental cytokine production pattern hallmarking this disease. However, this aspect will not be discussed here.
Cytokines and feto-placental tolerance
IL-10 and IL-4 are pleiotropic immunosuppressors, which display inhibitory effects on cytotoxic and inflammatory reactions. Their expression at the feto–maternal interface is suggested to help in counteracting the harmful effects of pro-inflammatory mediators on placental cells [141]. Remarkably, IL-10 also seems to be an important regulator of trophoblast survival, as it has been shown to selectively induces HLA-G expression in human cytotrophoblasts [142]. In line with these results, low IL-10 levels and diminished HLA-G expression have been observed in preeclampsia. Hence, deficient placental IL-10 expression might not only confer increased susceptibility to pro-inflammatory signals but also amplify inflammation-driven cell damage and NK cell-mediated trophoblast cell death. By opposition, increased trophoblast production of IFN-γ may account for the altered decidual lymphocyte subset distribution and increased number of CD8-positive T cells in the placental bed [143, 144]. IL-6 bears both pro- and anti-inflammatory actions, depending on the cell type and cellular microenvironment it acts on. In the placenta, IL-6 appears to be a positive regulator of pregnancy, as trophoblast-derived IL-6 acts in an autocrine manner on these cells to stimulate their secretion of the immunomodulatory hormone, human chorionic gonadotropin (hCG) [145]. However, serum levels of hCG are raised in preeclamptic patients despite decreased placental IL-6 production, and the role of IL-6 in fetal tolerance awaits elucidation.
Another possible player in promoting fetal tolerance is the subset of CD4+CD25+ regulatory T lymphocytes. These cells have been found to infiltrate the pregnant uterine wall and are, in fact, the second largest population of maternal immune cells present at the feto–maternal interface after uterine NK cells [146]. They function by dampening T cell mediated immune responses via the production of immunosuppressive cytokines [147], and they possibly play a role in the local maintenance of immune privilege [148]. Although preeclampsia is not associated with changes in the levels of regulatory T cells at the periphery [149], the cells may not perform adequately in this condition as TNFα, which is expressed in excess both at the feto–maternal interface and systemically, has been shown to inhibit their suppressive function in vitro [150]. Whether their numbers or activity is altered within the placentas of preeclamptic patients remain to be established.
Cytokines and the maternal symptoms
The endothelium plays a major role in several processes like the regulation of the coagulation cascade, the activation of platelets and leukocyte function, and is therefore an integral part of an inflammatory response. TNFα and IL-1 are the main pro-inflammatory cytokines that stimulate both structural and functional alterations in endothelial cells [151]. It has been suggested that placental IL-1 and TNFα could be potential mediators of maternal endothelial dysfunction in preeclampsia. The reasons for this are multiple. First, the circulating concentration of both cytokines is raised in pregnant women. In vitro, placentally produced IL-1 alters human umbilical vein endothelial cell proliferation and induces the secretion of soluble ICAM and IL-6 [152]. The latter are indeed found in increased levels in the peripheral blood of women with preeclampsia [153–155]. On the other hand, the elevated secretion of TNFα by placental villous tissue in response to hypoxia/reoxygenation causes a reduction of endothelial cell viability and up-regulates the expression of the adhesion molecule E-selectin by the endothelial cells [114]. Maternal serum from preeclamptic patients also stimulates the endothelial production of the vasoactive substances, endothelin-1 and prostacyclin, which might maintain or amplify maternal hypertension [156, 157]. According to recent research, hypertension might be initially triggered by the dysregulated placental production of the angiogenic factors vascular endothelial growth factor (VEGF), soluble fms-like tyrosine kinase (Flt)-1, and placental growth factor (PlGF) [158, 159]. Serum from women with preeclampsia also disrupts the membrane localization of cadherin-5, which may decrease the number of adhesion complexes at the cytoplasmic membrane and lead to vascular permeability [160]. It remains to be established whether these particular endothelial responses to serum are also mediated by cytokines.
Despite these numerous findings, some have questioned the idea that the maternal blood stream carries a placental “toxic” factor to the periphery which contributes to systemic maternal endothelial dysfunction, as they failed to find any effect of plasma factors on endothelial gene expression in vitro [161]. It has been suggested that the activation of maternal leukocytes during their uteroplacental passage may instead play an important part in the dissemination of an inflammatory response to the periphery [162]. Leukocyte activation could be triggered by their interaction with locally activated endothelium or placenta. In favor of this hypothesis, it has been shown that endothelial cells incubated with placental syncytiotrophoblast debris release factors, which can activate peripheral blood leukocytes in vitro [163]. It is interesting to note that placental syncytiotrophoblast debris and placentally derived IL-8 are also potent activators of neutrophils, in that they stimulate the formation of neutrophil extracellular traps (NET), made from DNA and granule proteins, by these cells [164]. Large numbers of NETs are indeed present in the intervillous space of preeclamptic placentas, demonstrating the importance of local neutrophil activation in this disease. Thus, two modes of propagation of the inflammatory reaction from the placental site into the maternal periphery, one encompassing soluble factors, among which cytokines, and the other being cell-mediated, are possibly at work in preeclampsia.
Conclusions
This review has attempted to describe how cytokine networks are affected by pregnancy, and how in return, these networks can affect the success of pregnancy. Cytokines are involved throughout pregnancy, from the implantation of the conceptus to parturition, including placentation, and the maintenance of fetal tolerance. A complete presentation of this large array of cytokine requirements was beyond the scope of the present work. We focused here on the role of placentally produced cytokines on the maternal and fetal events, which are substantially affected in preeclampsia, that is, placental function and maternal inflammatory response to pregnancy. It is clear that placental cytokines integrate into the general maternal cytokine networks to modulate these events, and it is most likely that positive feedback regulatory loops between placental and maternal cells amplify the load of cytokines present in the maternal body, and subsequently, the maternal syndromes. In conclusion, dysregulated cytokine expression by placental cells is certainly one facet of the elusive placental “toxic” factor, or factors, causing the maternal symptoms in preeclampsia.
References
Benirschke K, Kaufmann P (2000) Pathology of the human placenta, 4th edn. Springer, Berlin Heidelberg New York
Medawar PB (1961) Immunological tolerance. Nature 189:14–17
Redman CW, McMichael AJ, Stirrat GM, Sunderland CA, Ting A (1984) Class 1 major histocompatibility complex antigens on human extra-villous trophoblast. Immunology 52:457–468
Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, DeMars R (1990) A class I antigen, HLA-G, expressed in human trophoblasts. Science 248:220–223
Ishitani A, Sageshima N, Lee N, Dorofeeva N, Hatake K, Marquardt H, Geraghty DE (2003) Protein expression and peptide binding suggest unique and interacting functional roles for HLA-E, F, and G in maternal–placental immune recognition. J Immunol 171:1376–1384
Hunt JS, Petroff MG, McIntire RH, Ober C (2005) HLA-G and immune tolerance in pregnancy. FASEB J 19:681–693
Ishitani A, Sageshima N, Hatake K (2006) The involvement of HLA-E and -F in pregnancy. J Reprod Immunol 69:101–113
Bulmer JN, Lash GE (2005) Human uterine natural killer cells: a reappraisal. Mol Immunol 42:511–521
Saito S, Nishikawa K, Morii T, Enomoto M, Narita N, Motoyoshi K, Ichijo M (1993) Cytokine production by CD16–CD56 bright natural killer cells in the human early pregnancy decidua. Int Immunol 5:559–563
Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, Prus D, Cohen-Daniel L, Arnon TI, Manaster I, Gazit R, Yutkin V, Benharroch D, Porgador A, Keshet E, Yagel S, Mandelboim O (2006) Decidual NK cells regulate key developmental processes at the human fetal–maternal interface. Nat Med 12:1065–1074
Tabiasco J, Rabot M, Aguerre-Girr M, El Costa H, Berrebi A, Parant O, Laskarin G, Juretic K, Bensussan A, Rukavina D, Le Bouteiller P (2006) Human decidual NK cells: unique phenotype and functional properties—a review. Placenta 27(Suppl A):S34–S39
van der MA, Lukassen HG, van Lierop MJ, Wijnands F, Mosselman S, Braat DD, Joosten I (2004) Membrane-bound HLA-G activates proliferation and interferon-gamma production by uterine natural killer cells. Mol Hum Reprod 10:189–195
Hiby SE, Walker JJ, O’Shaughnessy KM, Redman CW, Carrington M, Trowsdale J, Moffett A (2004) Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J Exp Med 200:957–965
Wegmann TG, Lin H, Guilbert L, Mosmann TR (1993) Bidirectional cytokine interactions in the maternal–fetal relationship: is successful pregnancy a TH2 phenomenon? Immunol Today 14:353–356
Cadet P, Rady PL, Tyring SK, Yandell RB, Hughes TK (1995) Interleukin-10 messenger ribonucleic acid in human placenta: implications of a role for interleukin-10 in fetal allograft protection. Am J Obstet Gynecol 173:25–29
Roth I, Corry DB, Locksley RM, Abrams JS, Litton MJ, Fisher SJ (1996) Human placental cytotrophoblasts produce the immunosuppressive cytokine interleukin 10. J Exp Med 184:539–548
Moraes-Pinto MI, Vince GS, Flanagan BF, Hart CA, Johnson PM (1997) Localization of IL-4 and IL-4 receptors in the human term placenta, decidua and amniochorionic membranes. Immunology 90:87–94
Bennett WA, Lagoo-Deenadayalan S, Stopple JA, Barber WH, Hale E, Brackin MN, Cowan BD (1998) Cytokine expression by first-trimester human chorionic villi. Am J Reprod Immunol 40:309–318
Sacks GP, Clover LM, Bainbridge DR, Redman CW, Sargent IL (2001) Flow cytometric measurement of intracellular Th1 and Th2 cytokine production by human villous and extravillous cytotrophoblast. Placenta 22:550–559
Dungy LJ, Siddiqi TA, Khan S (1991) Transforming growth factor-beta 1 expression during placental development. Am J Obstet Gynecol 165:853–857
Graham CH, Lysiak JJ, McCrae KR, Lala PK (1992) Localization of transforming growth factor-beta at the human fetal–maternal interface: role in trophoblast growth and differentiation. Biol Reprod 46:561–572
Ando N, Hirahara F, Fukushima J, Kawamoto S, Okuda K, Funabashi T, Gorai I, Minaguchi H (1998) Differential gene expression of TGF-beta isoforms and TGF-beta receptors during the first trimester of pregnancy at the human maternal–fetal interface. Am J Reprod Immunol 40:48–56
Paulesu L, Romagnoli R, Cintorino M, Ricci MG, Garotta G (1994) First trimester human trophoblast expresses both interferon-gamma and interferon-gamma-receptor. J Reprod Immunol 27:37–48
Hanna N, Hanna I, Hleb M, Wagner E, Dougherty J, Balkundi D, Padbury J, Sharma S (2000) Gestational age-dependent expression of IL-10 and its receptor in human placental tissues and isolated cytotrophoblasts. J Immunol 164:5721–5728
Marzi M, Vigano A, Trabattoni D, Villa ML, Salvaggio A, Clerici E, Clerici M (1996) Characterization of type 1 and type 2 cytokine production profile in physiologic and pathologic human pregnancy. Clin Exp Immunol 106:127–133
Piccinni MP, Beloni L, Livi C, Maggi E, Scarselli G, Romagnani S (1998) Defective production of both leukemia inhibitory factor and type 2 T-helper cytokines by decidual T cells in unexplained recurrent abortions. Nat Med 4:1020–1024
Hill JA, Polgar K, Anderson DJ (1995) T-helper 1-type immunity to trophoblast in women with recurrent spontaneous abortion. JAMA 273:1933–1936
Raghupathy R (1997) Th1-type immunity is incompatible with successful pregnancy. Immunol Today 18:478–482
Chaouat G, Ledee-Bataille N, Dubanchet S, Zourbas S, Sandra O, Martal J (2004) TH1/TH2 paradigm in pregnancy: paradigm lost? Cytokines in pregnancy/early abortion: reexamining the TH1/TH2 paradigm. Int Arch Allergy Immunol 134:93–119
Sacks GP, Studena K, Sargent K, Redman CW (1998) Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol 179:80–86
Naccasha N, Gervasi MT, Chaiworapongsa T, Berman S, Yoon BH, Maymon E, Romero R (2001) Phenotypic and metabolic characteristics of monocytes and granulocytes in normal pregnancy and maternal infection. Am J Obstet Gynecol 185:1118–1123
Sacks G, Sargent I, Redman C (1999) An innate view of human pregnancy. Immunol Today 20:114–118
Kupferminc MJ, Peaceman AM, Wigton TR, Tamura RK, Rehnberg KA, Socol ML (1994) Immunoreactive tumor necrosis factor-alpha is elevated in maternal plasma but undetected in amniotic fluid in the second trimester. Am J Obstet Gynecol 171:976–979
Austgulen R, Lien E, Liabakk NB, Jacobsen G, Arntzen KJ (1994) Increased levels of cytokines and cytokine activity modifiers in normal pregnancy. Eur J Obstet Gynecol Reprod Biol 57:149–155
Sacks GP, Redman CW, Sargent IL (2003) Monocytes are primed to produce the Th1 type cytokine IL-12 in normal human pregnancy: an intracellular flow cytometric analysis of peripheral blood mononuclear cells. Clin Exp Immunol 131:490–497
Kameda T, Matsuzaki N, Sawai K, Okada T, Saji F, Matsuda T, Hirano T, Kishimoto T, Tanizawa O (1990) Production of interleukin-6 by normal human trophoblast. Placenta 11:205–213
Chen HL, Yang YP, Hu XL, Yelavarthi KK, Fishback JL, Hunt JS (1991) Tumor necrosis factor alpha mRNA and protein are present in human placental and uterine cells at early and late stages of gestation. Am J Pathol 139:327–335
Paulesu L, King A, Loke YW, Cintorino M, Bellizzi E, Boraschi D (1991) Immunohistochemical localization of IL-1 alpha and IL-1 beta in normal human placenta. Lymphokine Cytokine Res 10:443–448
Hu XL, Yang Y, Hunt JS (1992) Differential distribution of interleukin-1 alpha and interleukin-1 beta proteins in human placentas. J Reprod Immunol 22:257–268
Haynes MK, Jackson LG, Tuan RS, Shepley KJ, Smith JB (1993) Cytokine production in first trimester chorionic villi: detection of mRNAs and protein products in situ. Cell Immunol 151:300–308
King A, Jokhi PP, Smith SK, Sharkey AM, Loke YW (1995) Screening for cytokine mRNA in human villous and extravillous trophoblasts using the reverse-transcriptase polymerase chain reaction (RT-PCR). Cytokine 7:364–371
Kirwan JP, Hauguel-de Mouzon S, Lepercq J, Challier JC, Huston-Presley L, Friedman JE, Kalhan SC, Catalano PM (2002) TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes 51:2207–2213
Borzychowski AM, Sargent IL, Redman CW (2006) Inflammation and pre-eclampsia. Semin Fetal Neonatal Med 11:309–316
Sibai B, Dekker G, Kupferminc M (2005) Pre-eclampsia. Lancet 365:785–799
Redman CW, Sargent IL (2005) Latest advances in understanding preeclampsia. Science 308:1592–1594
von Dadelszen P, Magee LA, Roberts JM (2003) Subclassification of preeclampsia. Hypertens Pregnancy 22:143–148
Duckitt K, Harrington D (2005) Risk factors for pre-eclampsia at antenatal booking: systematic review of controlled studies. BMJ 330:565
van Dijk M, Mulders J, Poutsma A, Konst AA, Lachmeijer AM, Dekker GA, Blankenstein MA, Oudejans CB (2005) Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat Genet 37:514–519
Laivuori H (2007) Genetic aspects of preeclampsia. Front Biosci 12:2372–2382
Hahn S, Gupta AK, Troeger C, Rusterholz C, Holzgreve W (2006) Disturbances in placental immunology: ready for therapeutic interventions? Springer Semin Immunopathol 27:477–493
Sargent IL, Borzychowski AM, Redman CW (2006) Immunoregulation in normal pregnancy and pre-eclampsia: an overview. Reprod Biomed Online 13:680–686
Roberts JM, Taylor RN, Musci TJ, Rodgers GM, Hubel CA, McLaughlin MK (1989) Preeclampsia: an endothelial cell disorder. Am J Obstet Gynecol 161:1200–1204
Redman CW, Sargent IL (2003) Pre-eclampsia, the placenta and the maternal systemic inflammatory response—a review. Placenta 24(Suppl A):S21–S27
Roberts JM, Redman CW (1993) Pre-eclampsia: more than pregnancy-induced hypertension. Lancet 341:1447–1451
Hahn S, Holzgreve W (2002) Fetal cells and cell-free fetal DNA in maternal blood: new insights into pre-eclampsia. Hum Reprod Update 8:501–508
Goldman-Wohl DS, Ariel I, Greenfield C, Hochner-Celnikier D, Cross J, Fisher S, Yagel S (2000) Lack of human leukocyte antigen-G expression in extravillous trophoblasts is associated with pre-eclampsia. Mol Hum Reprod 6:88–95
Yie SM, Li LH, Li YM, Librach C (2004) HLA-G protein concentrations in maternal serum and placental tissue are decreased in preeclampsia. Am J Obstet Gynecol 191:525–529
Le Bouteiller P, Pizzato N, Barakonyi A, Solier C (2003) HLA-G, pre-eclampsia, immunity and vascular events. J Reprod Immunol 59:219–234
Parham P (2004) NK cells and trophoblasts: partners in pregnancy. J Exp Med 200:951–955
Reister F, Frank HG, Heyl W, Kosanke G, Huppertz B, Schroder W, Kaufmann P, Rath W (1999) The distribution of macrophages in spiral arteries of the placental bed in pre-eclampsia differs from that in healthy patients. Placenta 20:229–233
Redline RW (2001) Macrophages in the basal plate of pre-eclamptic placentae. Placenta 22:890–893
Burk MR, Troeger C, Brinkhaus R, Holzgreve W, Hahn S (2001) Severely reduced presence of tissue macrophages in the basal plate of pre-eclamptic placentae. Placenta 22:309–316
Reister F, Frank HG, Kingdom JC, Heyl W, Kaufmann P, Rath W, Huppertz B (2001) Macrophage-induced apoptosis limits endovascular trophoblast invasion in the uterine wall of preeclamptic women. Lab Invest 81:1143–1152
DiFederico E, Genbacev O, Fisher SJ (1999) Preeclampsia is associated with widespread apoptosis of placental cytotrophoblasts within the uterine wall. Am J Pathol 155:293–301
Robertson WB, Brosens I, Dixon G (1976) Maternal uterine vascular lesions in the hypertensive complications of pregnancy. Perspect Nephrol Hypertens 5:115–127
Graham CH, Burton GJ (2004) Oxygen and trophoblast behaviour—a workshop report. Placenta 25(Suppl A):S90–S92
Burton GJ, Caniggia I (2001) Hypoxia: implications for implantation to delivery—a workshop report. Placenta 22(Suppl A):S63–S65
Soleymanlou N, Jurisica I, Nevo O, Ietta F, Zhang X, Zamudio S, Post M, Caniggia I (2005) Molecular evidence of placental hypoxia in preeclampsia. J Clin Endocrinol Metab 90:4299–4308
Hung TH, Skepper JN, Burton GJ (2001) In vitro ischemia–reperfusion injury in term human placenta as a model for oxidative stress in pathological pregnancies. Am J Pathol 159:1031–1043
Sikkema JM, van Rijn BB, Franx A, Bruinse HW, de Roos R, Stroes ES, van Faassen EE (2001) Placenta l superoxide is increased in pre-eclampsia. Placenta 22:304–308
Many A, Hubel CA, Fisher SJ, Roberts JM, Zhou Y (2000) Invasive cytotrophoblasts manifest evidence of oxidative stress in preeclampsia. Am J Pathol 156:321–331
Wang Y, Walsh SW (2001) Increased superoxide generation is associated with decreased superoxide dismutase activity and mRNA expression in placental trophoblast cells in pre-eclampsia. Placenta 22:206–212
Vanderlelie J, Venardos K, Clifton VL, Gude NM, Clarke FM, Perkins AV (2005) Increased biological oxidation and reduced anti-oxidant enzyme activity in pre-eclamptic placentae. Placenta 26:53–58
Burton GJ, Jauniaux E (2004) Placental oxidative stress: from miscarriage to preeclampsia. J Soc Gynecol Investig 11:342–352
Vaughan JE, Walsh SW (2002) Oxidative stress reproduces placental abnormalities of preeclampsia. Hypertens Pregnancy 21:205–223
Leung DN, Smith SC, To KF, Sahota DS, Baker PN (2001) Increased placental apoptosis in pregnancies complicated by preeclampsia. Am J Obstet Gynecol 184:1249–1250
Ishihara N, Matsuo H, Murakoshi H, Laoag-Fernandez JB, Samoto T, Maruo T (2002) Increased apoptosis in the syncytiotrophoblast in human term placentas complicated by either preeclampsia or intrauterine growth retardation. Am J Obstet Gynecol 186:158–166
Levy R, Smith SD, Chandler K, Sadovsky Y, Nelson DM (2000) Apoptosis in human cultured trophoblasts is enhanced by hypoxia and diminished by epidermal growth factor. Am J Physiol Cell Physiol 278:C982–C988
Hung TH, Skepper JN, Charnock-Jones DS, Burton GJ (2002) Hypoxia–reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ Res 90:1274–1281
Huppertz B, Kingdom J, Caniggia I, Desoye G, Black S, Korr H, Kaufmann P (2003) Hypoxia favours necrotic versus apoptotic shedding of placental syncytiotrophoblast into the maternal circulation. Placenta 24:181–190
Redman CW, Sargent IL (2000) Placental debris, oxidative stress and pre-eclampsia. Placenta 21:597–602
Lo YM, Leung TN, Tein MS, Sargent IL, Zhang J, Lau TK, Haines CJ, Redman CW (1999) Quantitative abnormalities of fetal DNA in maternal serum in preeclampsia. Clin Chem 45:184–188
Zhong XY, Laivuori H, Livingston JC, Ylikorkala O, Sibai BM, Holzgreve W, Hahn S (2001) Elevation of both maternal and fetal extracellular circulating deoxyribonucleic acid concentrations in the plasma of pregnant women with preeclampsia. Am J Obstet Gynecol 184:414–419
Knight M, Redman CW, Linton EA, Sargent IL (1998) Shedding of syncytiotrophoblast microvilli into the maternal circulation in pre-eclamptic pregnancies. Br J Obstet Gynaecol 105:632–640
Goswami D, Tannetta DS, Magee LA, Fuchisawa A, Redman CW, Sargent IL, von Dadelszen P (2006) Excess syncytiotrophoblast microparticle shedding is a feature of early-onset pre-eclampsia, but not normotensive intrauterine growth restriction. Placenta 27:56–61
Saito S, Umekage H, Sakamoto Y, Sakai M, Tanebe K, Sasaki Y, Morikawa H (1999) Increased T-helper-1-type immunity and decreased T-helper-2-type immunity in patients with preeclampsia. Am J Reprod Immunol 41:297–306
Darmochwal-Kolarz D, Leszczynska-Gorzelak B, Rolinski J, Oleszczuk J (1999) T helper 1- and T helper 2-type cytokine imbalance in pregnant women with pre-eclampsia. Eur J Obstet Gynecol Reprod Biol 86:165–170
Rein DT, Schondorf T, Gohring UJ, Kurbacher CM, Pinto I, Breidenbach M, Mallmann P, Kolhagen H, Engel H (2002) Cytokine expression in peripheral blood lymphocytes indicates a switch to T(HELPER) cells in patients with preeclampsia. J Reprod Immunol 54:133–142
Sakai M, Tsuda H, Tanebe K, Sasaki Y, Saito S (2002) Interleukin-12 secretion by peripheral blood mononuclear cells is decreased in normal pregnant subjects and increased in preeclamptic patients. Am J Reprod Immunol 47:91–97
Wilczynski JR, Tchorzewski H, Glowacka E, Banasik M, Lewkowicz P, Szpakowski M, Zeman K, Wilczynski J (2002) Cytokine secretion by decidual lymphocytes in transient hypertension of pregnancy and pre-eclampsia. Mediators Inflamm 11:105–111
Orange S, Horvath J, Hennessy A (2003) Preeclampsia is associated with a reduced interleukin-10 production from peripheral blood mononuclear cells. Hypertens Pregnancy 22:1–8
Jonsson Y, Matthiesen L, Berg G, Ernerudh J, Nieminen K, Ekerfelt C (2005) Indications of an altered immune balance in preeclampsia: a decrease in in vitro secretion of IL-5 and IL-10 from blood mononuclear cells and in blood basophil counts compared with normal pregnancy. J Reprod Immunol 66:69–84
Holthe MR, Staff AC, Berge LN, Lyberg T (2004) Leukocyte adhesion molecules and reactive oxygen species in preeclampsia. Obstet Gynecol 103:913–922
Luppi P, Deloia JA (2006) Monocytes of preeclamptic women spontaneously synthesize pro-inflammatory cytokines. Clin Immunol 118:268–275
Kupferminc MJ, Peaceman AM, Wigton TR, Rehnberg KA, Socol ML (1994) Tumor necrosis factor-alpha is elevated in plasma and amniotic fluid of patients with severe preeclampsia. Am J Obstet Gynecol 170:1752–1757
Greer IA, Lyall F, Perera T, Boswell F, Macara LM (1994) Increased concentrations of cytokines interleukin-6 and interleukin-1 receptor antagonist in plasma of women with preeclampsia: a mechanism for endothelial dysfunction? Obstet Gynecol 84:937–940
Vince GS, Starkey PM, Austgulen R, Kwiatkowski D, Redman CW (1995) Interleukin-6, tumour necrosis factor and soluble tumour necrosis factor receptors in women with pre-eclampsia. Br J Obstet Gynaecol 102:20–25
Conrad KP, Miles TM, Benyo DF (1998) Circulating levels of immunoreactive cytokines in women with preeclampsia. Am J Reprod Immunol 40:102–111
Stallmach T, Hebisch G, Joller H, Kolditz P, Engelmann M (1995) Expression pattern of cytokines in the different compartments of the feto–maternal unit under various conditions. Reprod Fertil Dev 7:1573–1580
Kupferminc MJ, Peaceman AM, Aderka D, Wallach D, Socol ML (1996) Soluble tumor necrosis factor receptors and interleukin-6 levels in patients with severe preeclampsia. Obstet Gynecol 88:420–427
Kauma S, Takacs P, Scordalakes C, Walsh S, Green K, Peng T (2002) Increased endothelial monocyte chemoattractant protein-1 and interleukin-8 in preeclampsia. Obstet Gynecol 100:706–714
Adams KM, Mandel LS, Guthrie KA, Atkinson MW (2003) Interleukin-18 in the plasma of women with preeclampsia. Am J Obstet Gynecol 188:1234–1237
Bachmayer N, Rafik HR, Liszka L, Bremme K, Sverremark-Ekstrom E (2006) Aberrant uterine natural killer (NK)-cell expression and altered placental and serum levels of the NK-cell promoting cytokine interleukin-12 in pre-eclampsia. Am J Reprod Immunol 56:292–301
Jonsson Y, Ruber M, Matthiesen L, Berg G, Nieminen K, Sharma S, Ernerudh J, Ekerfelt C (2006) Cytokine mapping of sera from women with preeclampsia and normal pregnancies. J Reprod Immunol 70:83–91
Opsjon SL, Austgulen R, Waage A (1995) Interleukin-1, interleukin-6 and tumor necrosis factor at delivery in preeclamptic disorders. Acta Obstet Gynecol Scand 74:19–26
Agarwal R, Loganath A, Roy AC, Wong YC, Ng SC (2001) Expression profiles of interleukin-15 in early and late gestational human placenta and in pre-eclamptic placenta. Mol Hum Reprod 7:97–101
Wang Y, Walsh SW (1996) TNF alpha concentrations and mRNA expression are increased in preeclamptic placentas. J Reprod Immunol 32:157–169
Rinehart BK, Terrone DA, Lagoo-Deenadayalan S, Barber WH, Hale EA, Martin JN Jr, Bennett WA (1999) Expression of the placental cytokines tumor necrosis factor alpha, interleukin 1beta, and interleukin 10 is increased in preeclampsia. Am J Obstet Gynecol 181:915–920
Munno I, Chiechi LM, Lacedra G, Putignano G, Patimo C, Lobascio A, Loizzi P (1999) Spontaneous and induced release of prostaglandins, interleukin (IL)-1beta, IL-6, and tumor necrosis factor-alpha by placental tissue from normal and preeclamptic pregnancies. Am J Reprod Immunol 42:369–374
Banerjee S, Smallwood A, Moorhead J, Chambers AE, Papageorghiou A, Campbell S, Nicolaides K (2005) Placental expression of interferon-gamma (IFN-gamma) and its receptor IFN-gamma R2 fail to switch from early hypoxic to late normotensive development in preeclampsia. J Clin Endocrinol Metab 90:944–952
Benyo DF, Miles TM, Conrad KP (1997) Hypoxia stimulates cytokine production by villous explants from the human placenta. J Clin Endocrinol Metab 82:1582–1588
Malek A, Sager R, Schneider H (2001) Effect of hypoxia, oxidative stress and lipopolysaccharides on the release of prostaglandins and cytokines from human term placental explants. Placenta 22(Suppl A):S45–S50
Benyo DF, Smarason A, Redman CW, Sims C, Conrad KP (2001) Expression of inflammatory cytokines in placentas from women with preeclampsia. J Clin Endocrinol Metab 86:2505–2512
Hung TH, Charnock-Jones DS, Skepper JN, Burton GJ (2004) Secretion of tumor necrosis factor-alpha from human placental tissues induced by hypoxia–reoxygenation causes endothelial cell activation in vitro: a potential mediator of the inflammatory response in preeclampsia. Am J Pathol 164:1049–1061
Poston L, Briley AL, Seed PT, Kelly FJ, Shennan AH (2006) Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): randomised placebo-controlled trial. Lancet 367:1145–1154
Rumbold AR, Crowther CA, Haslam RR, Dekker GA, Robinson JS (2006) Vitamins C and E and the risks of preeclampsia and perinatal complications. N Engl J Med 354:1796–1806
Bowen RS, Gu Y, Zhang Y, Lewis DF, Wang Y (2005) Hypoxia promotes interleukin-6 and -8 but reduces interleukin-10 production by placental trophoblast cells from preeclamptic pregnancies. J Soc Gynecol Investig 12:428–432
Kauma SW, Wang Y, Walsh SW (1995) Preeclampsia is associated with decreased placental interleukin-6 production. J Soc Gynecol Investig 2:614–617
Hennessy A, Pilmore HL, Simmons LA, Painter DM (1999) A deficiency of placental IL-10 in preeclampsia. J Immunol 163:3491–3495
Rein DT, Breidenbach M, Honscheid B, Friebe-Hoffmann U, Engel H, Gohring UJ, Uekermann L, Kurbacher CM, Schondorf T (2003) Preeclamptic women are deficient of interleukin-10 as assessed by cytokine release of trophoblast cells in vitro. Cytokine 23:119–125
Caniggia I, Mostachfi H, Winter J, Gassmann M, Lye SJ, Kuliszewski M, Post M (2000) Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J Clin Invest 105:577–587
Caniggia I, Grisaru-Gravnosky S, Kuliszewsky M, Post M, Lye SJ (1999) Inhibition of TGF-beta 3 restores the invasive capability of extravillous trophoblasts in preeclamptic pregnancies. J Clin Invest 103:1641–1650
Zhou Y, Damsky CH, Fisher SJ (1997) Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J Clin Invest 99:2152–2164
Reister F, Kingdom JC, Ruck P, Marzusch K, Heyl W, Pauer U, Kaufmann P, Rath W, Huppertz B (2006) Altered protease expression by periarterial trophoblast cells in severe early-onset preeclampsia with IUGR. J Perinat Med 34:272–279
Kharfi A, Giguere Y, Sapin V, Masse J, Dastugue B, Forest JC (2003) Trophoblastic remodeling in normal and preeclamptic pregnancies: implication of cytokines. Clin Biochem 36:323–331
Staun-Ram E, Shalev E (2005) Human trophoblast function during the implantation process. Reprod Biol Endocrinol 3:56
Zhou Y, Genbacev O, Damsky CH, Fisher SJ (1998) Oxygen regulates human cytotrophoblast differentiation and invasion: implications for endovascular invasion in normal pregnancy and in pre-eclampsia. J Reprod Immunol 39:197–213
Meisser A, Chardonnens D, Campana A, Bischof P (1999) Effects of tumour necrosis factor-alpha, interleukin-1 alpha, macrophage colony stimulating factor and transforming growth factor beta on trophoblastic matrix metalloproteinases. Mol Hum Reprod 5:252–260
Meisser A, Cameo P, Islami D, Campana A, Bischof P (1999) Effects of interleukin-6 (IL-6) on cytotrophoblastic cells. Mol Hum Reprod 5:1055–1058
Librach CL, Feigenbaum SL, Bass KE, Cui TY, Verastas N, Sadovsky Y, Quigley JP, French DL, Fisher SJ (1994) Interleukin-1 beta regulates human cytotrophoblast metalloproteinase activity and invasion in vitro. J Biol Chem 269:17125–17131
Roth I, Fisher SJ (1999) IL-10 is an autocrine inhibitor of human placental cytotrophoblast MMP-9 production and invasion. Dev Biol 205:194–204
Lash GE, Otun HA, Innes BA, Bulmer JN, Searle RF, Robson SC (2005) Inhibition of trophoblast cell invasion by TGFB1, 2, and 3 is associated with a decrease in active proteases. Biol Reprod 73:374–381
Yui J, Garcia-Lloret M, Wegmann TG, Guilbert LJ (1994) Cytotoxicity of tumour necrosis factor-alpha and gamma-interferon against primary human placental trophoblasts. Placenta 15:819–835
Crocker IP, Barratt S, Kaur M, Baker PN (2001) The in-vitro characterization of induced apoptosis in placental cytotrophoblasts and syncytiotrophoblasts. Placenta 22:822–830
Bauer S, Pollheimer J, Hartmann J, Husslein P, Aplin JD, Knofler M (2004) Tumor necrosis factor-alpha inhibits trophoblast migration through elevation of plasminogen activator inhibitor-1 in first-trimester villous explant cultures. J Clin Endocrinol Metab 89:812–822
Renaud SJ, Postovit LM, Macdonald-Goodfellow SK, McDonald GT, Caldwell JD, Graham CH (2005) Activated macrophages inhibit human cytotrophoblast invasiveness in vitro. Biol Reprod 73:237–243
Crocker IP, Tansinda DM, Jones CJ, Baker PN (2004) The influence of oxygen and tumor necrosis factor-alpha on the cellular kinetics of term placental villous explants in culture. J Histochem Cytochem 52:749–757
Leisser C, Saleh L, Haider S, Husslein H, Sonderegger S, Knofler M (2006) Tumour necrosis factor-alpha impairs chorionic gonadotrophin beta-subunit expression and cell fusion of human villous cytotrophoblast. Mol Hum Reprod 12:601–609
Garcia-Lloret MI, Winkler-Lowen B, Guilbert LJ (2000) Monocytes adhering by LFA-1 to placental syncytiotrophoblasts induce local apoptosis via release of TNF-alpha. A model for hematogenous initiation of placental inflammations. J Leukoc Biol 68:903–908
Hauguel-de Mouzon S, Guerre-Millo M (2006) The placenta cytokine network and inflammatory signals. Placenta 27:794–798
Goodwin VJ, Sato TA, Mitchell MD, Keelan JA (1998) Anti-inflammatory effects of interleukin-4, interleukin-10, and transforming growth factor-beta on human placental cells in vitro. Am J Reprod Immunol 40:319–325
Moreau P, Adrian-Cabestre F, Menier C, Guiard V, Gourand L, Dausset J, Carosella ED, Paul P (1999) IL-10 selectively induces HLA-G expression in human trophoblasts and monocytes. Int Immunol 11:803–811
Stallmach T, Hebisch G, Orban P, Lu X (1999) Aberrant positioning of trophoblast and lymphocytes in the feto-maternal interface with pre-eclampsia. Virchows Arch 434:207–211
Wilczynski JR, Tchorzewski H, Banasik M, Glowacka E, Wieczorek A, Lewkowicz P, Malinowski A, Szpakowski M, Wilczynski J (2003) Lymphocyte subset distribution and cytokine secretion in third trimester decidua in normal pregnancy and preeclampsia. Eur J Obstet Gynecol Reprod Biol 109:8–15
Nishino E, Matsuzaki N, Masuhiro K, Kameda T, Taniguchi T, Takagi T, Saji F, Tanizawa O (1990) Trophoblast-derived interleukin-6 (IL-6) regulates human chorionic gonadotropin release through IL-6 receptor on human trophoblasts. J Clin Endocrinol Metab 71:436–441
Heikkinen J, Mottonen M, Alanen A, Lassila O (2004) Phenotypic characterization of regulatory T cells in the human decidua. Clin Exp Immunol 136:373–378
Read S, Powrie F (2001) CD4(+) regulatory T cells. Curr Opin Immunol 13:644–649
Zenclussen AC (2006) Regulatory T cells in pregnancy. Springer Semin Immunopathol 28:31–39
Paeschke S, Chen F, Horn N, Fotopoulou C, Zambon-Bertoja A, Sollwedel A, Zenclussen ML, Casalis PA, Dudenhausen JW, Volk HD, Zenclussen AC (2005) Pre-eclampsia is not associated with changes in the levels of regulatory T cells in peripheral blood. Am J Reprod Immunol 54:384–389
Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE (2006) TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood 108:253–261
Mantovani A, Bussolino F, Introna M (1997) Cytokine regulation of endothelial cell function: from molecular level to the bedside. Immunol Today 18:231–240
Rusterholz C, Gupta AK, Huppertz B, Holzgreve W, Hahn S (2005) Soluble factors released by placental villous tissue: interleukin-1 is a potential mediator of endothelial dysfunction. Am J Obstet Gynecol 192:618–624
Austgulen R, Lien E, Vince G, Redman CW (1997) Increased maternal plasma levels of soluble adhesion molecules (ICAM-1, VCAM-1, E-selectin) in preeclampsia. Eur J Obstet Gynecol Reprod Biol 71:53–58
Heyl W, Handt S, Reister F, Gehlen J, Mittermayer C, Rath W (1999) The role of soluble adhesion molecules in evaluating endothelial cell activation in preeclampsia. Am J Obstet Gynecol 180:68–72
Takacs P, Green KL, Nikaeo A, Kauma SW (2003) Increased vascular endothelial cell production of interleukin-6 in severe preeclampsia. Am J Obstet Gynecol 188:740–744
Scalera F, Schlembach D, Beinder E (2001) Production of vasoactive substances by human umbilical vein endothelial cells after incubation with serum from preeclamptic patients. Eur J Obstet Gynecol Reprod Biol 99:172–178
Winn HN, Todd HM, Amon E, al Malt A, Molnar M, Hertelendy F (1997) Effects of serum from preeclamptic women on prostacyclin production by human endothelial cells. J Matern Fetal Med 6:249–253
Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111:649–658
Levine RJ, Karumanchi SA (2005) Circulating angiogenic factors in preeclampsia. Clin Obstet Gynecol 48:372–386
Groten T, Kreienberg R, Fialka I, Huber L, Wedlich D (2000) Altered subcellular distribution of cadherin-5 in endothelial cells caused by the serum of pre-eclamptic patients. Mol Hum Reprod 6:1027–1032
Donker RB, Asgeirsdottir SA, Gerbens F, van Pampus MG, Kallenberg CG, te Meerman GJ, Aarnoudse JG, Molema G (2005) Plasma factors in severe early-onset preeclampsia do not substantially alter endothelial gene expression in vitro. J Soc Gynecol Investig 12:98–106
Mellembakken JR, Aukrust P, Olafsen MK, Ueland T, Hestdal K, Videm V (2002) Activation of leukocytes during the uteroplacental passage in preeclampsia. Hypertension 39:155–160
von Dadelszen P, Hurst G, Redman CW (1999) Supernatants from co-cultured endothelial cells and syncytiotrophoblast microvillous membranes activate peripheral blood leukocytes in vitro. Hum Reprod 14:919–924
Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S (2005) Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol 66:1146–1154
Acknowledgments
This work was supported in part by a grant form the Swiss National Science Foundation (No. 3200-107625).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rusterholz, C., Hahn, S. & Holzgreve, W. Role of placentally produced inflammatory and regulatory cytokines in pregnancy and the etiology of preeclampsia. Semin Immunopathol 29, 151–162 (2007). https://doi.org/10.1007/s00281-007-0071-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-007-0071-6