
Abstract
Transforming growth factor (TGF)-β signaling is involved in almost all major cell behaviors under physiological and pathological conditions, and its regulatory system has therefore been vigorously investigated. The fundamental elements in TGF-β signaling are TGF-β ligands, their receptors, and intracellular Smad effectors. The TGF-β ligand induces the receptors directly to phosphorylate and activate Smad proteins, which then form transcriptional complexes to control target genes. One of the classical questions in the field of research on TGF-β signaling is how this cytokine induces multiple cell responses depending on cell type and cellular context. Possible answers to this question include cross-interaction with other signaling pathways, different repertoires of Smad-binding transcription factors, and genetic alterations, especially in cancer cells. In addition to these genetic paradigms, recent work has extended TGF-β research into new fields, including epigenetic regulation and non-coding RNAs. In this review, we first describe the basic machinery of TGF-β signaling and discuss several factors that comprise TGF-β signaling networks. We then address mechanisms by which TGF-β induces several responses in a cell-context-dependent fashion. In addition to classical frames, the interaction of TGF-β signaling with epigenetics and microRNA is discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cytokines are small secreted proteins that are produced by numerous types of cells and that play important roles in intercellular communication to maintain order in the organism. They elicit biological effects by binding to the extracellular domains of specific transmembrane receptors in the outer membrane of cells. Cytokines mediate intercellular communication via the regulation of cell growth and differentiation and are thus crucial for maintaining the homeostasis of multicellular organisms. Aberrant regulation of cytokine signaling can therefore result in various diseases.
The transforming growth factor (TGF)-β family is particularly prominent among these signals (Blobe et al. 2000; Feng and Derynck 2005; Massagué 2008). TGF-β signaling controls a diverse set of cellular processes, including cell growth, differentiation, apoptosis, survival, and specification of developmental fate, during embryogenesis and in mature tissues (Ikushima and Miyazono 2010a; Moustakas and Heldin 2009). To control TGF-β-induced cell responses, numerous factors tightly regulate this signaling pathway under physiological conditions (Ikushima and Miyazono 2010b; Bierie and Moses 2006). Loss of balance of TGF-β signaling thus leads to several pathological conditions, including malignant tumors, fibrotic diseases, and abnormal immune reactions (Levy and Hill 2006; Varga and Pasche 2009; Flavell et al. 2010). Indeed, studies of clinical samples indicate that a distortion of TGF-β signaling is one of the major causes of several disorders. Here, we first discuss the way that (1) cells translate TGF-β signaling into cellular responses, and (2) TGF-β signaling and TGF-β-induced cell responses are tightly controlled. Possible and/or established mechanisms of the context-dependent diversity of TGF-β-induced cell responses are also addressed. In addition, recent research on TGF-β signaling has spread into novel fields, including epigenetics and non-coding RNAs. Thus, we also mention the involvement of epigenetic regulation and non-coding RNAs in the classical TGF-β signaling pathway.
Extracellular regulation of TGF-β signaling
Effects of TGF-β are mediated by three TGF-β ligands: TGF-β1, TGF-β2, and TGF-β3 (Feng and Derynck 2005; Shi and Massagué 2003). Although each of these ligands is produced by distinct genes, they exhibit approximately 70%–80% sequence similarity. The TGF-β ligand is first synthesized as a dimeric pro-protein (pro-TGF-β), which is then cleaved to form the mature disulfide-bridged TGF-β dimer. The pro-peptide has high affinity for the cleaved mature TGF-β ligand, which is secreted from cells as a small latent complex (ten Dijke and Arthur 2007). Since TGF-β in this form does not have the ability to interact with its receptor, the pro-peptide is termed the latency-associated protein (LAP). The LAP dimer is also bound to the latent TGF-β binding proteins (LTBPs) by disulfide bonds, and the tri-molecular complex is termed the large latent complex (Rifkin 2005). The dissociation of TGF-β from the complex is a critical regulatory event and is achieved by integrin, shear force, thrombospondin-1 (TSP-1), some enzymes including plasmin, changes in pH, heat treatment, radiation, and other agents. Among the four different LTBPs, LTBP-1, 3, and 4 bind to small latent complexes and play key roles in targeting the large latent complex to the extracellular matrix, where active TGF-β is released by proteolytic cleavage. Although the synthesis of TGF-β is regulated by a variety of factors at the level of transcription and/or mRNA stability, the generation of active TGF-β from its latent form is also subject to regulation.
TGF-β receptors
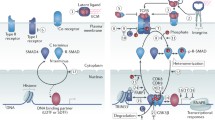
Activated TGF-β ligands transduce their effects through TGF-β type I and II receptors (Ikushima and Miyazono 2010b; Wrana et al. 2008). The TGF-β type II receptor (TβRII) is the specific receptor for TGF-β ligands. Both type II and type I receptors are comprised of an N-terminal extracellular ligand-binding domain, a transmembrane region, and a C-terminal intracellular serine/threonine kinase domain. TGF-β has high affinity for TβRII, and upon binding the ligand, the type I receptor forms a heteromeric complex consisting of two of each receptor type and is activated by the type II receptor (Fig. 1). The type I, but not type II, receptors contain a characteristic GS domain, located N-terminal to the kinase domain. Activation of the type I receptor involves the phosphorylation of its GS domain by the type II receptor. Although activin receptor-like kinase 5 (ALK5), also known as TβRI, mediates TGF-β signal transduction in most types of cells, ALK1 and other type I receptors also transduce TGF-β signaling in certain cells, including endothelial cells (Goumans et al. 2003; Daly et al. 2008).

Intracellular transforming growth factor-β (TGF-β) signal transduction. TGF-β signals are transduced by type II receptor (TβRII), type I receptor (TβRI), and their downstream Smad proteins (Smad2-4). Activated Smad complex interacts with DNA-binding transcription factors and co-activators/co-repressors and binds to the promoter regions of TGF-β target genes. Active TGF-β receptors also regulate signaling cascades other than the Smad pathway
TGF-β is also able to interact with proteins called TGF-β type III receptors, which do not have intrinsic kinase activity (Bernabeu et al. 2009). Betaglycan is a membrane-anchored proteoglycan that facilitates binding of TGF-β2 to TβRII (Gatza et al. 2010). Endoglin, a glycoprotein expressed at high levels in endothelial cells, binds to TβRII and is thought to act as an accessory protein for the receptor complex (ten Dijke et al. 2008). Although the function of endoglin in TGF-β signaling is still controversial, mutations of it have been linked to hereditary hemorrhagic telangiectasia (McAllister et al. 1994; Abdalla and Letarte 2006). In addition, endoglin produced in a soluble form is associated with the pathogenesis of preeclampsia (Venkatesha et al. 2006). These findings indicate the central roles of endoglin in controlling vascular homeostasis.
Intracellular signal transduction through Smad proteins
Once the functional TGF-β receptor complex is formed, it regulates the activation of downstream signaling pathways. Although several substrates for the type I receptor kinases have been identified, the most important ones for the transduction of TGF-β stimulation are members of the Smad family proteins (Massagué et al. 2005; Schmierer and Hill 2007; Derynck and Zhang 2003). Phosphorylation and activation of the type I receptor enable the recruitment of receptor-regulated Smads (R-Smads). The type I receptor then phosphorylates R-Smads, allowing them to form hetero-oligomeric complexes with the common-partner Smad (Co-Smad) and to move into the nucleus. Of the five R-Smads in mammals, Smad2 and Smad3 are activated by the TβRII–ALK5 complex, whereas Smad1, Smad5, and Smad8 are activated by the TβRII–ALK1 complex. Interestingly, Liu et al. (2009) have recently reported that ALK5 can directly activate Smad1/5 in certain types of cells. Smad4 is the only known Co-Smad in mammals. R-Smads consist of conserved Mad homology 1 (MH1) and MH2 domains, which are connected with a less-conserved linker region. The C-terminus of R-Smads has a characteristic SSXS (Ser-Ser-X-Ser) motif that is phosphorylated by active type I receptors. Smad4 contains MH1 and MH2 domains but lacks the C-terminal SSXS motif and, thus, is not phosphorylated by type I receptors. Smad complexes bind specific DNA sequences, namely 5′-AGAC-3′ or its reverse complement 5′-GTCT-3′, in the promoters or enhancers of target genes. They interact with other DNA-binding transcription factors, co-activators or co-repressors, and chromatin remodeling factors to the regulatory regions of target genes in order to regulate diverse TGF-β-induced cell responses. TGF-β stimulation also activates intracellular signals through non-Smad pathways, including mitogen-activated protein kinase, PI3K-Akt, and small GTPase pathways (Moustakas and Heldin 2005; Zhang 2009).
Context-dependent diversity of TGF-β-induced cell responses
At the core of this signaling pathway, TGF-β induces its membrane receptors directly to activate Smad proteins, which then form transcriptional complexes to control target genes. The aspect that makes this system complex is that these complexes activate or repress numerous target genes at the same time in a tightly regulated fashion. Furthermore, TGF-β stimulation induces numerous cell responses in a cellular context-dependent fashion (Roberts and Wakefield 2003; Bierie and Moses 2006). For example, TGF-β promotes cell proliferation in certain cellular contexts but inhibits it in most others (Ikushima and Miyazono 2010a). This cytokine plays crucial roles in the maintenance of the tumorigenic activities of some types of cancer stem cells (Ikushima et al. 2009; Peñuelas et al. 2009; Anido et al. 2010; Naka et al. 2010) but promotes the loss of tumorigenicity in others (Tang et al. 2007; Ehata et al. 2011). The cells making up one human body are all derived from a single cell, even if they are abnormal. However, they exhibit different responses to TGF-β because of slight but crucial differences. Moreover, even in the same type of cell, the cell responses mediated by TGF-β differ depending on environmental factors. Because of this inherent diversity, TGF-β-based therapeutic strategies are considered complex. Here, we discuss proposed or established mechanisms responsible for the chaotic diversity of TGF-β signaling.
Signal cross-talk
TGF-β is able to induce certain cell responses, under conditions including other types of signaling, but fails to induce the same responses without such signaling (Guo and Wang 2009). Cross-interaction with additional signaling is thus required for some TGF-β-induced cell responses (Fig. 2). Many signaling pathways have been reported to exhibit cross-talk with the TGF-β signaling pathway (Luo 2008; Zhang 2009); here, we discuss cross-talk with the Wnt, p53, and Ras signaling pathways.

“Signal cross-talk” model. In Context 1, but not in Context 2, Signal X is transduced in cells to modify downstream transducers of TGF-β signaling and induce a certain context-1-specific cell response
Wnt signaling plays diverse roles in regulating numerous cell responses, including cell proliferation, differentiation, migration, and survival (Kestler and Kühl 2008; Logan and Nusse 2004). Canonical Wnt signaling is mediated by β-catenin, which functions as a transcription co-factor and is also essential for the formation of adherence junctions between cells through its interaction with cadherins. In the absence of Wnt, cytoplasmic β-catenin is degraded through glycogen synthase kinase (GSK)-3β-mediated serial phosphorylation and subsequent polyubiquitination, which keeps the Wnt pathway in an "OFF" state. The binding of Wnt ligand to its receptor Frizzled (Fz) and co-receptor LRP5/6 leads to GSK-3β inactivation and β-catenin stabilization. The cytoplasmic accumulation of β-catenin promotes its translocation into the nucleus, where it binds the lymphocyte enhancer factor/T-cell transcription factor (Lef/TCF) family of transcription factors and turns the Wnt pathway “ON”. The most common format of cross-talk between the TGF-β and Wnt signaling pathways occurs in the nucleus, where the Smads and Lef/β-catenin synergistically regulate a set of shared target genes (Labbé et al. 2000, 2007; Hussein et al. 2003; Sasaki et al. 2003). These two pathways are also linked by protein interactions in the cytoplasm (Tang et al. 2008; Han et al. 2006; Liu et al. 2006; Edlund et al. 2005; Furuhashi et al. 2001).
Perturbations of TGF-β signaling have been strongly implicated in cancer progression. TGF-β can play both tumor-suppressive and tumor-promoting roles and is now generally accepted to act as an anti-oncogenic factor in the early phase of tumorigenesis, although it can be converted to a pro-oncogenic factor during cancer progression (Roberts and Wakefield 2003; Bierie and Moses 2006). This switching of TGF-β from an anti-oncogenic factor to a pro-oncogenic factor might be induced by various mechanisms. Adorno et al. (2009) have reported that additional mutation of p53 plays a role in this switching. In the early stages of tumorigenesis, TGF-β inhibits the proliferation of tumor cells in concert with wild-type p53 as an anti-oncogenic factor. In contrast, in the later stages, Smad complexes function cooperatively with mutant p53 to abrogate the abilities of p63 to suppress sharp-1 and cyclin G2 expression and to inhibit metastasis. Indeed, the expression of mutant p53 in noninvasive tumor cells enhances the pro-invasive and migratory effects of TGF-β, whereas the suppression of mutant p53 expression in aggressive tumors impairs their ability to metastasize.
TGF-β induces epithelial-mesenchymal transition (EMT), in which epithelial cells acquire mesenchymal characteristics (Thiery et al. 2009). Some transcription factors, including Snail, Slug, Twist, δEF1/ZEB1, and SIP1/ZEB2, are induced by TGF-β signaling and regulate the expression of E-cadherin and other EMT-related genes. In certain cells, oncogenic Ras and TGF-β signaling pathways have been shown to induce EMT cooperatively (Oft et al. 1996, 2002). TGF-β alone can only weakly induce the expression of Snail and repress that of E-cadherin; however, oncogenic Ras signaling enhances the expression of Snail induced by TGF-β and synergistically induces EMT (Horiguchi et al. 2009).
In this fashion, TGF-β-induced cell responses can be determined by cooperatively acting signaling pathways.
Co-factors
Since the affinity of the activated Smad complex for the Smad-binding element (SBE) is insufficient to support an association with promoters of target genes, Smad complexes are associated with other DNA-binding transcription factors to regulate gene expression. Furthermore, the combination of the direct interactions of Smads with DNA and with sequence-specific DNA-binding transcription factors yields the selectivity of interaction between Smad complexes and the regulatory promoter sequences. Various families of transcription factors, such as the forkhead, homeobox, zinc-finger, activator protein 1, Ets, and basic helix-loop-helix (bHLH) families, serve as Smad partners (Ikushima et al. 2008; Koinuma et al. 2009a, b). The juxtaposition of an SBE at variable distances from the sequence, to which the Smad-interacting transcription factor binds, allows selection of a subset of promoter sequences to which the Smad transcription complexes bind with high affinity. Each Smad-cofactor combination targets a particular set of genes, which is determined by the presence of cognate binding sequence element combinations in the regulatory regions of target genes. Gene responses induced by TGF-β are thus classified by groups of genes that are simultaneously regulated by a common Smad-cofactor combination. A group of genes jointly controlled by a given Smad-cofactor complex is denoted a “synexpression group”. Cells of different types or those exposed to different environments contain distinct repertoires of transcriptional partners for Smads and link their cellular context to their responses to TGF-β (Fig. 3).

“Cofactors” model. TGF-β target genes (A–I) are regulated by Smad proteins. Profiles of expression of cofactors of Smad proteins differ between Context 1 and Context 2, resulting in different responses to TGF-β stimulation
A novel negative regulator of TGF-β signaling, human homolog of maternal Id-like molecule (HHM), has been demonstrated to suppress TGF-β signaling in a cell-response-selective fashion (Ikushima et al. 2008; Seto et al. 2009). Among the several cell responses induced by TGF-β, cell cycle arrest is repressed by HHM, but EMT is not. HHM bins to DNA-binding transcription factor Olig1 (oligodendrocyte transcription factor 1), a novel Smad-binding cofactor, and abrogates the binding of Olig1 to Smad proteins. Olig1 and R-Smads interact with each other on chromosomes and synergistically promote the expression of TGF-β target genes whose promoter regions have Olig1-binding sequence(s) and Smad-binding sequence(s) in close vicinity. HHM interferes with the interaction between Olig1 and the activated Smad complex and, as a consequence, inhibits the gene expression of the Olig1-Smad synexpression group at the transcriptional level. Since HHM interacts with some but not all Smad-binding transcription factors, HHM abrogates only a subset of Smad-cofactor complexes, including the Olig1-Smad complex. HHM thus inhibits TGF-β-induced cell responses, which are controlled by Smad-cofactor synexpression groups targeted by HHM, but fails to affect cell responses, which are regulated by Smad-cofactor synexpression groups not targeted by HHM.
The transcriptional cooperativity of Smad complexes with a variety of DNA-binding transcription factors thus creates marked complexity in the transcriptional regulation of target genes.
Genetic alterations
Although all cells except immune cells have nearly identical blueprints, or genomes, under physiological conditions, cancer cells have a variety of genetic alterations conferring survival advantage on them. Deletion or amplification of TGF-β target genes in cancer cells alters their responsiveness to TGF-β stimulation (Fig. 4). Although TGF-β up-regulates the expression of p15Ink4b, one of the tumor suppressor genes, to inhibit cell proliferation (Hannon and Beach 1994), a subset of glioma cells sustains homozygous deletion of the p15Ink4b locus on chromosome 9p21 (Jen et al. 1994). Loss of p15Ink4b attenuates the anti-oncogenic effects of TGF-β, and glioma cells might benefit from host- and/or tumor-derived TGF-β stimulation.

“Genetic alterations” model. In Context 1, expression of a certain target gene is induced by TGF-β signaling. In Context 2, the gene is deleted at the chromosomal level, and TGF-β stimulation fails to induce its expression
Thus, genetic alterations of downstream genes modify the cell responses induced by TGF-β and contribute to the cellular context-specific plasticity of TGF-β signaling.
Epigenetics
Classical genetic processes are not sufficient to establish an organism. For proper development and cell functioning, epigenetic phenomena are absolutely required for the control of gene expression (Hirabayashi and Gotoh 2010; Ordovás and Smith 2010). In addition to genetic mechanisms, the gene expression and cell responses induced by TGF-β stimulation are regulated by epigenetic systems, including DNA methylation and post-translational histone modulation (Fig. 5).

“Epigenetics” model. In Context 1, promoter regions of certain TGF-β target genes adopt an “open conformation” and are exposed to the Smad complex. Conversely, in Context 2, promoter regions of the same target genes adopt a “closed conformation”, and the Smad complex fails to access the Smad-binding elements. This difference results in differential responses to TGF-β stimulation
DNA methylation is one of the most intensely studied epigenetic modifications in mammals and has a large impact on molecular pathophysiology and normal cell physiology (Esteller 2008; Suzuki and Bird 2008). Indeed, tumor cells are characterized by a different methylome from that of normal cells (Kulis and Esteller 2010). Interestingly, both hypo- and hypermethylation events can be observed in cancer. For instance, two cell-cycle-related genes, p16INK4a and p15INK4b, undergo DNA methylation-mediated silencing in various types of cancer, leading to tumor development (Kulis and Esteller 2010). On the other hand, a global decrease in methylated CpG content contributes to genomic instability and to the activation of silenced oncogenes.
The regulation of gene expression by TGF-β can be affected by DNA methylation status. TGF-β induces platelet-derived growth factor-B (PDGF-B) expression in glioblastoma U373MG cells but fails to affect it in another glioblastoma cell line, U87MG cells. TGF-β thus induces the proliferation of U373MG cells but inhibits that of U87MG cells (Bruna et al. 2007). This difference can be explained, at least in part, by the DNA methylation of SBEs of the PDGF-B promoter. In addition, hypomethylation of the PDGF-B promoter is associated with poor prognosis in glioma patients. DNA methylation status in cells can thus determine whether a certain cell response is controlled by TGF-β.
Covalent modification of conserved residues in core histones by acetylation, phosphorylation, methylation, ADP-ribosylation, ubiquitination, and sumoylation is a reversible post-translational modification and is thought to be an important mechanism by which cells regulate chromatin accessibility and the function of chromatin DNA (Rice and Allis 2001). Thus, epigenetic de-regulation involving histone-modifying complexes and histone marks might be an important mechanism underlying the development and progression of diseases (Sawan and Herceg 2010). Furthermore, recent research has demonstrated that different types of cells might have specific patterns of histone modifications (histone modification signatures), which cause cellular context-dependent behaviors of cells (Lee et al. 2010). Indeed, the modification of histones varies drastically during tumorigenesis, and the disruption of many chromatin-modifying proteins is associated with the formation of various malignant tumors (Esteller 2007).
Differences in the histone status of promoters and enhancers of target genes might lead to alterations in the TGF-β-mediated transcription profile, resulting in distinct TGF-β-induced cell responses. Regulatory T (Treg) cells function as a safeguard against autoimmunity and immune pathology (Sakaguchi et al. 2010), and TGF-β signaling plays important roles in the induction of Treg cells through the stimulation of the expression of the transcription factor Foxp3, which confers Treg cell function (Yoshimura et al. 2010). Di- and trimethylation of lysine 4 of histone H3 (H3K4me2 and −3) near the Foxp3 transcription start site and within the 5’ untranslated region is lost as a result of T cell receptor (TCR) stimulation and PI3K/Akt/mTOR activity, as a consequence of which the ability of TGF-β to induce Foxp3 expression is abrogated (Sauer et al. 2008). Post-translational histone modification status in cells can thus determine the ability of TGF-β to induce a certain cell response.
Non-coding RNA
Interactions of TGF-β signaling and non-coding RNA occur at various levels. microRNAs (miRNAs) are small non-coding RNAs that modulate diverse biological functions through the repression of target genes (Filipowicz et al. 2008; Winter et al. 2009). Recent studies have demonstrated that Smad complexes play a regulatory role in the processing of miRNA in the nucleus (Hata and Davis 2009). During the process of the maturation of miRNA, the first cleavage after the transcription of the miRNA gene is catalyzed by the RNase III enzyme Drosha, which generates precursor miRNA from primary miRNA (Davis-Dusenbery and Hata 2010). Davis et al. (2008, 2010) have showed that the knockdown of the R-Smads prevents the induction of mature miR-21 and pre-miR-21, although no alteration in pri-miR-21 transcription has been detected. Furthermore, co-immunoprecipitation and RNA-immunoprecipitation studies have confirmed that Smads are present in a complex with Drosha and the pri-miR-21 hairpin following TGF-β stimulation. The binding of Drosha to pri-miR-21 is also elevated following TGF-β treatment. These findings indicate that Smad complexes promote the association of Drosha with a subset of miRNA hairpins, resulting in the facilitation of the processing of the miRNAs, and that TGF-β can regulate gene expression not only through the direct transcriptional regulation of target genes, but also through miRNA processing.
Non-coding RNAs also contribute to the context-dependent diversity of TGF-β-induced cell responses (Singh and Settleman 2010). Cells of different cell types or cells exposed to different conditions express diverse repertoires of non-coding RNA (Lu et al. 2005), and TGF-β stimulation thus produces context-specific cell responses. Even when TGF-β stimulation activates promoter and/or enhancer regions to the same degree in two different contexts, differences in post-transcriptional regulation can result in differences in the levels of expression of proteins and hence in different cell responses to TGF-β stimulation (Fig. 6).

“Non-coding RNA” model. In Context 2, transcribed mRNAs of TGF-β target genes are negatively regulated by non-coding RNA (ncRNA). In Context 1, such ncRNA is not expressed, resulting in the translation of the mRNAs
Two miRNA clusters, miR-17-92 and miR-106b-25, have been reported to affect the TGF-β signaling pathway (Petrocca et al. 2008; Ventura et al. 2009). The miR-17-92 cluster is composed of miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a-1. Tumor-promoting roles have been suggested for it based on its frequent amplification and overexpression in small-cell lung carcinoma and diffuse large B cell lymphoma. The miR-106b-25 cluster contains the highly conserved miR-106b, miR-93, and miR-25, which accumulate in different types of cancer, such as neuroblastoma, gastric cancer, and multiple myeloma. Recent studies have unveiled the functional involvement of miR-17-92 and miR-106b-25 clusters in TGF-β-induced apoptosis and cell cycle arrest. They silence two main downstream effectors playing central roles in these cell responses: the pro-apoptotic gene Bim and the cyclin-dependent kinase inhibitor p21Waf1. Furthermore, overexpression of miR-25 inhibits TGF-β-induced apoptosis, and overexpression of miR-106b and miR-93 prevents TGF-β-mediated cell cycle arrest. These reports indicate that the profiles of expression of miR-17-92 and miR-106b-25 clusters can determine whether TGF-β signaling has tumor-suppressive effects.
The miR-17-92 cluster is also involved in the post-transcriptional regulation of some of the regulatory components in TGF-β signaling. This cluster targets Smad4 and TβRII and, as a result, shuts down this signaling pathway (Dews et al. 2010; Mestdagh et al. 2010). In addition, enforced expression of miR-17-92 has been demonstrated to result in impaired gene activation by TGF-β in glioblastoma cells (Dews et al. 2010) and neuroblastoma cells (Mestdagh et al. 2010).
TGF-β-induced EMT, in which epithelial cells acquire mesenchymal characteristics, has been reported to be regulated by the miRNA-200 family (miR-200a, miR-200b, miR-200c, miR-141, and miR-429; Gregory et al. 2008; Korpal et al. 2008; Burk et al. 2008; Park et al. 2008). These miRNAs cooperatively interfere with expression of δEF1/ZEB1 and SIP1/ZEB2, which are transcriptional repressors of E-cadherin induced by TGF-β and involved in EMT. Manipulation of miR-200 family expression suppresses EMT and induces the opposite change, namely mesenchymal-epithelial transition. Since the levels of expression of the miR-200 family might vary from cell to cell, they determine, at least in part, at downstream gene levels whether TGF-β induces EMT. TGF-β has also been demonstrated to induce miR-155 expression through the Smad pathway, which in turn regulates epithelial plasticity by targeting RhoA and promotes TGF-β-mediated EMT as a result of the dissolution of tight junctions (Kong et al. 2008).
TGF-β-induced miRNAs also play important roles in cancer stem cells. TGF-β up-regulates miR-181 at the post-transcriptional level in breast cancer cells. miR-181 targets a tumor suppressor (ataxia telangiectasia mutaed, ATM) and maintains the breast cancer stem cell population (Wang et al. 2011).
PDGF-BB antagonizes the effects of TGF-β in certain cells, including smooth muscle cells, and Chan et al. (2010) have reported that this antagonism is mediated in part via the function of miR-24. However, PDGF-BB induces the expression of miR-24, which in turn down-regulates Tribbles-like protein-3 (Trb3). Trb3 has been shown to induce the degradation of Smurf1 (Chan et al. 2007), and repression of Trb3 by miR-24 therefore results in the reduced expression of Smad proteins and the attenuation of TGF-β and bone morphogenetic protein signaling.
The interaction of TGF-β signaling and miRNAs also contributes to the regulation of renal function. TGF-β activates prosurvival PI3K-Akt signaling in glomerular mesangial cells by inducing the expression of miR-216a and miR-217, which target the phosphatase and tensin homolog (PTEN; Kato et al. 2009).
Concluding remarks and perspectives
TGF-β has been studied with regard to the regulation of intercellular communication for over three decades. The intracellular TGF-β signal transduction pathway has also been vigorously investigated, and a large number of studies have elucidated its simple but well-organized mode of transmission. At the core of this signaling pathway, TGF-β induces its membrane receptors directly to activate Smad proteins, which then form transcriptional complexes to control target genes. One crucial question concerning the TGF-β signaling pathway is how such a simple signal transduction pathway triggers multiple behaviors in cellular context-dependent fashion, i.e., how does TGF-β induce different responses in two different types of cells, despite their derivation from a single cell and possession of identical genetic makeup?
This question has been answered in part in terms of the classical frames: cross-interaction with other signaling pathways, different repertoires of Smad-binding transcription factors, and genetic alterations, especially in cancer cells. Nevertheless, the question remains largely unanswered, and recent research has added new frames to the field of intracellular TGF-β signal transduction.
The importance of epigenetic regulation in the development and maintenance of the human body is indicated by its disturbance in several types of diseases. Not surprisingly, gene expression and cell responses induced by TGF-β stimulation are regulated by epigenetic systems. Dynamic epigenetic changes determine an “open conformation” or “closed conformation” of chromatin status on TGF-β target genes; this is directly reflected in the induction of certain cell responses by TGF-β. Thus, differences in the epigenetic map can, at least in part, explain the cellular context-dependent diversity of TGF-β-induced cell responses.
Another new frame of intracellular signal transduction is its regulation by non-coding RNAs. The subtraction of transcribed mRNAs has added a novel paradigm to the regulation of TGF-β signal transduction, and recent research has demonstrated that interactions of TGF-β signaling and non-coding RNA occur at various levels. In addition to changes in non-coding RNA repertories by TGF-β stimulation at the transcriptional level, the TGF-β-Smad pathway is involved in the process of maturation of miRNAs. On the other hand, TGF-β-mediated cell responses, including cell proliferation and EMT, are affected by non-coding RNAs through direct and/or indirect modulation of TGF-β signaling.
The field of research into TGF-β signaling is thus still spreading. In addition, recent research has added new dimensions to the TGF-β field. Further work is needed to obtain a complete TGF-β map for the elucidation of the mechanisms of TGF-β-related diseases and for the development of TGF-β-based therapeutic strategies.
References
Abdalla SA, Letarte M (2006) Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 43:97–110
Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, Parenti AR, Rosato A, Bicciato S, Balmain A, Piccolo S (2009) A mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137:87–98
Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, Rodón L, Folch G, Carmona MA, Prieto-Sánchez RM, Barba I, Martínez-Sáez E, Prudkin L, Cuartas I, Raventós C, Martínez-Ricarte F, Poca MA, García-Dorado D, Lahn MM, Yingling JM, Rodón J, Sahuquillo J, Baselga J, Seoane J (2010) TGF-β receptor inhibitors target the CD44high/Id1high glioma-initiating cell population in human glioblastoma. Cancer Cell 18:655–668
Bernabeu C, Lopez-Novoa JM, Quintanilla M (2009) The emerging role of TGF-β superfamily coreceptors in cancer. Biochim Biophys Acta 1792:954–973
Bierie B, Moses HL (2006) Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 6:506–520
Blobe GC, Schiemann WP, Lodish HF (2000) Role of transforming growth factor β in human disease. N Engl J Med 342:1350–1358
Bruna A, Darken RS, Rojo F, Ocaña A, Peñuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, Seoane J (2007) High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11:147–160
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 9:582–589
Chan MC, Nguyen PH, Davis BN, Ohoka N, Hayashi H, Du K, Lagna G, Hata A (2007) A novel regulatory mechanism of the bone morphogenetic protein (BMP) signaling pathway involving the carboxyl-terminal tail domain of BMP type II receptor. Mol Cell Biol 27:5776–5789
Chan MC, Hilyard AC, Wu C, Davis BN, Hill NS, Lal A, Lieberman J, Lagna G, Hata A (2010) Molecular basis for antagonism between PDGF and the TGFβ family of signalling pathways by control of miR-24 expression. EMBO J 29:559–573
Daly AC, Randall RA, Hill CS (2008) Transforming growth factor β-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol 28:6889–6902
Davis BN, Hilyard AC, Lagna G, Hata A (2008) SMAD proteins control DROSHA-mediated microRNA maturation. Nature 454:56–61
Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A (2010) Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell 39:373–384
Davis-Dusenbery BN, Hata A (2010) Mechanisms of control of microRNA biogenesis. J Biochem 148:381–392
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425:577–584
Dews M, Fox JL, Hultine S, Sundaram P, Wang W, Liu YY, Furth E, Enders GH, El-Deiry W, Schelter JM, Cleary MA, Thomas-Tikhonenko A (2010) The myc-miR- 17~92 axis blunts TGFβ signaling and production of multiple TGFβ-dependent antiangiogenic factors. Cancer Res 70:8233–8246
Edlund S, Lee SY, Grimsby S, Zhang S, Aspenström P, Heldin CH, Landström M (2005) Interaction between Smad7 and β-catenin: importance for transforming growth factor β-induced apoptosis. Mol Cell Biol 25:1475–1488
Ehata S, Johansson E, Katayama R, Koike S, Watanabe A, Hoshino Y, Katsuno Y, Komuro A, Koinuma D, Kano MR, Yashiro M, Hirakawa K, Aburatani H, Fujita N, Miyazono K (2011) Transforming growth factor-β decreases the cancer-initiating cell population within diffuse-type gastric carcinoma cells. Oncogene 30:1693–1705
Esteller M (2007) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 8:286–298
Esteller M (2008) Epigenetics in cancer. N Engl J Med 358:1148–1159
Feng XH, Derynck R (2005) Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol 21:659–693
Filipowicz W, Bhattacharyya SN, Sonenberg N (2008) Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9:102–114
Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P (2010) The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol 10:554–567
Furuhashi M, Yagi K, Yamamoto H, Furukawa Y, Shimada S, Nakamura Y, Kikuchi A, Miyazono K, Kato M (2001) Axin facilitates Smad3 activation in the transforming growth factor β signaling pathway. Mol Cell Biol 21:5132–5141
Gatza CE, Oh SY, Blobe GC (2010) Roles for the type III TGF-β receptor in human cancer. Cell Signal 22:1163–1174
Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P (2003) Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol Cell 12:817–828
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601
Guo X, Wang XF (2009) Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res 19:71–88
Han G, Li AG, Liang YY, Owens P, He W, Lu S, Yoshimatsu Y, Wang D, ten Dijke P, Lin X, Wang XJ (2006) Smad7-induced β-catenin degradation alters epidermal appendage development. Dev Cell 11:301–312
Hannon GJ, Beach D (1994) p15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature 371:257–261
Hata A, Davis BN (2009) Control of microRNA biogenesis by TGFβ signaling pathway—a novel role of Smads in the nucleus. Cytokine Growth Factor Rev 20:517–521
Hirabayashi Y, Gotoh Y (2010) Epigenetic control of neural precursor cell fate during development. Nat Rev Neurosci 11:377–388
Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M (2009) Role of Ras signaling in the induction of Snail by transforming growth factor-β. J Biol Chem 284:245–253
Hussein SM, Duff EK, Sirard C (2003) Smad4 and β-catenin co-activators functionally interact with lymphoid-enhancing factor to regulate graded expression of Msx2. J Biol Chem 278:48805–48814
Ikushima H, Miyazono K (2010a) Cellular context-dependent "colors" of transforming growth factor-β signaling. Cancer Sci 101:306–312
Ikushima H, Miyazono K (2010b) TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer 10:415–424
Ikushima H, Komuro A, Isogaya K, Shinozaki M, Hellman U, Miyazawa K, Miyazono K (2008) An Id-like molecule, HHM, is a synexpression group-restricted regulator of TGF-β signalling. EMBO J 27:2955–2965
Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K (2009) Autocrine TGF-β signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 5:504–514
Jen J, Harper JW, Bigner SH, Bigner DD, Papadopoulos N, Markowitz S, Willson JK, Kinzler KW, Vogelstein B (1994) Deletion of p16 and p15 genes in brain tumors. Cancer Res 54:6353–6358
Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, Rossi JJ, Natarajan R (2009) TGF-β activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol 11:881–889
Kestler HA, Kühl M (2008) From individual Wnt pathways towards a Wnt signalling network. Philos Trans R Soc Lond B Biol Sci 363:1333–1347
Koinuma D, Tsutsumi S, Kamimura N, Taniguchi H, Miyazawa K, Sunamura M, Imamura T, Miyazono K, Aburatani H (2009a) Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor β signaling. Mol Cell Biol 29:172–186
Koinuma D, Tsutsumi S, Kamimura N, Imamura T, Aburatani H, Miyazono K (2009b) A promoter-wide analysis of Smad4 binding sites in human epithelial cells. Cancer Sci 100:2133–2142
Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, Cheng JQ (2008) MicroRNA-155 is regulated by the transforming growth factor β/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol 28:6773–6784
Korpal M, Lee ES, Hu G, Kang Y (2008) The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283:14910–14914
Kulis M, Esteller M (2010) DNA methylation and cancer. Adv Genet 70:27–56
Labbé E, Letamendia A, Attisano L (2000) Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signaling by the transforming growth factor-β and Wnt pathways. Proc Natl Acad Sci USA 97:8358–8363
Labbé E, Lock L, Letamendia A, Gorska AE, Gryfe R, Gallinger S, Moses HL, Attisano L (2007) Transcriptional cooperation between the transforming growth factor-β and Wnt pathways in mammary and intestinal tumorigenesis. Cancer Res 67:75–84
Lee JS, Smith E, Shilatifard A (2010) The language of histone crosstalk. Cell 142:682–685
Levy K, Hill CS (2006) Alteration in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev 17:41–58
Liu W, Rui H, Wang J, Lin S, He Y, Chen M, Li Q, Ye Z, Zhang S, Chan SC, Chen YG, Han J, Lin SC (2006) Axin is a scaffold protein in TGF-β signaling that promotes degradation of Smad7 by Arkadia. EMBO J 25:1646–1658
Liu IM, Schilling SH, Knouse KA, Choy L, Derynck R, Wang XF (2009) TGFβ-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGFβ switch. EMBO J 28:88–98
Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR (2005) MicroRNA expression profiles classify human cancers. Nature 435:834–838
Luo K (2008) Regulation of the Smad pathway by signaling cross-talk. In: Derynck R, Miyazono K (eds) The TGF-β family. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, pp 439–459
Massagué J (2008) TGFβ in cancer. Cell 134:215–230
Massagué J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19:2783–2810
McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, McCormick MK, Pericak-Vance MA, Heutink P, Oostra BA, Haitjema T, Westerman CJJ, Porteous ME, Guttmacher AE, Letarte M, Marchuk DA (1994) Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 8:345–351
Mestdagh P, Boström AK, Impens F, Fredlund E, Van Peer G, De Antonellis P, Stedingk K von, Ghesquière B, Schulte S, Dews M, Thomas-Tikhonenko A, Schulte JH, Zollo M, Schramm A, Gevaert K, Axelson H, Speleman F, Vandesompele J (2010) The miR-17-92 microRNA cluster regulates multiple components of the TGF-β pathway in neuroblastoma. Mol Cell 40:762–773
Moustakas A, Heldin CH (2005) Non-Smad TGF-β signals. J Cell Sci 118:3573–3584
Moustakas A, Heldin CH (2009) The regulation of TGFβ signal transduction. Development 136:3699–3714
Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N, Hirao A (2010) TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 463:676–680
Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E (1996) TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev 10:2462–2477
Oft M, Akhurst RJ, Balmain A (2002) Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat Cell Biol 4:487–494
Ordovás JM, Smith CE (2010) Epigenetics and cardiovascular disease. Nat Rev Cardiol 7:510–519
Park SM, Gaur AB, Lengyel E, Peter ME (2008) The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22:894–907
Peñuelas S, Anido J, Prieto-Sánchez RM, Folch G, Barba I, Cuartas I, García-Dorado D, Poca MA, Sahuquillo J, Baselga J, Seoane J (2009) TGF-β increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 15:315–327
Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, Martino I de, Iliopoulos D, Pilozzi E, Liu CG, Negrini M, Cavazzini L, Volinia S, Alder H, Ruco LP, Baldassarre G, Croce CM, Vecchione A (2008) E2F1-regulated microRNAs impair TGFβ-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell 13:272–286
Rice JC, Allis CD (2001) Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr Opin Cell Biol 13:263–273
Rifkin DB (2005) Latent transforming growth factor-β (TGF-β) binding proteins: orchestrators of TGF-β availability. J Biol Chem 280:7409–7412
Roberts AB, Wakefield LM (2003) The two faces of transforming growth factor β in carcinogenesis. Proc Natl Acad Sci USA 100:8621–8623
Sakaguchi S, Miyara M, Costantino CM, Hafler DA (2010) FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10:490–500
Sasaki T, Suzuki H, Yagi K, Furuhashi M, Yao R, Susa S, Noda T, Arai Y, Miyazono K, Kato M (2003) Lymphoid enhancer factor 1 makes cells resistant to transforming growth factor β-induced repression of c-myc. Cancer Res 63:801–806
Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O'Connor E, Shokat KM, Fisher AG, Merkenschlager M (2008) T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA 105:7797–7802
Sawan C, Herceg Z (2010) Histone modifications and cancer. Adv Genet 70:57–85
Schmierer B, Hill CS (2007) TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 8:970–982
Seto A, Ikushima H, Suzuki T, Sato Y, Fukai S, Yuki K, Miyazawa K, Miyazono K, Ishitani R, Nureki O (2009) Crystallization and preliminary X-ray diffraction analysis of GCIP/HHM transcriptional regulator. Acta Crystallogr F Struct Biol Cryst Commun 65:21–24
Shi Y, Massagué J (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113:685–700
Singh A, Settleman J (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29:4741–4751
Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9:465–476
Tang B, Yoo N, Vu M, Mamura M, Nam JS, Ooshima A, Du Z, Desprez PY, Anver MR, Michalowska AM, Shih J, Parks WT, Wakefield LM (2007) Transforming growth factor-β can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res 67:8643–8652
Tang Y, Liu Z, Zhao L, Clemens TL, Cao X (2008) Smad7 stabilizes β-catenin binding to E-cadherin complex and promotes cell-cell adhesion. J Biol Chem 283:23956–23963
ten Dijke P, Arthur HM (2007) Extracellular control of TGF-β signalling in vascular development and disease. Nat Rev Mol Cell Biol 8:857–869
ten Dijke P, Goumans MJ, Pardali E (2008) Endoglin in angiogenesis and vascular diseases. Angiogenesis 11:79–89
Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139:871–890
Varga J, Pasche B (2009) Transforming growth factor β as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol 5:200–206
Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, Stillman IE, Roberts D, D'Amore PA, Epstein FH, Sellke FW, Romero R, Sukhatme VP, Letarte M, Karumanchi SA (2006) Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 12:642–649
Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T (2009) Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132:875–886
Wang Y, Yu Y, Tsuyada A, Ren X, Wu X, Stubblefield K, Rankin-Gee EK, Wang SE (2011) Transforming growth factor-β regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene 30:1470–1480
Winter J, Jung S, Keller S, Gregory RI, Diederichs S (2009) Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11:228–234
Wrana JL, Ozdamar B, Le Roy C, Benchabane H (2008) Signaling receptors of the TGF-β family. In: Derynck R, Miyazono K (eds) The TGF-β family. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, pp 179–202
Yoshimura A, Wakabayashi Y, Mori T (2010) Cellular and molecular basis for the regulation of inflammation by TGF-β. J Biochem 147:781–792
Zhang YE (2009) Non-Smad pathways in TGF-β signaling. Cell Res 19:128–139
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by KAKENHI (Grant-in-aid for Scientific Research) and the Global COE Program (Integrative Life Science Based on the Study of Biosignaling Mechanisms) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by a Grant-in-Aid for Cancer Research for the Third-Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan. H.I. was supported by a Tetsumon scholarship for the Ph.D.-M.D. program of the University of Tokyo.
Rights and permissions
About this article
Cite this article
Ikushima, H., Miyazono, K. TGF-β signal transduction spreading to a wider field: a broad variety of mechanisms for context-dependent effects of TGF-β. Cell Tissue Res 347, 37–49 (2012). https://doi.org/10.1007/s00441-011-1179-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-011-1179-5