
Abstract
Neurotrophic factors are operationally defined as molecules that promote the survival and differentiation of neurons. Chemically, they belong to divergent classes of molecules but most of the classic neurotrophic factors are proteins. Together with stem cells, viral vectors and genetically engineered cells, they constitute important tools in neuroprotective and regenerative neurobiology. Protein neurotrophic molecules signal through receptors located on the cell membrane. Their downstream signaling exploits pathways that are often common to chemically different factors and frequently target a relatively restricted set of transcription factors, RNA interference and diverse molecular machinery involved in the life vs. death decisions of neurons. Application of neurotrophic factors with the aim of curing or, at least, improving the outcome of neurodegenerative diseases requires (1) profound knowledge of the complex molecular pathology of the disease, (2) the development of animal models as closely as possible resembling the human disease, (3) the identification of target cells to be addressed, (4) intense efforts in chemical engineering to ensure the stability of molecules or to design carriers and small analogs with the ability to cross the blood–brain barrier and (5) scrutinity with regard to possible side effects. Last, but not least, engineering efforts to optimize administration, e.g., by designing the right canulae and infusion devices, are important for the successful translation of preclinical advances into clinical benefit. This article presents selected examples of neurotrophic factors that are currently being tested in animal models or developed for transfer to the clinic, with a major focus on factors with the potential of becoming applicable in various forms of retinal degeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The concept of neurotrophic factors
The concept of neurotrophic factors can be traced back to the discoveries of a phenomenon and a molecule, i.e., ontogenetic neuron death and the first neurotrophic factor, nerve growth factor (NGF; Hamburger and Oppenheim 1982; Cowan 2001; Aloe 2004). The neurotrophic hypothesis postulates that developing post-mitotic neurons in the peripheral nervous system are generated in excess and compete during well-defined time windows for survival-promoting molecules, which are synthesized in limited amounts in the neuronal target tissue. These survival-ensuring molecules bind to specific receptors on the neuronal membrane, are internalized into axon terminals and transported retrogradely to the cell body, where they regulate translational and transcriptional events resulting in survival and differentiation. As a consequence of the limited availability of the factors in the peripheral targets, a defined number of neurons undergoes apoptotic death (Oppenheim 1991; Lewin and Barde 1996; Perez-Polo 2006; Pong Ng et al. 2006). However, competition for neurotrophic factors as a survival-determining mechanism of developing neurons does not seem to account for neurons of the central nervous system (CNS) in general. For example, the developmental death of cortical interneurons, which originate far from the cerebral cortex, has been shown to be an intrinsically determined process that occurs without interference from the target (Southwell et al. 2012).
Discoveries made during the past two decades have not only added many new facets to the classic neurotrophic factor hypothesis but also revealed an initially unexpected complexity in the number of neurotrophically acting molecules, their receptors and diversity of functions. As shown in Fig. 1, anterograde trophic signaling and local actions of neurotrophic factors have been added to the classic retrograde pathway for mediating trophic effects (Korsching 1993). Glial cells have been acknowledged as important sources and targets of neurotrophic factors and mediators of indirect neurotrophic effects (von Bohlen und Halbach and Unsicker 2013). Similarly, neurons have been recognized as a site of origin of neurotrophic factors and different neurotrophic factors have been shown to exert overlapping, yet distinct patterns of activity and divergent functions when acting in different contexts (Korsching 1993). The actions of neurotrophic factors on synaptic efficacy and the regulation of their expression by electrical activity have indicated that they play crucial roles in regulating neuronal connectivity (Lewin and Barde 1996). Perhaps most strikingly, endogenous NGF has been shown to act as a death-promoting factor when signaling through the non-tyrosine kinase receptor p75, a member of the tumor necrosis factor (TNF) receptor and Fas family (Frade et al. 1996; Lu et al. 2005). In a similar vein, the neurotrophin receptors TrkA (tropomyosin-related kinase A) and TrkC but not TrkB, have been revealed to cause neuronal death, thus explaining why developing sympathetic and sensory neurons become trophic-factor-dependent for survival (Nikoletopoulou et al. 2010). In summary, the broad repertoire of functions that neurotrophic factors possess and their contextualities in cooperation with other factors have to be taken into account when considering their application in therapies of neurodegenerative disease.

Sources and directions of signaling of neurotrophic molecules. A The “classic” retrograde signaling pathway involves a target cell (here: a neuron) as the source of the molecule, the release of the molecule, its binding to an axon terminal and its retrograde transport to the neuronal perikaryon. B Glial cells, including Schwann cells, astrocytes, oligodendrocytes and microglia, can synthesize and release neurotrophic factors. C Autocrine mode of neurotrophic factor stimulation. D Anterograde signaling of neurotrophic factors. Modified from Korsching (1993)
The spectrum of neurotrophic factors
Neurotrophins and their receptors
Neurotrophins, the classic neurotrophic factor family, comprise NGF, brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3) and NT-4 (also known as NT-5), NT-6 and NT-7 (Huang and Reichardt 2001). They probably arose through successive duplications of the genome of an ancestral chordate (Hallbrock 1999). The NT-6 and NT-7 genes are only found in fish and do not seem to have mammalian orthologs. The structures of neurotrophins have been solved and several features of their structures, e.g., the cystine knot, are present in other growth factors (McDonald et al. 1991; McDonald and Chao 1995). A receptor now named p75 was the first identified neurotrophin receptor. It was initially believed to be a low-affinity receptor for NGF but was later shown to bind all neurotrophins with similar affinities (Rodriguez-Tebar et al. 1991). The cytoplasmic domain of p75 contains a “death” domain shared with other members of the TNF receptor family, thereby eliciting death when activated separate from the trk neurotrophin receptors. The Trk family of neurotrophin receptors comprises TrkA, which is preferentially activated by NGF, TrkB, the key receptor for BDNF and NT-4 and TrkC, preferentially used for NT-3 signaling (Reichardt 2006). Phosphorylation of the tyrosines within the Trk kinase domain and docking of adaptor proteins are indispensible for triggering several intracellular signaling pathways, including the Ras-mitogen-activated protein kinase, phosphatidyl inositol 3 (PI3)-kinase and phospholipase C-gamma pathways (Fig. 2; cf. review by Pong Ng et al. 2006).

Representation of neurotrophin (NT) signaling (Trk tropomyosin-related kinase, PI3K phosphatidyl inositol 3-kinase, Shc Src homology 2 domain containing, Gab2 GRB2-associated binding protein 2, Shp2 tyrosine phosphatase, Akt protein kinase B, Ras rat sarcoma protein, Raf rapidly accelerated fibrosarcoma protein, Rap Ras-related protein, MEK mitogen-activated protein kinase kinase, ERK extracellular-signal-regulated kinase, PLC phospholipase C)
BDNF is the most abundant neurotrophin in the brain (Barde et al. 1982) and seems to exist as pro-BDNF and “mature” BDNF. “Mature” BDNF is the predominant form and is crucially involved in physiological processes in the intact adult brain, including morphological and functional synaptic plasticity, long-term potentiation, learning and memory. Because of its wide distribution in the CNS, it is the neurotrophin with the largest therapeutic potential in CNS disorders, such as Alzheimer’s, Parkinson’s and Huntinton’s disease, stroke, depression and metabolic disorders (reviewed by Nagahara and Tuszynski 2011).
BDNF and NGF in the treatment of neurodegenerative diseases
Stroke is the most common neurological disorder affecting mostly the cerebral cortex. The grafting of a BDNF-expressing cell line or the intravenous systemic injection of BDNF has been shown to reduce neuron death after middle cerebral artery occlusion and photothrombotic ischemia, respectively (Ferrer et al. 2001; Müller et al. 2008). However, major obstacles to treatment, as for other widespread brain lesions, are the delivery of sufficiently high concentrations of stable BDNF to the lesioned areas over long periods of time and the putative adverse effects. Drugs, e.g., antidepressants and AMPA (2-amino-3-[3-hydroxy-5-methyl-isoxazol-4-yl]propanoic acid) receptor agonists, which increase the endogenous expression of BDNF, might turn out to be useful in BDNF therapies. Moreover, small molecule BDNF mimetics that bind to TrkB and trigger downstream signaling have been shown to be useful therapeutic drugs in initial studies (Massa et al. 2010; Jang et al. 2010; Monteggia 2011). Whether gene therapy as a tool to deliver BDNF can become an established method and a reliable therapeutic alternative in widespread CNS lesion paradigms remains to be investigated.
Treatment of Alzheimer’s disease with BDNF basically implies similar challenges concerning delivery. Both BDNF gene delivery and protein administration to the entorhinal cortex, a key region in the initiation of Alzheimer’s disease (Braak and Braak 1991; Kordower et al. 2001), have been successfully used in rodent and primate models of the disease. Therapeutic improvements include those to memory, cell survival, cell body size and extracellular-signal-regulated kinase (ERK) signaling and normalized gene expression but no change in amyloid plaques (Nagahara et al. 2009; Nagahara and Tuszynski 2011). Significant improvement in the rate of cognitive decline has been reported in six patients suffering from Alzheimer’s disease following implantation of autologous fibroblasts genetically modified to express human NGF into the forebrain (Tuszynski et al. 2005). No adverse effects have been observed in this study. A link between beneficial neural stem cell transplantation and BDNF secretion by the grafted hippocampal neural stem cells in a transgenic model of Alzheimer’s disease has been reported by Blurton-Jones et al. (2009). Grafts rescue spatial learning and memory deficits, effects that are mimicked by recombinant BDNF and abolished when BDNF is depleted from the neural stem cells. Together, these results are sufficiently encouraging for the further exploration of the therapeutic potential of BDNF and NGF and the collection of additional sets of data to support their efficacy and safety profile with the goal of initiating clinical trials.
Parkinson’s disease is a chronic progressive neurodegenerative disease that afflicts more than five million people worldwide. Degeneration of midbrain dopaminergic neurons in the substantia nigra and subsequent depletion of dopamine in the striatum, together with the presence of Lewy bodies containing alpha-synuclein in the surviving substantia nigra dopaminergic neurons, are pathological hallmarks of the disease. Current understanding of the “idiopathic” disease is incomplete, despite considerable advances in understanding the pathogenesis of familial forms of Parkinson’s disease. Although both genetic and lesion-based animal models are available, models that recapitulate the chronic disease in detail are still lacking (however, see Decressac et al. 2011, 2012a, b). Replacement of dopamine is the currently available symptomatic therapy. Although physiological roles of neurotrophic factors in dopaminergic neurons of the aging midbrain are not comprehensively understood, several members of the neurotrophin family and other growth factor families have been tested in animal models and administered in patients (Aron and Klein 2011). BDNF is one of several growth factors that promote the survival of both cultured and in-vivo-lesioned midbrain dopaminergic neurons (Hyman et al. 1991). Substantial evidence suggests that BDNF infused or released from genetically engineered fibroblasts protects dopaminergic neurons in the substantia nigra from toxic insults (Levivier et al. 1995; Frim et al. 1994; Tsukahara et al. 1995). Whether inhibitors of the type B monoamine oxidase (MAO), namely rasagiline, selegiline and (−)deprenyl, prevent dopaminergic neuron loss by increasing the expression of BDNF and putatively of other neurotrophic factors is the subject of controversy (Maruyama and Naoi 2012; Weinreb et al. 2007). As shown by Flannery et al. (2010), selegiline increases BDNF content in the striatum by 32% but this change does not reach statistical significance.
Details of the application of BDNF in other neurodegenerative diseases such as Huntingtons’s disease, amyotrophic lateral sclerosis (ALS) and spinal cord injury, can be found in Nagahara and Tuszynski (2011).
Neurotrophins in ocular disease
In the normal adult retina, the NGF receptor TrkA is expressed in retinal ganglion cells (RGCs), whereas p75 is found in Müller glia (Bai et al. 2010). Injury of the retina causes the upregulation of TrkA, p75 and NGF. However, neither endogenous NGF nor exogenously administered NGF can ultimately protect degenerating RGCs. The selective activation of TrkA with a mutant NGF or the prevention of endogenous NGF and pro-NGF from binding to p75 protect RGCs in glaucoma or following optic nerve transection. Selective activation of p75 causes RGC death in normal eyes and accelerates RGC death after injury. The activation of p75 during glaucoma-driven retinal degeneration has been suggested to enhance the synthesis and release of neurotoxic molecules, e.g., TNF-α (Bai et al. 2010). Although the complete details of the retinal NGF and NGF receptor networks are currently not understood, increasing evidence indicates that a fragile balance of ligand and receptors needs to be achieved for the protection of RGCs (Lebrun-Julien et al. 2009; Sposato et al. 2008; Coassin et al. 2008). Somewhat surprisingly, NGF administered to the cornea has been shown to protect RGC in models of glaucoma and diabetic retinopathy (Colafrancesco et al. 2011; for a review of NGF in ocular diseases, see Lambiase et al. 2011). Whereas such results are certainly spectacular, they should be rigorously tested not only with regard to the transport of iodinated NGF by using the full spectrum of available molecular and cell biological methodologies.
RGCs also express TrkB (Fischer 2012) and so do several other neuronal cell types in the retina. A vast literature documents that BDNF delays RGC death after axotomy and in several other lesion models. Many modes of application have been used, including stem-cell-based delivery (Harper et al. 2011; S.H. Park et al. 2010; H.Y. Park et al. 2012), recombinant adeno-associated viral vectors (Rodger et al. 2012; Ren et al. 2012; Isenmann et al. 1998), adenovirus-infected Müller cells (Di Polo et al. 1998) and combinatorial treatment with other factors, e.g., the leucine-rich repeat protein LINGO-1 (Fu et al. 2009). TrkB-mediated protection from light has also been documented following the use of N-acetylserotonin (Shen et al. 2012). BDNF gene delivery has also been shown to protect the structure and function of light-damaged photoreceptors (Gauthier et al. 2005). Expression of TrkB by RGC also indicates that NT-4 is a candidate for protecting against retina injury (Peinado-Ramon et al. 1996; Watanabe et al. 1997). However, we need to take into account that, when administered for therapeutic purposes, BDNF/TrkB signaling not only stimulates the growth of axonal branches of RGC (Sawai et al. 1996) but can also profoundly refine RGC dendritic arborization and connectivities (Liu et al. 2007). Finally, two more caveats should be pointed out: first, the manipulation of growth factor expression almost inevitably affects the expression of other growth factors, as exemplified by the heterozygote BDNF+/− retina with its significantly increased levels of glial-cell-line-derived neurotrophic factor (GDNF; Wilson et al. 2007); second, BDNF is anterogradely transported by RGC to the superior colliculus and lateral geniculate (Caleo et al. 2000; Bartheld 1998) and therefore, its overexpression in the eye will probably have an impact on these structures and further distal parts of the visual pathway.
NT-3 has been less extensively studied in the retina and RGCs, which express the cognate receptor trkC (Fischer 2012). During development, NT-3 and trkC expression is widespread in retinal neuroepithelial progenitor cells (Das et al. 2000). Later, in a mouse strain carrying the reporter gene LacZ, NT-3 is transiently restricted to a small subset of cells in the inner nuclear and ganglion cell layers, whereas in the adult mouse eye, NT-3 expression is confined to the corneal epithelium (Bennet et al. 1999). In a model of open angle glaucoma, levels of retinal NT-3, in contrast to trkC and NGF, remain unchanged (Rudzinski et al. 2004). Peinado-Ramon et al. (1996) have found that the intraocular administration of NT-3 does not modify the survival of RGCs after injury. Together, these data do not suggest that NT-3 is a promising candidate for the treatment of RGC injury.
As a general note, for the screening of molecules with a potential neuroprotective capacity for RGCs, retinal explant models have been developed and can be used for efficient medium-throughput screening (Bull et al. 2011).
Neuroregulatory cytokines—the ciliary neurotrophic factor (“neurokine”) family
Ciliary neurotrophic factor (CNTF) was the first purified and cloned non-neurotrophin neurotrophic factor (Stöckli et al. 1989; for a review, see Halvorsen and Knaur 2006). It was the founding member of a gene family that shares a gp130 receptor subunit with other neuroregulatory cytokines, including leukemia inhibitory factor (LIF), interleukin-6 (IL-6), cardiotrophin-1 and −2 (CT-1, CT-2), oncostatin-M and neuropoietin (Fig. 3). Their biological actions are diverse and include prominent roles in the hematopoietic system and in the nervous system. Their signaling is mediated through the Janus tyrosine kinase and activator of transcription (JAK/STAT) pathway. Whereas none of the family members appears to be essential by itself, the deletion of each of the individual receptor subunits is not compatible with life. CNTF and LIF gene knockouts result in modest motoneuron defects, which are potentiated in CNTF/LIF-double and CNTF/LIF/CT-1 triple knockouts (cf. Holtmann et al. 2005).

Members of the ciliary neurotrophic factor (CNTF) family and their cognate receptors (LIF leukemia inhibitory factor, IL interleukin, CLC cardiotrophin-like cytokine, NP neuropoietin, CT cardiotrophin, OSM oncostatin-M, R receptor). Modified from Halvorsen and Knaur (2006)
Neuroregulatory cytokines in the treatment of neurodegenerative diseases
CNTF and related cytokines play many roles in neurodegenerative diseases and trauma. They can be induced or can promote the injury response including the survival of injured cells. Hippocampal and entorhinal cortex lesions are accompanied by the increased expression of CNTF and CNTF-α-receptor by astrocytes (C.K. Park et al. 2000; Lee et al. 1997). Several animal models of motor neuron disease, including pmn and Wobler mice, have revealed responses to exogenous and endogenous neuroregulatory cytokines (Sendtner et al. 1992, 1997; Mitsumoto et al. 1994). Hopes that CNTF might be effective in ALS have not been substantiated (ALS study group 1996). However, encouraging data have been reported from experiments in an Alzheimer’s disease mouse model with recombinant cells secreting CNTF encapsulated in alginate polymers (Garcia et al. 2010).
CNTF and related molecules in ocular disease
The neurokine family and its receptors are well represented in the retina. CNTF is expressed in pigment epithelium and Müller glia (Finn and Nishi 1996; Walsh et al. 2001). RGCs express gp130, CNTF receptor alpha and LIF receptor β (Fischer 2012), suggesting that they are responsive to the corresponding ligands. In culture, CNTF and LIF delay rod cell development (Kirsch et al. 1998) and CNTF redirects rods to bipolar, amacrine and Müller glia cells (Ezzedine et al. 1997). The effect of intraocularly administered CNTF on the viability of RGCs is controversial (Cui et al. 1999; Barnstable and Tombran-Tink 2006). Despite a lack of effect on RGC survival following optic nerve transection and insertion of a sciatic nerve graft, as reported in this study, the intravitreal application of CNTF substantially enhances regeneration (Cui et al. 1999). CNTF also remarkably prevents RGC loss in a rat model of posterior ischemic optic neuropathy (Wang et al. 2012). In combination with lens injury or intravitreal application of zymosan, RGCs switch into an active regenerative state. The promoting effect on survival and axon regeneration is mediated by CNTF (Leibinger et al. 2009). The axon-growth-promoting effect of lens crystallins is also generated by enhancing the production of CNTF (Thanos et al. 2012). In a search for mechanisms underlying the axon-regenerative capacity of CNTF, the investigation of a possible involvement of STAT3, which has been shown to act locally in axons of motoneurons to modify the tubulin cytoskeleton (Selvaraj et al. 2012), will be important. Interestingly, exogenous CNTF also induces endogenous CNTF expression in glial cells (Müller et al. 2009). In agreement with the established effects of CNTF on photoreceptors (see above), intravitreal injections of a human CNTF analog (Axokine, Regeneron) significantly delays photoreceptor loss in a feline model of rod-cone dystrophy (Chong et al. 1999). Several recent reports corroborate the notion of CNTF being a potent trophic molecule for photoreceptors. End-stage photoreceptor degeneration has been successfully treated with CNTF in many animal models and in humans (Wen et al. 2012a, b). Application by encapsulated cell technology might be preferred following the documentation of delivery over a period of 2 years and a favorable pharmacokinetic profile (Kauper et al. 2012). With a perspective towards using CNTF therapeutically, we should however note that long-term gene therapy has been reported to cause aberrant dendritic RGC morphology and stratification (Rodger et al. 2012). With regard to the efficacy of other members of the neurokine family on RGCs, IL-6, which is upregulated in injured RGCs in experimental glaucoma, potently stimulates axon regeneration of RGCs in culture (Caraci et al. 2012).
The fibroblast growth factor family
The fibroblast growth factor (FGF) family comprises 23 members each of which signals via four tyrosine kinase receptors (FGFR1-FGFR4) that exist in many splice variants (for a review, see Unsicker et al. 2006). Since the first reports of the neurotrophic functions of FGF-2 in the late 1980s, numerous other functions have been discovered for FGFs in the nervous system in which more than a dozen FGF family members are expressed. FGF-2 is primarily synthesized by astrocytes, whereas other FGF members (FGF-1, -5, -8, -9, -10, -15, -18) are predominantly synthesized by neurons. Similar to the postulated functions of other growth factor families, the neural functions of FGFs are often extrapolations from pharmacological experiments and applications of exogenous FGFs. Although a loss of cortical neurons in FGF-2-deficient mice might suggest a role of this FGF as a neurotrophic factor (Dono et al. 1998), the activities of FGF-2 in relation to the generation and migration of neurons place a strong caveat on such an interpretation. This also applies to another widely popularized function of FGF-2, i.e., its assumed role in adult neurogenesis (Werner et al. 2011).
FGFs in neurodegenerative disease
Mitogenic and differentiative effects on astrocytes and neurotrophic survival-promoting effects are the earliest described neural functions of FGFs, most notably of FGF-2 (cf. Bieger and Unsicker 1996; Unsicker et al. 1987). Several distinct mechanisms seem to account for the neurotrophic functions of FGF-2, including interference with neurotransmitter receptors and combinatorial actions with other growth factors (Unsicker et al. 2006). The in vivo capacity of FGFs to protect lesioned neurons and to orchestrate responses to lesions has been widely documented (Unsicker et al. 2006). Probably the most intensely studied neurons protected by FGFs are midbrain dopaminergic neurons in the substantia nigra (Otto and Unsicker 1990, 1993a, b; von Bohlen und Halbach and Unsicker 2013). Within the abundance of reports documenting enhanced survival of impaired neurons as a prominent function of FGFs, the loss of FGF-2 accounting for the prolonged survival and milder impairment of motor function in an SOD1 ALS mouse model has come as a surprise (Thau et al. 2012), underscoring the complexity of the growth factor networks involved in neurodegenerative and regenerative events. Several neurodegenerative diseases, as exemplified by Huntington’s disease, seem to benefit from the neuroprotective and neuroproliferative capacities of FGF-2 (Jin et al. 2005). Even the most complex psychiatric disorders, e.g., major depression, seem to result from the imbalanced expression of FGFs and apparently respond to endogenous and exogenous FGF (Riva et al. 2005; Jarosik et al. 2011; Turner et al. 2012).
FGFs in ocular disease
FGF has well-documented protective effects on the viability of photoreceptors and RGCs (Cui et al. 1999; Joly et al. 2007). The capacity of FGF-1 and FGF-2 to protect RGCs following transection of the optic nerve was the first neural lesion paradigm in which the neurotrophic potential of these FGFs was documented in vivo (Sievers et al. 1987). Mechanistically, FGF-2 has been shown to increase Bcl-2 and to decrease Bax in RGCs by activating the ERK pathway (Rios-Munoz et al. 2005). In addition to FGF-1 and FGF-2, other members of the FGF family have established their potential to rescue photoreceptors and RGCs in models of retinal degeneration. For example, FGF-5 and FGF-18 rescue photoreceptors from apoptosis in a recombinant adeno-associated virus-mediated paradigm of retinitis pigmentosa (Green et al. 2001). Similarly, FGF-19 has been shown to protect adult mammalian photoreceptors (Siffroi-Fernandez et al. 2008). When considering FGFs for therapy, small molecules, e.g., Enreptin, which activate both FGF receptors and the FGF co-receptor NCAM (neural cell adhesion molecule), might have distinct advantages (Ernevoldsen et al. 2012). However, when administering FGF for therapeutic purposes, the mitogenic and angiogenic properties of FGF always constitute obstacles that can probably only be overcome by developing FGF agonists lacking these side effects.
The transforming growth factor family
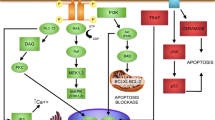
The transforming growth factor-β family (TGF-β) comprises more than 30 secreted proteins that regulate numerous developmental processes, control tissue homeostasis and mediate repair (Böttner et al. 2000; De Robertis and Kuroda 2004; Derynck and Zhang 2003;Derynck and Akhurst 2007; Krieglstein et al. 2011). A major function of TGF-β related to this Special Issue is the control of neuron survival (Unsicker and Krieglstein 2002; Krieglstein 2006). However, since both neurons and glial cells express TGF-βs and the congnate receptors, functions of TGF-βs are extremely diverse and difficult to extrapolate (Krieglstein 2006). More recently, their involvement in synaptic functions have begun to come to light (Krieglstein et al. 2011). TGF-β members play crucial roles in diseases including neurodegenerative disorders (Flanders et al. 1998); as typical, contextually acting, growth factors, they do so by diverse cross-talk with other growth factors (Ikushima and Miyazono 2012; Massagué 2012). TGF-βs are almost ubiquitously expressed, with all three isoforms TGF-β1, β2 and β3 being found in the CNS. Furthermore, members of several TGF-β subfamilies, e.g., bone morphogenetic proteins (BMPs), growth/differentiation factors (GDFs), activins/inhibins and GDNFs (GDNF, neurturin, artemin, persephin), are widespread in the nervous system. With the exception of GDNF and its relatives, which employ the tyrosine kinase Ret and alpha receptors (GFLalpha1–4) for signaling, they communicate via membrane serine/threonine kinase complexes and use both Smad and non-Smad signaling pathways (Peterziel and Strelau 2006; Heldin et al. 2009; Heldin and Moustakas 2012; Mu et al. 2012). TGF-βs activate numerous immediate early genes that have been implicated in regulating apoptosis and cell cycle, e.g., Klf10 and Klf11 (Spittau and Krieglstein 2012) and TAK1, a TGF-β activated mitogen-activated protein-3 kinase involved in at least five signaling cascades that modulate ischemic brain damage (Ridder and Schwaninger 2012). The signaling network activated by TGF-β is depicted in Fig. 4.

Network of TGF-β-induced genes. Modified from Wendt et al. (2012)
TGF-βs in neurodegenerative diseases
Since TGF-βs are pleiotropic growth factors with neurotrophic and immunosuppressive properties targeting the entire spectrum of neuronal, glial and inflammatory cells, they unsurprisingly play a leading role in all neurodegenerative diseases including Alzheimer’s, Parkinson’s and Huntington’s disease, multiple sclerosis, stroke, neurodepressive disorders and others (Dobolyi et al. 2012). This is best underscored by studying responses to neural lesions in mice lacking distinct TGF-βs (cf. Makwana et al. 2007). In the human Alzheimer brain, levels of TGF-β receptor type II (TβRII) typically expressed on neurons are reduced and correlate with the pathological hallmarks of the disease (Tesseur et al. 2006). Impairment of TGF-β signaling in Alzheimer’s disease implies that the targeting of the neuroprotective TGF-β in this disease might slow down the neurodegenerative processes (Caraci et al. 2012; Wyss-Coray 2006). Midbrain dopaminergic neurons are established target cells for the trophic actions of members of the TGF-β family, most notably the GDNF family ligands (Krieglstein 2006; Roussa et al. 2009; Aron and Klein 2011; Sidorova et al. 2010; Airkasinen and Saarma 2002). Current translational studies are focusing on the development of neurturin for the treatment of Parkinson’s disease (Marks et al. 2010; Bartus et al. 2011, 2012; Bartus 2012). Many studies have revealed the involvement of TGF-βs in the de- and regenerative processes following brain ischemia and stroke (Beck and Schachtrup 2012).
TGF-βs in ocular disease
Among the most stunning effects exerted by TGF-βs is the induction of developmental neuron death in the peripheral nervous system and CNS, including the chick and mouse retina (Krieglstein et al. 2000; Dünker et al. 2001; Dünker and Krieglstein 2003). Immunoneutralization of all three TGF-β isoforms in the chick embryo or deletion of the TGF-β2 and –β3 genes in mice results in the lack of cell death in the peripheral ganglia, spinal cord and retina. Furthermore, neuron death following limb bud ablation in the chick embryo is markedly reduced (Krieglstein et al. 2000). The relevance of these observations in chick and mouse embryos for the biology of adult normal and diseased retinal neurons remains to be explored. As has long been known, detachment of the retina induces the synthesis and secretion or TGF-β2, thereby increasing levels of TGF-β and TβR in Müller cells (Guérin et al. 2001). This might result in the retinal gliotic response and subsequent RGC death. Elevated levels of TGF-β2 are also found in the aqueous humor and reactive optic nerve astrocytes in primary open angle glaucoma (Fuchshofer and Tamm 2012). Available evidence indicates that TGF-β2 contributes to changes in the ECM of the trabecular meshwork and optic nerve head and possibly also to compression of optic nerve axons.
GDNF has been shown to attenuate significantly the degeneration of RGCs in vivo in a dose-dependent fashion (Yan et al. 1999; see also Cui et al. 1999); GDNF also highly effectively rescues photoreceptor function and survival in an animal model of retinal degeneration (Frasson et al. 1999). With regard to underlying mechanisms, photoreceptors interestingly lack the GDNF receptors GFRalpha1 and RET, which, however, are expressed on Müller glial cells. RGCs express the receptors for the GDNF family members neurturin and artemin. The trophic effect of GDNF signaling results from the induction of FGF-2 in Müller glia (Hauck et al. 2006) providing an example for the importance of glia in providing neurotrophic factors. In this context, investigations seem worthwhile into whether the robust survival-promoting effect of GDNF on RGCs can be mediated by a similar indirect mechanism (Yan et al. 1999).
Insulin-like growth factor family
Members of the insulin-like growth factor (IGF) family including IGF-1 and IGF-2 promote cell growth and survival by regulating carbohydrate, lipid and protein metabolism (Bondy et al. 2006). IGF-1 and its receptors are highly expressed in the developing brain and their deletion retards brain growth, diminishes cell size, dendritic arbors and numbers of synapses and causes mental retardation in humans. Exogenously applied IGF-1 can protect injured neurons from cell death. The anabolic effects of IGF-1 are mediated through the PI3K (phosphatidyl inositol 3-kinase)-Akt (also known as protein kinase B) signaling cascade.
The neuroprotective role of IGF-1 has been challenged by the recent analysis of IGF-1-receptor-deficient mice (Torres Aleman 2012). Even so, IGF-1 has been shown to ameliorate hippocampal neurodegeneration in an animal model of temporal lobe epilepsy (Miltiadous et al. 2011) and motor neuron death in type III SMA mice (Tsai et al. 2012). Furthermore, IGF-1 exerts robust protection of cultured neurons against prion peptide-induced death by blocking ROS formation and the translocation of Bax to mitochondria (Y.G. Park et al. 2012). Additional mechanistic studies are required for defining the precise roles of IGF-1 in neurodegenerative diseases.
Several studies have indicated that IGF-1 can protect retinal photoreceptors and axotomized RGCs from death in a variety of lesion paradigms. For example, transcorneal electrical stimulation has been shown to rescue axotomized RGCs by activating the endogenous retinal IGF-1 system (Morimoto et al. 2005). Physiological protection of rod photoreceptors against light stress employs the activation of the IGF-1 receptor and the Akt survival pathway (Dilly and Rajala 2008). In the rd10 mouse model of retinitis pigmentosa, IGF-1 is able to attenuate photoreceptor cell death (Arroba et al. 2011). The pivotal role of IGF-1 in the retina is also underscored by its capacity to induce the increased secretion of vascular endothelial growth factor by the retinal pigment epithelium underlying choroidal neovascularization in age-related macular degeneration (Economous et al. 2008; Cordeiro et al. 2010).
Other growth factor with putative relevance for neuroprotection of RGCs
Additional growth factors and their receptors are expressed in the retina including RGCs but their functions and benefit for neuroprotection have not been systematically studied. For example, RGCs express the hepatocyte growth factor receptor c-Met and the granulocyte macrophage colony stimulating factor alpha receptor; both mediate protection of RGC when activated with their respective ligands (Fischer 2012). In addition, an increasing list of growth factors, e.g., platelet-derived growth factor (Tang et al. 2010) and pituitary adenylate cyclase activiating polypeptide (Atlasz et al. 2010; Seaborn et al. 2011) are being shown to protect against apoptosis in ischemia-induced retinal damage. Beyond the field of growth factors for which receptors have been shown to be expressed in the retina, other factors for which receptors have not been identified are emerging and give stunning neuroprotection in other lesion models. Examples for this category of growth factors that should be mentioned here are the mesencephalic astrocyte-derived neurotrophic factor (MANF; Lindholm et al. 2008) and the conserved dopamine neurotrophic factor (CDNF; Lindholm et al. 2007).
Concluding remarks
In view of the available evidence, cautious optimism is probably the correct attitude to be taken towards the applicability of growth factors for the treatment of neurodegenerative diseases. Among the more than 50 growth factors expressed in the nervous system (Johnson and Tuszynski 2008), only a few have emerged as promising candidates for potential treatments in neurodegenerative diseases. These include BDNF, the TGF-βs GDNF and neurturin and IGF-1. However, significant gaps still have to be filled, from understanding disease pathologies to generating optimal animal models and applicable drugs. Pitfalls along the road to effective growth factor therapies are frequent and even occur on what is considered to be firm ground: a recent example is the observation that GDNF fails to exert neuroprotection in a rat alpha-synuclein model of Parkinson’s disease, a finding that brings into question all previous interpretions of results obtained in preclinical models of this disease (Decressac et al. 2011, 2012a, b). Finally, therapeutic break-throughs can hardly be expected from the administration of a single growth factor or its mimetics: apparently, biology does not work this way. Hence, combinatorial approaches have to be explored more systematically and have to involve survival factors, axon growth promoting molecules and agents to suppress adverse side effects (McCall et al. 2012).
References
Airkasinen MS, Saarma M (2002) The GDNF family: signaling, biological functions and therapeutic value. Nat Rev Neurosci 3:383–394
Aloe L (2004) Rita Levi-Montalcini: the discovery of nerve growth factor and modern neurobiology. Trends Cell Biol 14:395–399
ALS CNTF treatment study group (1996) A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. Neurology 46:1244–1249
Aron L, Klein R (2011) Repairing the Parkinsonian brain with neurotrophic factors. Trends Neurosci 34:88–100
Arroba AI, Alvarez-Lindo N, van Rooijen N, de la Rosa EJ (2011) Microglia-mediated IGF-1 neuroprotection in the red10 mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 52:9124–9130
Atlasz T, Szabadfi K, Kiss P, Tamas A, Toth G, Reglodi D, Gabriel R (2010) Evaluation of the protective effects of PACAP with cell-specific markers in ischemia-induced retinal degeneration. Brain Res Bull 81:497–504
Bai Y, Dergham P, Nedev H, Xu J, Galan A, Rivera JC, ZhiHua S, Mehta HM, Woo SB, Sarunic MV, Neet KE, Saragovi HU (2010) Chronic and acute models of retinal neurodegeneration TrkA activity are neuroprotective whereas p75NTR activity is neurotoxic through a paracrine mechanism. J Biol Chem 285:39392–39400
Barde Y-A, Edgar D, Thoenen H (1982) Purification of a new neurotrophic factor from mammalian brain. EMBO J 1:549–553
Bartheld CS (1998) Neurotrophins in the developing and regenerating visual system. Histol Histopathol 13:437–459
Bartus RT (2012) Translating the therapeutic potential of neurotrophic factors to clinical “proof of concept”: a personal saga achieving a career-long quest. Neurobiol Dis 48:153–178
Bartus RT, Brown L, Wilson A, Kruegel B, Siffert J, Johnson EM Jr, Kordower JH, Herzog CD (2011) Properly scaled and targeted AAV2-NRTN (neurturin) to the substantia nigra is safe, effective and causes no weight loss: support for nigral targeting in Parkinson's disease.Neurobiol Dis 44:38-52
Bartus RT, Baumann TL, Brown L, Kruegel BR, Ostrove JM, Herzog CD (2012) Advancing neurotrophic factors as treatments for age-related neurodegenerative diseases: developing and demonstrating "clinical proof-of-concept" for AAV-neurturin (CERE-120) in Parkinson's disease. Neurobiol Aging 34:35–61
Barnstable CJ, Tombran-Tink J (2006) Molecular mechanisms of neuroprotection in the eye. Adv Exp Med Biol 572:292–295
Beck K, Schachtrup C (2012) Vascular damage in the central nervous system: a multifaceted role for vascular-derived TGF-β. Cell Tissue Res 347:187–201
Bennet JL, Zeiler SR, Jones KR (1999) Patterned expression of BDNF and NT-3 in the retina and anterior segment of the developing mammalian eye. Invest Ophthalmol Vis Sci 40:2996–3005
Bieger S, Unsicker K (1996) Functions of fibroblast growth factors (FGFs) in the nervous system. In: Bell C (ed) Chemical factors in neural growth, degeneration, and repair. Elsevier, Amsterdam, pp 339-375
Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM (2009) Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer’s disease. Proc Natl Acad Sci USA 106:13594–13599
Böttner M, Krieglstein K, Unsicker K (2000) The transforming growth factor-βs: structure, signalling, and roles in nervous system development and functions. J Neurochem 75:2227–2240
von Bohlen und Halbach O, Unsicker K (2013) Storage and release of non-transmitter signalling molecules from macroglia. In: Kettenmann H, Ransom B (eds) Neuroglia, 3rd edn. Oxford University Press, New York, chapter 18, pp 212–222
Bondy C, Cheng C, Zhong J, Lee WH (2006) IGF-1 in brain growth and repair processes. In: Lajtha A (ed) Handbook of neurochemistry and molecular neurobiology. Neuroactive proteins and peptides. Springer, Berlin, pp 143–165
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 82:239–259
Bull ND, Johnson TV, DeKorver HW, Tomarev SI, Martin KR (2011) Use of an adult retinal explant model for screening of potential retinal ganglion cell neuroprotective therapies. Invest Ophthalmol Vis Sci 52:3309–3320
Caleo M, Menna E, Chierzi S, Cenni MC, Maffei L (2000) Brain derived neurotrophic factor is an anterograde survival factor in the rat visual system. Curr Biol 10:1155–1161
Caraci F, Spampinato S, Sortino MA, Bosco P, Battaglia G, Bruno V, Drago F, Nicoletti F, Chidlow G, Wood JP, Ebneter A, Casson RJ (2012) Interleukin-6 is an efficacious marker of axonal transport disruption during experimental glaucoma and stimulates neuritogenesis in cultured retinal ganglion cells. Neurobiol Dis 48:568–581
Chong NA, Alexander RA, Waters L, Barnett KC, Bird AC, Luthert PJ (1999) Repeated injections of a ciliary neurotrophic factor analogue leading to long-term photoreceptor survival in hereditary retinal degeneration. Invest Ophthalmol Vis Sci 40:1298–1305
Coassin M, Lambiase A, Sposato V, Micera A, Bonini S, Aloe L (2008) Retinal p75 and bax overexpression is associated with retinal ganglion cells apoptosis in a rat model of glaucoma. Graefes Arch Clin Exp Ophthalmol 246:1743–1749
Colafrancesco V, Coassin M, Rossi S, Aloe L (2011) Effect of eye NGF administration on two animal models of retinal ganglion cells degeneration. Ann Ist Super Sanita 47:284–289
Cordeiro S, Seyler S, Stindl J, Milenkovic VM, Strauss O (2010) Heat-sensitive TRPV channels in retinal pigment epithelial cells: regulation of VEGF-A secretion. Invest Ophthalmol Vis Sci 51:6001–6008
Cowan WM (2001) Victor Hamburger and Rita Levi-Montalcini; the path to the discovery of nerve growth factor. Annu Rev Neurosci 24:551–600
Cui Q, Lu Y, So KF, Yip HK (1999) CNTF, not other trophic factors promotes axonal regeneration of axotomized retinal ganglion cells in adult hamsters. Invest Ophthalmol Vis Sci 40:760–766
Das I, Sparrow JR, Lin MI, Shih E, Mikawa T, Hempstead BL (2000) TrkC signalling is required for retinal progenitor cell proliferation. J Neurosci 20:2887–2895
De Robertis EM, Kuroda H (2004) Dorsal-ventral patterning and neural induction in Xenopus embryos.Annu Rev Cell Dev Biol 20:285–308
Decressac M, Ulusoy A, Mattsson B, Georgievska B, Romero-Ramos M, Kirik D, Björklund A (2011) GDNF fails to exert neuroprotection in a rat alpha-synuclein model of Parkinson’s disease. Brain 134:2302–2311
Decressac M, Mattsson B, Björklund A (2012a) Comparison of the behavioural and histological characteristics of the 6-OHDA and α-synuclein rat models of Parkinson’s disease. Exp Neurol 235:306–315
Decressac M, Mattsson B, Lundblad M, Weikop P, Björklund A (2012b) Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of α-synuclein in midbrain dopamine neurons. Neurobiol Dis 45:939–953
Derynck R, Akhurst RJ (2007) Differentiation plasticity regulated by TGF-beta family proteins in development and disease.Nat Cell Biol 9:1000–1004
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425:577–584
Dilly AK, Rajala RV (2008) Insulin growth factor 1 receptor/PI3K/AKT survival pathway in outer segment membranes of rod photoreceptors. Invest Ophthalmol Vis Sci 49:4765–4773
Di Polo A, Aigner LJ, Dunn RJ, Bray GM, Aguayo AJ (1998) Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected Müller cells temporarily rescues injured retinal ganglion cells. Proc Natl Acad Sci USA 95:3978–3983
Dobolyi A, Vincze C, Pal G, Lovas G (2012) The neuroprotective functions of transforming growth factor beta proteins. Int J Mol Sci 13:8219–8258
Dono R, Texido G, Dussel R, Ehmke H, Zeller R (1998) Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J 17:4213–4225
Dünker N, Krieglstein K (2003) Reduced programmed cell death in the retina and defects in lens and cornea of TGFbeta2(−/−) TGFbeta3(−/−) double deficient mice. Cell Tissue Res 313:1-10
Dünker N, Schuster N, Krieglstein K (2001) TGF-beta modulates programmed cell death in the retina of the developing chick embryo. Development 128:1933–1942
Economous MA, Wu J, Vasilcanu D, Rosengren L, All-Ericsson C, van der Ploeg I, Menu E, Girnita L, Axelson M, Larsson O, Seregard S, Kvanta A (2008) Inhibition of VEGF secretion and experimental choroidal neovascularization by picropodophyllin (PPP), an inhibitor of the insulin-like growth factor-1 receptor. Acta Ophthalmol 86:4–49
Ernevoldsen MN, Kochoyan A, Jurgenson M, Jaako K, Dmytriyeva O, Walmod PS, Nielsen JD, Nielsen J, Li S, Korshunova I, Klementiev B, Novikova T, Zharovsky A, Berezin V, Boch E (2012) Neuroprotective and memory enhancing properties of a dual agonist of the FGF receptor and NCAM. Neurobiol Dis 48:533–545
Ezzedine ZD, Yanag XJ, Dechiara T, Yancopoulos G, Cepko CL (1997) Postmitotic cells fated to become rod photoreceptors can be respecified by CNTF treatment of the retina. Development 124:1055–1067
Ferrer I, Krupinski J, Goutan E, Martí E, Ambrosio S, Arenas E (2001) Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol 101:229–238
Finn TP, Nishi R (1996) Expression of ciliary neurotrophic factor in targets of ciliary ganglion neurons during and after the cell death phase. J Comp Neurol 366:559–571
Fischer D (2012) Stimulating axonal regeneration of mature retinal ganglion cells and overcoming inhibitory signalling. Cell Tissue Res 349:79–85
Flanders KC, Ren RF, Lippa CF (1998) Transforming growth factor-βs in neurodegenerative disease. Prog Neurobiol 54:71–85
Flannery JG, Gyarfas T, Kuuttila J, Lindholm P, Rantamäki T, Castrén E (2010) Regulation of brain-derived neurotrophic factor (BDNF) and cerebral dopamine neurotrophic factor (CDNF) by anti-Parkinonian drug therapy in vivo. Cell Mol Neurobiol 30:361–368
Frade JM, Rodriguez-Tébar A, Barde Y-A (1996) Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383:166–168
Frasson M, Picaud S, Léveillard T, Simonutti M, Mohand-Said S, Dreyfus H, Hicks D, Sabel J (1999) Glial cell line-derived neurotrophic factor induces histologic and functional protection of rod photoreceptors in the rd/rd mouse. Invest Ophthalmol Vis Sci 40:2724–2734
Frim DM, Uhler TA, Galpern WR, Beal MF, Breakefield XO, Isacson O (1994) Implanted fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevent 1-methyl-4-phenylpyridinium toxicity to dopaminergic neurons in the rat. Proc Natl Acad Sci USA 24:5104–5108
Fu QL, Li X, Yip HK, Shao Z, Wu W, Mi S, So KF (2009) Combined effect of brain-derived neurotrophic factor and LINGO-1 fusion protein on long-term survival of retinal ganglion cells in chronic glaucoma. Neuroscience 162:375–382
Fuchshofer R, Tamm E (2012) The role of TGF-β in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res 347:279–290
Garcia P, Youssef I, Utvik JK, Florent-Béchard S, Barthélémy V, Malaplaste-Armand C, Kriem B, Stenger C, Koziel V, Olivier JL, Escanye MC, Hanse M, Allouche A, Desbène C, Yen FT, Bjerkvig R, Oster T, Nicou SP, Pillot T (2010) Ciliary neurotrophic factor cell-based delivery prevents synaptic impairment and improves memory in mouse models of Alzheimer’s disease. J Neurosci 30:7516–7527
Gauthier R, Joly S, Pernet V, Lachapelle P, Di Polo A (2005) Brain-derived neurotrophic factor gene delivery to Müller glia preserves structure and function of light-damaged photoreceptors. Invest Ophthalmol Vis Sci 46:3383–3392
Green ES, Rendahl KG, Zhou S, Ladner M, Coyne M, Srivastava R, Manning WC (2001) Two animal models of retinal degeneration are rescued by recombinant adeno-associated virus-mediated production of FGF-5 and FGF-18. Mol Ther 3:507–515
Guérin CJ, Hu L, Scicli G, Scicli AG (2001) Transforming growth factor beta in experimentally detached retina and periretinal membranes. Exp Eye Res 73:753–764
Hallbrock F (1999) Evolution of the vertebrate neurotrophin and Trk receptor gene families. Curr Opin Neurobiol 9:616–621
Halvorsen SW, Knaur N (2006) CNTF and related neurokines. In: Lajtha A (ed) Handbook of neurochemistry and molecular neurobiology. Neuroactive proteins and peptides. Volume Editor: R. Lim. pp 43–68
Hamburger V, Oppenheim RW (1982) Naturally occuring cell death in vertebrates. Neurosci Comment 1:39–55
Harper MM, Gozdanic SD, Blits B, Kuehn MH, Zamzow D, Buss JE, Kardon RH, Sakaguchi DS (2011) Transplantation of BDNF-secreting mesenchymal stem cells provides neuroprotection in chronically hypertensive rats. Invest Ophthalmol Vis Sci 52:4506–4515
Hauck SM, Kinkl N, Deeg CA, Swiatek-de Lange M, Schöffman S, Ueffing M (2006) GDNF family ligands trigger indirect neuroprotective signalling in retinal glial cells. Mol Cell Biol 26:2746–2757
Heldin C-H, Moustakas A (2012) Role of Smads in TGFβ signalling. Cell Tissue Res 347:21–36
Heldin C-H, Landström M, Moustakas A (2009) Mechanism of TGF-β signalling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol 21:166–176
Holtmann B, Wiese S, Samsan M, Grohmann K, Pennica D, Martini R, Sendtner M (2005) Triple knock-out of CNTF, LIF and CT-1 defines cooperative and distinct roles of these neurotrophic factors for motoneuron maintenance and function. J Neurosci 25:1778–1787
Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24:677–736
Hyman C, Hofer M, Barde YA, Juhasz M, Yancopoulos GD, Squinto SP, Lindsay RM (1991) BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 350:230–232
Ikushima H, Miyazono K (2012) TGF-β signal transduction spreading to a wider field: a broad variety of mechanisms for context-dependent effects of TGF-β. Cell Tissue Res 347:37–49
Isenmann S, Klöcker N, Gravel C, Bähr M (1998) Short communication: protection of axotomized retinal ganglion cells by adenovirally delivered BDNF in vivo. Eur J Neurosci 10:2751–2756
Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K (2010) A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci USA 107:2687–2692
Jarosik J, Legutko B, Werner S, Unsicker K, von Bohlen und Halbach O (2011) Roles of exogenous and endogenous FGF-2 in animal models of depression. Restor Neurol Neurosci 29:153–165
Jin K, LaFevre-Bernt M, Sun Y, Chen S, Crippen D, Logvinova A, Ross CA, Greenberg DA, Ellerby LM (2005) FGF-2 promotes neurogenesis and neuroprotection and prolongs survival in a transgenic mouse model of Huntington’s disease. Proc Natl Acad Sci USA 102:18189–18194
Johnson EM Jr, Tuszynski MH (2008) Neurotrophic factors. In: Kordower JH, Tuszynski MA (eds) CNS regeneration. Academic Press, San Diego, pp 95–144
Joly S, Pernet V, Chemtob S, DiPolo A, Lachapelle P (2007) Neuroprotection in the juvenile rat model of light-induced retinopathy: evidence suggesting a role for FGF-2 and CNTF. Invest Ophthalmol Vis Sci 48:2311–2320
Kauper K, McGovern C, Sherman S, Heatherton P, Rapoza R, Stabila P, Dean B, Lee A, Borges S, Bouchard B, Tao W (2012) Two-year intraocular delivery of ciliary neurotrophic factor by encapsulated cell technology implants in patients with chronic retinal degenerative disease. Invest Ophthalmol Vis Sci 53:7484–7491
Kirsch M, Schulz-Key S, Wiese A, Furhmann S, Hofmann D (1998) Ciliary neurotrophic factor blocks rod photoreceptor differentiation from postmitotic precursor cells in vitro. Cell Tissue Res 291:207–216
Kordower JH, Chu Y, Stebbins GT, DeKosky ST, Cochran EJ, Bennett D, Mufson EJ (2001) Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol 49:202–213
Korsching S (1993) The neurotrophic factor concept: a re-examination. J Neurosci 13:2739–2748
Krieglstein K (2006) Transforming growth factor-βs in the brain. In: Lajtha A (ed) Handbook of neurochemistry and molecular neurobiology. Neuroactive proteins and peptides. Volume Editor: R. Lim. pp 123–142
Krieglstein K, Richter S, Farkas L, Schuster N, Dünker N, Oppenheim RW, Unsicker K (2000) Reduction of endogenous transforming growth factors β prevents ontogenetic neuron death. Nat Neurosci 3:1085–1090
Krieglstein K, Zheng F, Unsicker K, Alzheimer C (2011) More than being protective: functional roles for TGF-β/activin signaling pathways at central synapses. Trends Neurosci 34:421–429
Lambiase A, Mantelli F, Sacchetti M, Rossi S, Aloe L, Bonini S (2011) Clinical applications of NGF in ocular diseases. Arch Ital Biol 149:283–292
Lebrun-Julien F, Morquette B, Douillette A, Saragovi HU, Di Polo A (2009) Inhibition of p75(NTR) in glia potentiates TrkA-mediated survival of injured retinal ganglion cells. Mol Cell Neurosci 40:410–420
Lee MY, Naumann T, Kirsch M, Frotscher M, Hofmann HD (1997) Transient upregulation of ciliary neurotrophic factor receptor-alpha mRNA in axotomized rat septal neurons. Eur J Neurosci 9:622–626
Leibinger M, Müller A, Andreadaki A, Hauk TG, Kirsch M, Fischer D (2009) Neuroprotective and axon growth-promoting effects following inflammatory stimulation on mature retinal ganglion cells in mice depend on ciliary neurotrophic factor and leukaemia inhibitory factor. J Neurosci 29:14334–14341
Levivier M, Przedborski S, Bensics C, Kang UJ (1995) Intrastriatal implantation of fibroblasts genetically engineered to produce brand-derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson’s disease. J Neurosci 15:7810–7820
Lewin GR, Barde Y-A (1996) Physiology of the neurotrophins. Annu Rev Neurosci 19:289–317
Lindholm P, Voutilainen MH, Laurén J, Peränen J, Leppänen VM, Andressoo JO, Lindahl M, Janhunen S, Kalkkinen N, Timmusk T, Tuominen RK, Saarma M (2007) Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 448:73–77
Lindholm P, Peränen J, Andressoo JO, Kalkkinen N, Kokaia Z, Lindvall O, Timmusk T, Saarma M (2008) MANF is widely expressed in mammalian tissues and differently regulated after ischemic and epileptic insults in rodent brain. Mol Cell Neurosci 39:356–371
Liu X, Grishanin RN, Tolwani RJ, Rentería RC, Xu B, Reichardt LF, Copenhagen DR (2007) Brain-derived neurotrophic factor and TrkB modulate visual experience-dependent refinement of neuronal pathways in the retina. J Neurosci 27:7256–7267
Lu B, Pang PT, Woo NH (2005) The yin and yang of neurotrophin action. Nat Rev Neurosci 6:603–614
Makwana M, Jones LL, Cuthill D, Heuer H, Bohatschek M, Hristova M, Friedrichsen S, Ormsby I, Buereinger D, Koppius A, Bauer K, Doetschmann T, Raivich G (2007) Endogenous transforming growth factor beta 1 suppresses inflammation and promotes survival in adult CNS. J Neurosci 27:11201–11213
Marks WJ, Bartus RT, Siffert J, Davis CS, Lozano A, Bouli N, Vitek J, Stacy M, Turner D, Verhagen L, Bakay R, Watts R, Guthrie B, Jankovic J, Simpson R, Tagliati M, Alterman R, Stern M, Baltuch G, Starr PA, Larson PS, Ostrem JL, Nutt J, Kieburtz K, Kordower JH, Olanow CW (2010) Gene delivery of AAV2-neurturin for Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol 9:1164–1172
Maruyama W, Naoi M (2012) “70th birthday Professor Riederer” Induction of glial cell line-derived and brain-derived neurotrophic factors by rasagiline and (−)deprenyl: a way to a disease-modifiying therapy? J Neural Transm 120:83–89
Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, Longo FM (2010) Small molecule BDNF mimetics activate TrkB signalling and prevent neuronal degeneration in rodents. J Clin Invest 120:1774–1785
Massagué J (2012) TGFβ signalling in context. Nat Rev Mol Cell Biol 13:616–630
McCall J, Weidner N, Blesch A (2012) Neurotrophic factors in combinatorial approaches for spinal cord regeneration. Cell Tissue Res 349:27–37
McDonald NQ, Lapatto R, Murray-Rust J, Gunning J, Wlodawer A, Blundell TL (1991) New protein fold revealed by a 2.3-Å resolution crystal structure of nerve growth factor. Nature 354:411–414
McDonald NQ, Chao MV (1995) Structural determinants of neurotrophin action. J Biol Chem 270:19669–19672
Miltiadous P, Stamatakis A, Koutsoudaki PN, Tiniakos DG, Stylianopoulou F (2011) IGF-1 ameliorates hippocampal neurodegeneration and protects against cognitive deficits in an animal model of temporal lobe epilepsy. Exp Neurol 231:223–235
Mitsumoto H, Ikeda K, Klinkosz B, Cedarbaum JM, Wong V, Lindsay RM (1994) Arrest of motor neuron disease in wobbler mice cotreated with CNTF and BDNF. Science 19:1107–1110
Monteggia LM (2011) Toward neurotrophin-based therapeutics. Am J Psychiatry 168:114–116
Morimoto T, Miyoshi T, Matsuda S, Tano Y, Fujikado T, Fukuda Y (2005) Transcorneal electrical stimulation rescues axotomized retinal ganglion cells by activating endogenous retinal IGF-1 system. Invest Ophthalmol Vis Sci 46:2147–2155
Mu Y, Kumar S, Landström M (2012) Non-Smad signaling pathways. Cell Tissue Res 347:11–20
Müller A, Hauk TG, Leibinger M, Marienfeld R, Fischer D (2009) Exogenous CNTF stimulates axon regeneration of retinal ganglion cells partially via endogenous CNTF. Mol Cell Neurosci 41:233–246
Müller HD, Hanumanthiah KM, Diederich K, Schwab S, Schäbitz WR, Sommer C (2008) Brain-derived neurotrophic factor but not forced arm use improves long-term outcome after photothrombotic stroke and transiently upregulates binding densities of excitatory glutamate receptors in the rat brain. Stroke 39:1012–1021
Nagahara AH, Tuszynski MH (2011) Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discovery 10:209–216
Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15:331–337
Nikoletopoulou V, Lickert H, Frade JM, Rencurel C, Giallonardo P, Zhang L, Bibel M, Barde Y-A (2010) Neurotrophin receptors TrkA and TrkC cause neuronal death, whereas TrkB does not. Nature 467:59–63
Oppenheim RW (1991) Cell death during development of the nervous system. Annu Rev Neurosci 14:453–501
Otto D, Unsicker K (1990) Basic FGF reverses chemical and morphological deficits in the nigrostriatal system of MPTP-treated mice. J Neurosci 10:1912–1921
Otto D, Unsicker K (1993a) FGF-2-mediated protection of cultured mesencephalic dopaminergic neurons against MPTP and MPP+: specificity and impact of culture conditions, non-dopaminergic neurons, and astroglial cells. J Neurosci Res 34:382–393
Otto D, Unsicker K (1993b) FGF-2 modulates dopamine and dopamine-related striatal transmitter systems in the intact and MPTP-lesioned mouse. Eur J Neurosci 5:927–932
Park CK, Ju WK, Hofmann HD, Kirsch M, Kang JK, Chun MH, Lee MY (2000) Differential regulation of ciliary neurotrophic factor and its receptor in the rat hippocampus following transient global ischemia. Brain Res 861:345–353
Park HY, Kim JH, Sun Kim H, Park CK (2012) Stem cell-based delivery of brain-derived neurotrophic factor gene in the rat retina. Brain Res 1469:10–23
Park SH, Doh J, Park SI, Lim JY, Kim SM, Youn JI, Jin HT, Seo SH, Song MY, Sung SY, Kim M, Hwang SJ, Choi JM, Lee SK, Lee HY, Lim CL, Chung YJ, Yang D, Kim HN, Lee ZH, Choi KY, Jeun SS, Sung YC (2010) Branched oligomerization of cell-permeable peptides markedly enhances the transduction efficiency of adenovirus into mesenchymal stem cells. Gene Ther 17:1052–1061
Park YG, Jeong JK, Moon MH, Lee JH, Seol JW, Kim SJ, Kang SJ, Park SY (2012) Insulin-like growth factor-1 protects against prion peptide-induced cell death in neuronal cells via inhibition of Bax translocation. Int J Mol Med 30:1069–1074
Peinado-Ramon P, Salvador M, Villegas-Perez MP, Vidal-Sanz M (1996) Effects of axotomy and intraocular administration of NT-4, NT-3 and brain-derived neurotrophic factor on the survival of adult rat retinal ganglion cells. Invest Ophthalmol Vis Sci 37:489–500
Perez-Polo JR (2006) Nerve growth factor and related proteins. In: Lajtha A (ed) Handbook of neurochemistry and molecular neurobiology. Neuroactive proteins and peptides. Volume Editor: R. Lim. pp 2–9
Peterziel H, Strelau J (2006) GDNF and related proteins. In: Lajtha A (ed) Handbook of neurochemistry and molecular neurobiology. Neuroactive proteins and peptides. Volume Editor: R. Lim. pp 69–91
Pong Ng Y, Yip LO K, Cheung ZH, Ip NY (2006) Signaling through the neurotrophin receptors. In: Lajtha A (ed) Handbook of neurochemistry and molecular neurobiology. Neuroactive proteins and peptides. Volume Editor: R. Lim. pp 12–41
Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361:1545–1564
Ren R, Li Y, Liu Z, Liu K, He S (2012) Long-term rescue of rat retinal ganglion cells and visual function by AAV-mediated BDNF expression after acute elevation of intraocular pressure. Invest Ophthalmol Vis Sci 53:1003–1011
Ridder DA, Schwaninger M (2012) TAK1 inhibition for treatment of cerebral ischemia. Exp Neurol 239C:68–72
Rios-Munoz W, Soto I, Duprey-Diaz MV, Blagburn J, Blanco RE (2005) Fibroblast growth factor 2 applied to the optic nerve after axotomy increases Bcl-2 and decreases Bax in ganglion cells by activating the extracellular signal-regulated kinase signalling pathway. J Neurochem 93:1422–1433
Riva MA, Molteni R, Bedogni F, Racagni G, Fumagalli F (2005) Emerging role of the FGF system in psychiatric disorders. Trends Pharmacol Sci 26:228–231
Rodger J, Drummond ES, Hellström M, Robertson D, Harvey AR (2012) Long-term gene therapy causes transgene-specific changes in the morphology of regenerating retinal ganglion cells. PLoS One 7:e31061
Rodriguez-Tebar A, Dechant G, Barde Y-A (1991) Neurotrophins: structural relatedness and receptor interactions. Philos Trans R Soc Lond B Biol Sci 331:225–228
Roussa E, von Bohlen und Halbach O, Krieglstein K (2009) TGF-beta in dopamine neuron development, maintenance and neuroprotection. Adv Exp Med Biol 651:81–90
Rudzinski M, Wong TP, Saragovi HU (2004) Changes in retinal expression of neurotrophins and neurotrophin receptors induced by ocular hypertension. J Neurobiol 58:341–354
Sawai H, Clarke DB, Kittlerova P, Bray GM, Aguayo AJ (1996) Brain-derived neurotrophic factor and neurotrophin-4/5 stimulate growth of axonal branches from regenerating retinal ganglion cells. J Neurosci 16:3887–3894
Seaborn T, Masmoudi-Kouli O, Fournier A, Vadry H, Vaudry D (2011) Protective effects of pituitary adenylate cyclase-activating polypeptide (PACAP) against apoptosis. Curr Pharm Des 17:204–214
Selvaraj BT, Frank N, Bender FL, Asan E, Sendtner M (2012) Local axonal function of STAT3 rescues axon degeneration in the pmn model of motoneuron disease. J Cell Biol 199:437–451
Sendtner M, Schmalbruch H, Stockli K, Kreutzberg PCG, Thoenen H (1992) Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive neuronopathy. Nature 358:502–504
Sendtner M, Götz R, Holtmann B, Thoenen H (1997) Endogenous ciliary neurotrophic factor is a lesion factor for axotomized motoneurons in adult mice. J Neurosci 17:6999–7006
Shen J, Ghai K, Sompol P, Lui X, Cao X, Iuvone PM, Ye K (2012) N-acetyl serotonin derivatives as potent neuroprotectants for retinas. Proc Natl Acad Sci USA 109:3540–3545
Sidorova YA, Mätlik K, Paveliev M, Lindahl M, Piranen M, Milbrandt J, Arumäe U, Saarma M, Bespalov MM (2010) Persephin signalling through GFRalpha1: the potential for the treatment of Parkinson’s disease. Mol Cell Neurosci 44:223–232
Sievers J, Hausmann B, Unsicker K, Berry M (1987) Fibroblast growth factors promote the survival of adult rat retinal ganglion cells after transection of the optic nerve. Neurosci Lett 76:157–162
Siffroi-Fernandez S, Felder-Schmittbuhl MP, Khanna H, Swaroop A, Hicks D (2008) FGF19 exhibits neuroprotective effects on adult mammalian photoreceptors in vitro. Invest Ophthalmol Vis Sci 49:1696–1704
Southwell DG, Parades MF, Galvao RP, Jones DL, Froemke RC, Sebe JY, Alfaro-Cervello C, Tang Y, Garcia-Verdugo JM, Rubenstein JL, Baraban SC, Alvarez-Buylla A (2012) Intrinsically determined cell death of developing cortical interneurons. Nature 491:109–115
Spittau B, Krieglstein K (2012) Klf10 and Klf11 as mediators of TGF-beta superfamily signalling. Cell Tissue Res 347:65–72
Sposato V, Bucci MG, Coassin M, Russo MA, Lambiase A, Aloe L (2008) Reduced NGF level and TrkA protein and TrkA gene expression in the optic nerve of rats with experimentally induced glaucoma. Neurosci Lett 446:20–24
Stöckli KA, Lottspeich F, Sendtner M, Masiakowski P, Carroll P, Götz R, Lindholm D, Thoenen H (1989) Molecular cloning, expression, and regional distribution of rat ciliary neurotrophic factor. Nature 342:920–923
Tang Z, Arjunan P, Lee C, Li Y, Kumar A, Hou X, Wang B, Wardega P, Zhang F, Dong L, Zhang Y, Zhang SZ, Ding H, Fariss RN, Becker KG, Lennartsson J, Nagai N, Caao Y, Li X (2010) Survival effect of PDGF-CC rescues neurons from apoptosis in both brain and retina by regulating GSK3beta phosphorylation. J Exp Med 207:867–880
Tesseur I, Zou K, Esposito L, Bard F, Berber E, von Can J, Lin AH, Crews L, Tremblay P, Mathews P, Mucke L, Masliah E, Wysso-Coray T (2006) Deficiency in neuronal TGF-β signaling promotes neurodegeneration and Alzheimer’s pathology. J Clin Invest 116:3060–3069
Thanos S, Böhm MR, Schallenberg M, Oellers P (2012) Traumatology of the optic nerve and contribution of crystallins to axonal regeneration. Cell Tissue Res 349:49–69
Thau N, Jungnickel J, Knippenberg S, Ratzka A, Dengler R, Petri S, Grothe C (2012) Prolonged survival and milder impairment of motor function in the SOD1 ALS mouse model devoid of fibroblast growth factor 2. Neurobiol Dis 47:248–257
Torres Aleman I (2012) Insulin-like growth factor-1 and central neurodegenerative diseases. Endocrinol Metab Clin North Am 41:395–408
Tsai LK, Chen YC, Cheng WC, Ting CH, Dodge JC, Hwu WL, Cheng SH, Passini MA (2012) IGF-1 delivery to CNS attenuates motor neuron cell death but does not improve motor function in type III SMA mice. Neurobiol Dis 45:272–279
Tsukahara T, Takeda M, Shimohama S, Ohara O, Hashimoto N (1995) Effects of brain-derived neurotrophic factor on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in monkeys. Neurosurgery 37:733–739
Turner CA, Watson SJ, Akil H (2012) The fibroblast growth factor family: neuromodulation of affective behavior. Neuron 76:160–174
Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, Tong G, Potkin SG, Fallon J, Hansen L, Mufson EJ, Kordower JH, Gall C, Conner J (2005) A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 11:551–555
Unsicker K, Krieglstein K (2002) TGF-βs and their roles in the regulation of neuron survival. In: Alzheimer C (ed) Molecular and cellular biology of neuroprotection in the CNS. Springer, Berlin, pp 353-374
Unsicker K, Reichert-Preibsch H, Schmidt R, Pettmann B, Labourdette G, Sensenbrenner M (1987) Astroglial and fibroblast growth factors have neurotrophic functions for cultured peripheral and central nervous system neurons. Proc Natl Acad Sci USA 84:5459–5463
Unsicker K, Reuss B, von Bohlen und Halbach O (2006) Fibroblast growth factors in brain functions. In: Lajtha A (ed) Handbook of neurochemistry and molecular neurobiology. Neuroactive proteins and peptides. Volume Editor: R. Lim. pp 93–121
Walsh N, Valter K, Stone J (2001) Cellular and subcellular patterns of expression of bFGF and CNTF in the normal and light stressed adult rat retina. Exp Eye Res 72:495–501
Wang Y, Brown DP, Duan Y, Kong W, Watson BD, Goldberg JL (2012) A novel rodent model of posterior ischemic optic neuropathy. Arch Ophthalmol 8:1–11
Watanabe M, Sawei H, Fukuda Y (1997) Survival of axotomized retinal ganglion cells in adult mammals. Clin Neurosci 4:233–239
Weinreb O, Amit T, Bar-Am O, Youdim MB (2007) Induction of neurotrophic factors GDNF and BDNF associated with the mechanaism of neurorescue action of rasagiline and ladostigil: new insights and implications for therapy. Ann N Y Acad Sci 1122:155–168
Wen R, Tao W, Huang D, Kauper K, Stabila P, LaVail MM, Laties AM, Li Y (2012a) Regeneration of cone outer segments induced by CNTF. Adv Exp Med Biol 723:93–99
Wen R, Tao W, Li Y, Sieving PA (2012b) CNTF and retina. Prog Retin Eye Res 31:136–151
Wendt MK, Tian M, Schiemann WP (2012)Deconstructing the mechanisms and consequences of TGF-β-induced EMT during cancer progression. Cell Tissue Res 347:85–101
Werner S, Unsicker K, von Bohlen und Halbach O (2011) Fibroblast growth factor-2 deficiency causes defects in adult hippocampal neurogenesis, which are not rescued by exogenous fibroblast growth factor-2. J Neurosci Res 89:1605–1617
Wilson RB, Kunchithapautham K, Rohrer B (2007) Paradoxical role of BDNF: BDNF+/− retinas are protected against light damage-mediated stress. Invest Ophthalmol Vis Sci 48:2877–2886
Wyss-Coray T (2006) Tgf-beta pathway as a potential target in neurodegeneration and Alzheimer’s. Curr Alzheimer Res 3:191–195
Yan Q, Wang J, Matheson CR, Ulrich JL (1999) Glial cell line-derived neurotrophic factor (GDNF) promotes the survival of axotomized retinal ganglion cells in adult rats: comparison to and combination with brain-derived neurotrophic factor (BDNF). J Neurobiol 38:382–390
Acknowledgement
I am grateful to Dr. Andreas Schober for help with the figures and to Dr. Ernst Tamm for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
The work of the author described in this article was supported by the Deutsche Forschungsgemeinschaft.
Rights and permissions
About this article
Cite this article
Unsicker, K. Neurotrophic molecules in the treatment of neurodegenerative disease with focus on the retina: status and perspectives. Cell Tissue Res 353, 205–218 (2013). https://doi.org/10.1007/s00441-013-1585-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-013-1585-y