
Abstract
Recent advances in genome-editing techniques have made it possible to modify any desired DNA sequence by employing programmable nucleases. These next-generation genome-modifying tools are the ideal candidates for therapeutic applications, especially for the treatment of genetic disorders like sickle cell disease (SCD). SCD is an inheritable monogenic disorder which is caused by a point mutation in the β-globin gene. Substantial success has been achieved in the development of supportive therapeutic strategies for SCD, but unfortunately there is still a lack of long-term universal cure. The only existing curative treatment is based on allogeneic stem cell transplantation from healthy donors; however, this treatment is applicable to a limited number of patients only. Hence, a universally applicable therapy is highly desirable. In this review, we will discuss the three programmable nucleases that are commonly used for genome-editing purposes: zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9). We will continue by exemplifying uses of these methods to correct the sickle cell mutation. Additionally, we will present induction of fetal globin expression as an alternative approach to cure sickle cell disease. We will conclude by comparing the three methods and explaining the concerns about their use in therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sickle cell disease (SCD) is a generic term used for various inheritable genetic disorders which result in malformation of the hemoglobin protein structure, leading to abnormal sickle-shaped red blood cells and a wide range of other clinical pathologies. SCD affects 70,000 to 100,000 people in the USA (Hassell 2010) and approximately 300,000 neonates worldwide every year (Piel 2016), making it one of the most common monogenic diseases. The median lifespan of SCD patients in the USA is 42 years for females and 38 years for males (Lanzkron et al. 2013). Since the identification of the causal monogenic mutation in 1956 by Ingram (1956), genetics and regulation of the implicated genes have been an area of intensive research.
SCD is caused by inheriting two abnormal copies of the β-globin (HBB) gene, at least one of which is the hemoglobin S (HbS) variant. The presence of two copies of the HbS gene (HbSS) causes sickle cell anemia, the most severe case compared to compound heterozygosity (Frenette and Atweh 2007). The HbS variant is a result of a single nucleotide substitution from A to T in the codon for the sixth amino acid in the β-globin protein, a subunit of the oxygen-carrying tetrameric hemoglobin protein (α2β2) in red blood cells (Frenette and Atweh 2007). This point mutation converts a hydrophilic glutamic acid to a hydrophobic valine at position six in β-globin, leading to abnormal hemoglobin folding. The resulting HbS hemoglobin has a tendency to polymerize and aggregate, changing red blood cells into stiff, sickle-shaped cells (Ashley-Koch et al. 2000). These inflexible cells tend to stick to each other and to the walls of blood vessels, causing vaso-occlusion, which slows down blood flow and decreases oxygen delivery to tissues. Such poor blood circulation can cause damage to organs and lead to complications of pain crises, acute chest syndrome, stroke, splenic and renal dysfunction, and susceptibility to infections (Ashley-Koch et al. 2000). Sickle cells also have a tendency to hemolyze, shortening their lifespan and leading to chronic anemia. Detailed reviews on the genetics (Steinberg and Sebastiani 2012) and physiopathology (Paradowski 2015; Ballas 2015) of SCD are available for further reading.
Despite the high carrier frequency of HbS gene in various areas of the world such as sub-Saharan Africa, Middle East or Indian sub-continent (Weatherall and Clegg 2001), unfortunately there is no universal cure yet, although supportive treatments to help reduce disease complications are available. These treatments include blood transfusions, preventive therapies such as penicillin prophylaxis and pneumococcal vaccination, and hydroxyurea therapy, which decreases HbS polymerization by increasing fetal hemoglobin (HbF) levels (Aliyu et al. 2006). Considering the fact that all blood cells are derived from hematopoietic stem cells (HSCs), the only curative treatment available for SCD has been allogeneic hematopoietic stem cell transplantation (HSCT) from healthy donors to replenish the patient’s body with healthy blood cells (Shenoy 2011). However, even though HSCT is a promising treatment strategy with a success rate of 85–90 % (Locatelli and Pagliara 2012), this method is not available for every patient because of the rare availability of matched donors and associated side effects including long-term toxicities such as infertility and endocrinopathies.
In principle, autologous transplantation of patient-derived HSCs after in vitro correction for the SCD could offer an invaluable cure for patients without a compatible donor (Fig. 1). Although it is not the focus of our review, it is worth pointing out that gene transfer therapy has been an important approach for the treatment of various hemaglobinopathies including SCD. Briefly, the main focus of previous efforts has been on delivery of either γ-globin or an antisickling β-globin protein to inhibit polymerization of the HbS protein (Pawliuk et al. 2001; Levasseur et al. 2003; Sadelain et al. 2004; Pestina et al. 2009) However, stable protein expression requires utilization of viral vectors for efficient gene delivery and such vectors still hold long-term concerns such as immunogenic response or carcinogenesis due to random insertional mutagenesis (Check 2002; Baum et al. 2003; Thomas et al. 2003; Woods et al. 2006; Hacein-Bey-Abina et al. 2008). Strategies to reduce insertional mutagenesis risk without compromising gene transfer efficiency have been an extensive research area and excellent reviews about the recent advances in gene transfer therapy of SCD can be found in other papers (Dong et al. 2013; Chandrakasan and Malik 2014).

Potential gene therapy-based treatment for sickle cell disease (SCD) patients. In theory, both hematopoietic stem cells (HSCs) and induced pluripotent stem cells (iPSCs) can be used for therapy. In an ideal case, HSCs isolated from patients can be corrected for the mutation in vitro, followed by transplantation into the patient (the procedure is indicated with solid arrows). As an alternative, somatic cells such as skin or blood cells from patients can first be reprogrammed into iPSCs, followed by correction of the cells and differentiation into HSCs. These corrected HSCs can then be transplanted back into the patient to create healthy blood cells. The procedure for iPSCs is indicated with dashed arrows. Even though both strategies are theoretically viable, there are various issues to be addressed before use of HSCs or iPSCs for therapy. For further discussion, please see “Efficiency”
To cure monogenic diseases like SCD for which the causative mutation is already identified, another promising approach would be directly correcting the mutation at the endogenous locus without any need for expression of an exogenous gene. Such a precise gene-editing-based therapy could offer a safer alternative to the current transgene expression-based therapies. This review will focus on strategies in the field of genome editing to treat SCD by employing programmable nucleases: zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and RNA-guided endonucleases (RGENs), commonly known as clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9). The functionality of these programmable nucleases will be explained briefly, followed by describing the experimental approaches applied to correct the sickle mutation in relevant model systems. Moreover, therapeutic strategies based on induction of fetal hemoglobin (HbF) expression, which has been shown to rescue the disease phenotype of SCD by preventing HbS polymerization, will be described. We will conclude with an overview of the current limitations to be addressed before translation of the genome-editing techniques for clinical applications.
Gene targeting as a strategy for sickle cell therapeutics
Traditional gene targeting by homologous recombination
Gene targeting is the modification of the chromosome by utilizing homologous recombination (HR) between the genomic DNA and an exogenous targeting vector (Bollag et al. 1989). The targeting vector, or the “donor DNA”, contains an insert DNA to be added to a specific region in the genome and this insert is flanked by sequences homologous to the target locus, which are referred to as 5′ and 3′ homology arms (Fig. 2a). Precise integration of the insert DNA utilizes homology-directed repair (HDR) of spontaneous DNA double-strand breaks (DSBs), which normally uses sister chromatid as repair template (Pfeiffer et al. 2000). After transfection of a donor DNA with sequences homologous to the locus to be modified, the HR between the chromosomal and exogenous DNA can also be utilized to repair spontaneous DSBs, leading to integration of the desired insert into the target site (Fig. 2a; Smithies et al. 1985).

Homologous recombination and logic of using programmable nucleases. a Gene targeting is the insertion of an exogenous DNA into a desired locus by homologous recombination (HR). For modification of the genomic DNA (gDNA) by HR, cells need to be transfected with a donor DNA which contains the desired insert, and preferably a positive selection marker, all of which are flanked by 3′ and 5′ homology arms (HAs). The insert is added to the genome by HR between HAs and their corresponding homologous sequences in the genome. If a selection marker is co-inserted, its removal after positive selection is desirable. The excision of the marker can be performed by methods such as Cre-loxP system or piggyBac-excision. b Double-strand breaks generated by programmable nucleases (indicated by red triangle) can be repaired by either non-homologous end joining (NHEJ) or HR. NHEJ can cause frameshift mutations, leading to gene knockout. In the presence of a donor DNA, repair by HR can create gene deletion, replacement or insertion. Creating a DSB at the target site can increase the efficiency of gene targeting by HR
Gene targeting can be a useful approach for the treatment of SCD since the treatment would only require replacement of the mutant sequence with the correct one through HR and this would also preserve the natural environment of the HBB gene. For gene correction, various strategies have been used in mammalian cells. In selection-free systems, a DSB repair template with only the corrected mutation is introduced into the cells. The repair template could be in the form of a plasmid, or a short single-stranded oligonucleotide for small modifications (Ochiai 2015). Some novel donor DNAs such as integration-deficient lentiviral vectors (IDLVs), adenoviruses (AdVs) or adeno-associated viruses (AAV) could be used as well (Hendrie and Russell 2005; Khan et al. 2011; Holkers et al. 2014). As another strategy, a positive selection marker together with the corrected mutation site can be introduced into the genome to select for the cells with gene correction (Ochiai 2015). However, the presence of a transgene in the genome is undesirable and this strategy would require a second round of modifications to excise the selection cassette out of the genome (Fig. 2a).
One of the first studies showing that HR between a chromosomal region and an exogenous DNA can happen was actually performed by targeting the HBB locus, considering its potential use for gene correction in SCD or thalassemia patients (Smithies et al. 1985). Later, gene targeting was shown to be successful in mouse embryonic stem cells (mESCs) (Thomas and Capecchi 1987), followed by its use in mESCs harboring human HbS gene (Chang et al. 2006; Wu et al. 2006) to replace the HbS with normal human HBB gene. HR-mediated modification of the HBB locus was demonstrated in human cells as well (Goncz et al. 2002).
Following the development of induced pluripotent stem cell (iPSC) technologies in 2006 (Takahashi and Yamanaka 2006), the therapeutic potential of gene targeting in iPSCs was demonstrated, including for hematological diseases (Kim 2014; Focosi et al. 2014; Singh et al. 2015). In one example, Hanna et al. derived somatic cells from a humanized sickle cell anemia mouse model, reprogrammed the cells into iPSCs, corrected the sickle mutation in iPSCs by HR and differentiated iPSCs into hematopoietic progenitors to be transplanted back into the mouse (Hanna et al. 2007). The suggested strategy was able to rescue the mice from disease complications, although it is important to note that ectopic HoxB4 expression, which was used in this paper to generate repopulating cells from iPSCs, cannot be applied therapeutically due to the significant risk of leukemogenesis (Larochelle and Dunbar 2008; Zhang et al. 2008). Moreover, HoxB4 overexpression does not show the same effect in human pluripotent cells (Wang et al. 2005). Thus, iPSC-dependent strategies suffer from lack of efficient methods to derive HSCs from human iPSCs. A later study was able to correct the sickle mutation in patient-derived human iPSCs using a helper-dependent adenoviral vector as a targeting DNA (Li et al. 2011b). Nevertheless, further characterization of the methodology and development of efficient methods to derive repopulating hematopoietic progenitors from human iPSCs will be necessary before clinical applications in humans. Issues related to the use of iPSCs for therapeutics that are not directly genome editing related are out of the scope of this paper; however, more detailed discussions can be found in other reviews (Lengerke and Daley 2010; Slukvin 2013).
A disadvantage of using HR for gene targeting in human cells is its low efficiency. One reason is that this method relies on rare DSBs within the target region, requiring the use of extensive homology arms to capture the DSBs (Deng and Capecchi 1992). Additionally, HR is not the preferred pathway for repairing DSBs in mammalian cells, resulting in an overall frequency of less than one event per 105 cells (Bollag et al. 1989; Vasquez et al. 2001), which is too low to be used in the clinic and makes it difficult to identify correctly targeted clones. However, several proof-of-principle experiments suggested that introduction of a DSB at a specific site in the genome can increase HR rates at the desired region (Choulika et al. 1995; Johnson and Jasin 2001). An attractive method for inducing site-specific DSBs at any region of interest is the use of programmable nucleases. In “Use of programmable nucleases to mediate correction of the sickle mutation by gene targeting”, we will explain how programmable nucleases were utilized to improve gene-targeting efficiency in the HBB locus.
Use of programmable nucleases to mediate the correction of the sickle mutation by gene targeting
Programmable nucleases can significantly increase gene-targeting efficiency, making them very promising tools for the treatment of monogenic SCD. ZFNs, TALENs and CRISPR/Cas9 are the commonly used genome-editing tools which can create a desired mutation in a wide variety of cell types and eukaryotic models. In brief, gene editing by programmable nucleases is based on recruitment of an endonuclease to a user-specified genomic site for induction of a site-specific DNA DSB (Fig. 2b). The desired modification is introduced to the genome as the DSB is being repaired by the cellular DNA repair mechanisms. One way the DSB can be repaired is error-prone non-homologous end joining (NHEJ), which causes gene knockout as a result of insertion/deletion mutations. In the presence of a donor DNA with suitable homology arms, the damage can also be repaired through HR, enabling gene insertion, replacement or deletion (Fig. 2b).
The potential of these three programmable nucleases for therapeutic applications has been demonstrated in previous proof-of-concept studies (Cox et al. 2015; Abil et al. 2015; Xiao-Jie et al. 2015; LaFountaine et al. 2015). In the following subsections, we will evaluate each programmable nuclease individually in terms of the experimental approaches used for correction of the sickle mutation by gene targeting.
Zinc finger nucleases (ZFNs)
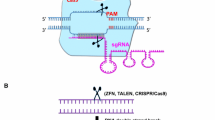
ZFNs were the first programmable nucleases used for gene-editing purposes (Bibikova et al. 2001, 2002). These artificial proteins have two functional domains: a DNA-binding domain (DBD) based on the eukaryotic zinc finger transcription factors and a FokI endonuclease domain (Fig. 3a; Urnov et al. 2010). DBD consists of tandem repeats of three to six zinc finger proteins, each of which can recognize three base pairs (bp). Zinc finger proteins have been engineered so that they could recognize most of the three bp combinations (Urnov et al. 2010). Thus, each ZFN monomer can recognize a desired DNA sequence of 9–18 bp. Changing the zinc finger proteins in ZFNs can modify the recognition site for each monomer. To introduce a DSB to a specific locus, the DNA cleavage domain of the type IIS restriction endonuclease FokI is fused to the DNA-binding domain. FokI functions only when it is dimerized (Wah et al. 1998), which is the reason why ZFNs work in pairs for DSB induction. The dimer recognizes a total of 18–36 bp sequence and binds to the DNA in a tail-to-tail orientation with a spacer in between the binding sites of each monomer, allowing FokI dimerization (Fig. 3a). The DSB is induced in the region between two ZFN binding sites. Several strategies have been developed to assemble a DBD of multiple zinc finger proteins recognizing a user-defined region, which were discussed in other reviews (Cathomen and Joung 2008; Urnov et al. 2010; Carroll 2011).

Schematic representation of the three programmable nucleases. a DNA-binding domain (DBD) of zinc finger nucleases (ZFNs) is composed of tandem repeats of zinc finger proteins (ZFPs) at the amino terminus (N). Each ZFP recognizes three nucleotides. ZFNs have a FokI nuclease (light purple) in their carboxyl terminus. A ZFN heterodimer bound in a tail-to-tail orientation at the target site induces a site-specific DNA double-strand break within the spacer between two ZFN-binding sites (indicated as lowercase letters). b The DBD of transcription activator-like effector nucleases (TALENs) consists of repeats of ~34 amino acids and each repeat (shown as green, red, blue or yellow boxes) recognizes one nucleotide. Similar to ZFNs, DBD is fused to FokI nuclease and the heterodimer induces a double-strand break within the spacer region. c Shown is the Cas9 nuclease from S. pyogenes (yellow) which is recruited to the target DNA by a guide RNA (gRNA) containing a 20 nucleotide spacer sequence (highlighted in green), followed by a scaffold RNA. Target recognition is based on Watson–Crick base pairing between the spacer and the target DNA. For proper recognition, the DNA target must be directly upstream of the protospacer adjacent motif (PAM, highlighted in blue) 5′-NGG-3′. Double-strand break is induced ~3 bp upstream of PAM (red triangle)
Being the first programmable nuclease to be discovered, initial studies started with ZFNs. In one of the first demonstrations of their use in mammalian cells, a ZFN that can target a region surrounding the sickle mutation at the β-globin locus was designed, although it was not directly tested at the β-globin locus, but instead in a GFP reporter system (Porteus 2006). An indication for the therapeutic potential of ZFNs in SCD came from the studies in which ZFNs were used to correct sickle mutation in patient-derived iPSCs (Sebastiano et al. 2011). Use of site-specific nucleases enabled more efficient gene targeting to correct sickle mutation in human iPSCs, which was otherwise very difficult to achieve. To enhance the identification of the correct clones with a successful gene-targeting event, a drug-resistance cassette was also included in the donor construct. With this method, a mean of 9.8 % targeting efficiency was achieved. To create transgene-free cells, Cre-recombinase-mediated excision of the selection marker was utilized. However, this method still leaves a loxP site behind as a “scar” in the genome, which can affect β-globin expression. An encouraging finding in the studies of Sebastiano et al. was the lack of off-target mutagenesis at potential off-target sites, although a genome-wide analysis would be more informative to check for any possible off-target mutations. In another report using iPSCs derived from sickle cell anemia patients, a similar approach was used for gene correction (Zou et al. 2011). They reported correction of one of the HbS alleles and successful differentiation of the iPSCs into erythroid cells, although the corrected β-globin gene showed reduced expression in erythroids. The reduced expression can be a result of the remaining loxP site after excision of the selection marker. Despite the use of hsv-TK-negative selection marker to reduce the frequency of false-positive clones due to random integration of the whole plasmid, the targeting efficiency was not high (1 in 300 drug-resistant clones). Low HR efficiency could be due to the fact that the HBB gene is silent in iPSCs and silent genes are generally more difficult to target by HR (Hockemeyer et al. 2009). Moreover, difference in efficiencies reported by Sebastiano et al. and Zou et al. could be due to different designs of ZFNs and donors used in each study.
A recent use of ZFNs was performed in CD34+ hematopoietic stem and progenitor cells (HSPCs), instead of iPSCs (Hoban et al. 2015). Electroporation of a ZFN mRNA led to high cleavage efficiency of 35 to 65 %. A comprehensive analysis of off-target DSBs using IDLV capture method showed high specificity of ZFNs. Another distinctive part of that study was the use of IDLVs or short oligonucleotides as the donor DNA. Lentiviral vectors have an advantage of high transduction efficiency in stem cells, whereas short oligonucleotide donor templates are easier to use with low cost. The modified HSPCs were able to be engrafted into immunocompromised mice and they differentiated into erythroid, myeloid and lymphoid cells, both in vivo and in vitro (Hoban et al. 2015). Despite the promising results, it is important to note that although bulk-edited human HSPCs had a gene modification level of 10–20 % before transplantation, gene correction level was significantly lower in human cells in the spleen and bone marrow of the mice analyzed 16 weeks after engraftment. Thus, low gene correction rate in long-term repopulating HSCs was still a major hurtle.
Transcription activator-like effector nucleases (TALENs)
Similar to ZFNs, TALENs bind to specific DNA sequences and function in heterodimers to induce a site-specific DSB (Carlson et al. 2012). A TALEN monomer comprises a DBD fused to a FokI nuclease domain (Fig. 3b; Christian et al. 2010; Li et al. 2011a). The DBD is derived from the transcription activator-like effectors (TALEs) of the plant pathogenic bacteria Xanthomonas, which injects TALEs into the plant cells to act as transcriptional activators in the nucleus (Boch and Bonas 2010). The DBD consists of tandem repeats of 33–35 amino acids (Boch et al. 2009; Moscou and Bogdanove 2009). Typically, TALENs are designed to have 14–31 repeats. These nearly identical repeats differ only at the 12th and 13th amino acid residues, which are known as repeat variable diresidue (RVD). Each repeat recognizes a single nucleotide and the RVDs determine the base specificity (Boch et al. 2009; Moscou and Bogdanove 2009). The typically used code is: NI recognizes adenine, NG recognizes thymine, HD recognizes cytosine, NH recognizes guanine and NN recognizes both guanine and adenine. A TALEN pair is required for the formation of a site-specific DSB since TALENs also utilize the DNA cleavage domain of FokI nuclease. Numerous TALEN synthesis platforms, all of which are dependent on the assembly of the repeats in a pre-determined order, are available (Liang et al. 2014; Kim et al. 2013; Sun and Zhao 2013).
After discovery of TALENs, their therapeutic potential in SCD was also explored. In an initial study, a pair of TALENs with optimized architecture and high efficiency was designed to induce a DSB at a sequence close to the sickle mutation in the HBB locus (Sun et al. 2012). The improved TALEN architecture allowed targeting the sites without a preceding 5′T, which is normally a requirement for TALEN binding. This finding increases the number of possible sites that can be targeted by TALENs, giving more flexibility. This study was followed by targeting the endogenous HBB locus in patient-derived human iPSCs with an efficiency of more than 60 % in drug-resistant clones (Sun and Zhao 2014), showing an improvement in gene-targeting efficiency compared to the efficiencies reported in ZFN studies. Seamless correction of the sickle mutation was achieved by using piggyBac transposon instead of the previously used Cre-loxP system. It would be interesting to see whether piggyBac transposon alleviated issues with β-globin expression due to the use of the Cre-loxP system. Absence of off-target events in sites with high sequence similarity was also promising, although a genome-wide analysis would be necessary. A different study reported use of highly active TALENs in β-globin locus with 19 % targeting efficiency even without any drug selection (Voit et al. 2014). However, this study was done in K562 cells and further analysis of the methodology in clinically relevant cells would be desired. Considering the low HR rates in human stem cells, a selection-free system might not be feasible in these cell types. Another study in the same year demonstrated the use of TALENs in patient-specific iPSCs, although the Cre-loxP system was used for excision of the selection marker (Ramalingam et al. 2014). Erythroid cells derived from the corrected iPSCs expressed the wild-type allele to the levels of 30–40 %. Similar to other studies, only one allele was corrected, whereas the other allele still had the sickle mutation. Since heterozygotes of HbA with HbS do not show disease symptoms (Serjeant 2013), correction of one allele can be therapeutically sufficient. Findings of Ramalingam et al. also confirmed no off-target effect at similar sites. Later, a more comprehensive analysis of off-target effects by whole-genome sequencing of several gene-corrected human iPSC clones supported that TALENs led to only a few mutations in the genome (Suzuki et al. 2014). Overall, the above papers suggest that TALENs could be efficient and specific tools for correction of sickle mutation in iPSCs, although its demonstration in HSCs is still missing.
Clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (CRISPR/Cas9)
Currently, CRISPR/Cas9 is the newest and most popular genome-editing tool due to its ease of use and cost-effectiveness. CRISPR and CRISPR-associated (Cas) proteins were first identified as part of the immune system in bacteria against invading viral or plasmid DNA, and more detailed reviews on CRISPR systems can be found elsewhere (Barrangou et al. 2007; Wright et al. 2016). Among the six CRISPR systems identified, the currently used CRISPR/Cas9 technology for gene editing is based on the type II system (Wright et al. 2016). Unlike ZFNs and TALENs, DNA recognition is based on RNA–DNA interaction, rather than protein–DNA interaction. The two major components of CRISPR/Cas9 are a guide RNA (gRNA) and a Cas9 endonuclease. The gRNA contains a user-defined 20-nucleotide targeting sequence and can recruit the Cas9 protein, associated with the scaffold of gRNA, to any DNA sequence (Fig. 3c). One requirement for this system is that the target sequence must be immediately upstream of a protospacer adjacent motif (PAM). The most commonly used Cas9 protein is from Streptococcus pyogenes and it requires a PAM sequence of 5′-NGG-3′. Upon binding to the target, Cas9 induces a DSB ~3 nucleotides upstream of the PAM (Ran et al. 2013b).
A recent study in human iPSCs found that the CRISPR/Cas9 system had better cutting efficiency at the HBB locus compared to ZFNs or TALENs (Huang et al. 2015). In the same study, CRISPR/Cas9 was used for correction of iPSCs from adult SCD patients. The CRISPR-corrected stem cells were able to differentiate into erythrocytes and produce β-globin protein from the corrected allele. Efficient erythrocyte maturation and β-globin expression could be due to either the use of improved culture conditions or of blood-derived iPSCs. Another study also found that CRISPR/Cas9 had better cutting efficiency than TALENs at the HBB locus (Cottle et al. 2015). Cottle et al. observed a dose-dependent activity of nucleases after microinjection into K562 cells. When co-injected with a GFP donor DNA, they observed a homology-directed repair rate of ~10.5 % for CRISPR and ~1.6 % for TALEN. However, since HR efficiencies can change from cell to cell, a direct comparison of efficiencies in clinically useful cells would be more informative. In an effort for rational design of CRISPRs, a detailed bioinformatics analysis of the HBB locus for suitable CRISPR target sites showed that targeting introns could have less off-target effects compared to exons (Luo et al. 2015). An additional reason why introns in HBB locus were suggested as a better target was the finding that they contained less single nucleotide polymorphisms (SNPs), making a designed CRISPR applicable to more people. However, it is important to note that although this finding could be true for the HBB locus, introns may not have fewer SNPs than exons in other loci. In a striking study, Liang and his colleagues took the CRISPR technology one step further and demonstrated its use in human zygotes, although it led to many ethical debates (Liang et al. 2015b). Unfortunately, HR efficiency at the HBB locus of zygotes was low (4 in 54 embryos) and off-target mutations were detected. Another problem was the preference of HBD locus, which has high sequence homology to HBB gene, as DSB repair template instead of the injected short oligonucleotide donor. Although the CRISPR/Cas9 tool has been rapidly accepted by scientists, another study demonstrated that a number of CRISPRs designed to target HBB locus had substantial off-target mutations (Cradick et al. 2013). These evidences highlight the importance of improving the specificity of CRISPR/Cas9 before any human applications. A more detailed discussion about off-target effects can be found in “Off-target effects and toxicity”.
Despite specificity concerns, a recent study showed significant improvement in HDR efficiency in HSCs when CRISPR/Cas9 was used to correct HSPCs with sickle mutation (DeWitt et al. 2016). As opposed to previous attempt for ZFN-mediated gene targeting in HSCs, which resulted in efficiencies below the clinically relevant levels (Hoban et al. 2015), DeWitt et al. showed that CRISPR-mediated gene correction rate in re-populating HSCs was at clinically beneficial levels. This important observation could be due to the preferred delivery method of CRISPR/Cas9 as a ribonucleoprotein complex or it may be due to CRISPR/Cas9 itself. A systematic analysis could help understand the underlying reason for improved efficiency, which can be used as a guideline for the development of an optimum treatment strategy for SCD. This recent report by DeWitt et al. shows that given its reported high gene-targeting efficiency in HSCs, CRISPR/Cas9 may have an advantage in the treatment of hematopoietic diseases.
Comparison of the three programmable nucleases
An advantage of ZFNs is the relatively small size of the coding sequence for a ZFN monomer. In contrast to 1 kb ZFN coding sequence, each TALEN monomer requires around 3 kb, whereas the Cas9-gRNA cassette is around 4.3 kb (Table 1). The large size of TALEN and Cas9 coding DNA could be a problem for some viral delivery methods due to packaging limits. One disadvantage of ZFNs is that the number of sequences that can be effectively targeted by them is limited. There is no collection of all 64 possible zinc finger proteins to target every possible triplet bases (Isalan 2012). Changing ZFN target specificity might require extensive protein engineering. Moreover, targeting efficiency is not high for all designed ZFNs which can be due to the fact that ZFNs mostly have a preference for G-rich sequences (Isalan 2012). In contrast to ZFNs, it is possible to design TALENs that can target any region in the genome due to their high modularity. The reported success of improved TALENs to target sites without a preceding 5′-T suggests that theoretically any region can be targeted by TALENs (Sun et al. 2012). Moreover, recent developments in cloning techniques to assemble TALEN repeats in a desired order made it simpler to produce site-specific TALENs (Liang et al. 2014; Kim et al. 2013; Sun and Zhao 2013). The targeting flexibility is also true for CRISPRs, although it requires a following PAM sequence after the target site (Table 1). Changing targeting specificity of CRISPRs is much simpler than TALENs, since it only requires synthesis of a new oligonucleotide for each new target site and its subsequent cloning by standard methods. Although TALENs and CRISPR/Cas9 can offer more flexibility in choosing a target sequence, site-dependent differences in editing efficiency is true for all programmable nucleases and further studies would be necessary to understand the underlying reason.
Genome editing to induce fetal hemoglobin expression
HbF (α2γ2) constitutes the majority of hemoglobin during fetal stage, however, HbF expression is substantially reduced in adults making only about 0.1–1 % of the total hemoglobin. There are five common haplotypes of the HbS mutation which are identical in the E6V mutation, but differ with regard to γ-globin expression (Bauer et al. 2012). In fact, haplotypes associated with higher HbF levels have a milder clinical course. These observations were further bolstered by studies investigating the mode of action of hydroxyurea (HU), the only FDA-approved pharmacologic treatment for SCD whose therapeutic benefits are partially due to increased HbF expression (Lanzkron et al. 2008). Biochemical studies have shown that in the presence of HbF, HbS polymerization is delayed and solubility of deoxygenated HbS is substantially increased (Sunshine et al. 1978). Hence, HbF has emerged as a crucial target whose expression can be modulated to alleviate complications associated with SCD. Although hydroxyurea is commonly used in SCD therapy, such γ-globin inducing compounds were shown to have variable effects (Steinberg et al. 1997); thus, researchers have been interested in finding more efficient and universal ways of increasing γ-globin levels for the treatment of SCD. In this section we will describe novel uses of programmable nucleases to induce HbF expression.
HbF induction by artificial transcription factors
In addition to being used as site-specific nucleases, zinc fingers, TALEs and CRISPRs can also be used as artificial transcription factors when fused with suitable effector domains such as activation domains or epigenetic modifiers (Fig. 4). By taking advantage of the customizability of these transcription factors and the compensatory nature of HbF expression in SCD, Gräslund et al. designed a zinc finger-based protein called gg1-VP64 (Gräslund et al. 2005). This protein consisted of a DBD, recognizing a specific sequence proximal to the −117 position of the γ-globin promoter, and a transcriptional activator domain VP64. Gg1-VP64 expressing cells showed up to 16-fold higher level of HbF compared to the wild-type K562 cells. Although K562 cells mainly express embryonic hemoglobin Hb Gower 1 and Hb Portland, this study was the first of its kind to demonstrate HbF induction with artificial transcription factors as a viable therapeutic strategy which can be extended to other erythroid cell systems. Gg1-VP64 was then tested in early erythroid progenitors derived from cytokine-mobilized adult peripheral blood CD34+ cells and this artificial transcription factor was able to increase HbF levels from 2 to 20 % of the total hemoglobin which can be therapeutically applicable (Wilber et al. 2010). Costa et al. further showed five-fold induction of HbF in gg1-VP64 β-YAC double-transgenic (bigenic) mice, demonstrating its efficacy in vivo (Costa et al. 2012).

Genome-editing strategies for induction of fetal hemoglobin (γ-globin). The β-globin locus comprises a locus control region (LCR) which regulates various hemoglobins in a developmental stage-specific manner. Embryonic, fetal and adult hemoglobins are sequentially expressed in erythroid cells as a result of interaction with LCR via chromatin looping. The figure depicts therapeutic strategies to induce γ-globin. In one strategy based on artificial transcription factors, an activation domain is tethered to a designer zinc finger protein which recognizes the γ-globin promoter region (Gräslund et al. 2005). Another novel strategy of HbF induction based on forced chromatin looping of LCR onto developmentally silenced γ-globin gene is also presented (Deng et al. 2014). We also showed some of the important regulators of γ-globin expression. Based on recent studies, BCL11A is found to be a crucial repressor of γ-globin. c-MYB also indirectly represses γ-globin by activating KLF1, which then activates BCL11A. Several other known regulators of γ-globin expression are also represented (Wilber et al. 2011b). Inset describes a strategy based on disruption of coding or regulatory sequences of the depicted regulators
Human β-globin locus control region (LCR) is responsible for hemoglobin switching in erythroid cells in a developmental stage-specific manner (Noordermeer and de Laat 2008). In a seminal study, Deng et al. induced looping of the LCR onto the silent γ-globin promoter to reactivate transcription of the developmentally silenced γ-globin gene (Deng et al. 2014). Forced LCR–promoter looping resulted in reactivation of the γ-globin expression to 85 % of the total globin synthesis while reducing adult β-globin expression. To induce chromatin looping, Deng et al. utilized Ldb1 protein, which is a self-dimerizing transcription cofactor involved in forming long-range chromatin contacts in erythroid cells. Forced looping was achieved by fusing artificial zinc finger proteins, targeting γ-globin promoter, to the self-association (SA) domain of Ldb1 (Fig. 4). This study demonstrated yet another innovative therapeutic strategy for SCD based on programmable DNA-binding proteins. Targeting the LCR to the γ-globin locus has an advantage of reducing HbS expression as a result of reduced contact between LCR and β-globin promoter. Thus, this strategy can be more advantageous compared to activation domain-based HbF induction which does not affect HbS levels. Nevertheless, HbF induction by forced looping can be further improved by fine-tuning of the targeted tethering technologies and exploration of additional nuclear factors involved in chromatin looping. Alternatively, to achieve concerted increment in levels of HbF in SCD patients, combinatorial therapy integrating more than one of the approaches described above can also prove to be beneficial as was shown for the case of erythroid progeny of β-thalassemic CD34+cells (Wilber et al. 2011a). As a final note, development of safer delivery methods for long-term expression of these artificial transcription factors will greatly enhance the applicability of these novel therapeutic strategies for clinical translation.
HbF induction by targeting regulators of γ-globin gene expression
β-globin cluster has always been the benchmark model for studying gene expression and regulation. Recent progress in our understanding of transcriptional regulation of β-globin locus has unveiled the fact that γ-globin gene expression is under strong negative transcriptional control in adult erythroid cells, which is mediated by several transcription factors and epigenetic modifications. Novel experimental approaches like genome-wide association studies (GWAS) identified two important loci which play a crucial role in γ-globin regulation (Galarneau et al. 2010). The findings across numerous studies involving subjects with and without SCD and of diverse ethnic backgrounds have been strikingly consistent. BCL11A on chromosome 2 and the HBS1L-MYB intergenic interval on chromosome 6 account for a substantial fraction of variation in HbF levels (Thein et al. 2007; Menzel et al. 2007; Uda et al. 2008; Sedgewick et al. 2008). Knockdown studies of BCL11A and c-Myb were able to induce HbF significantly, but the major concern of these studies was the essential role of these transcription factors in native and erythroid-independent context. For instance, although BCL11A is a potent HbF silencer, Bcl11a knockout mice are perinatal lethal, possibly due to the important role of Bcl11a in lymphoid and neuronal development (Liu et al. 2003; Kuo et al. 2010; John et al. 2012). Moreover, BCL11A knockout HSCs are deficient in lymphopoiesis (Tsang et al. 2015). Thus, instead of complete knockout of the BCL11A gene, approaches focusing on the deletion of BCL11A-binding sequences in the proximity of γ-globin gene or erythroid-specific repression of BCL11A can be promising (Fig. 4). In a recent study to repress erythroid BCL11A expression, Canver and colleagues employed CRISPR/Cas9 gRNA libraries for saturation mutagenesis of erythroid-specific BCL11A enhancer in human HSCs to screen for critical minimal features of the enhancer (Canver et al. 2015). After saturation mutagenesis, the cells were allowed to mature into red blood cells and the amount of HbF produced was determined. This led to the identification of a specific location in the BCL11A enhancer, whose deletion resulted in the production of substantially high levels of HbF in primary human progenitor cells as compared to the wild-type cells. BCL11A expression was reduced substantially, although not completely, in primary erythroid precursors only. These data suggest that targeted editing of BCL11A enhancer in HSCs could be an attractive approach to increase HbF level without the obvious neurological and immunological toxicities associated with whole gene knockout of BCL11A.
Similar to BCL11A, c-Myb is also ubiquitously expressed in HSCs and progenitor cells (Bianchi et al. 2010). A crucial role of c-Myb in the maintenance and differentiation of HSCs raises concerns whether it can be a valid therapeutic target in SCD without affecting erythroid maturation. To circumvent these issues, approaches similar to those described in the case of BCL11A can be employed. In addition to BCL11A and c-Myb, other negative regulators of HbF such as the transcription factors Erythroid Kruppel-like factor (KLF1) (Borg et al. 2010; Zhou et al. 2010), SOX6 (Xu et al. 2010), TR2/TR4 DR erythroid-definitive complex (Collins et al. 1985; Berry et al. 1992), Mi2β, FOP (van Dijk et al. 2010) and COUP-TFII (Filipe et al. 1999); epigenetic regulators like EHMT1/2 (Renneville et al. 2015) and HDACs; and activators such as NF-E4 (Jane et al. 1995; Zhou et al. 2004) can also be potential therapeutic targets (Wilber et al. 2011b). However, a comprehensive investigation into their mechanisms and functions is mandatory before moving onto clinical applications. For instance, EHMT1/2 catalyze gene silencing by mono- and dimethylation of lysine 9 on histone 3 (H3K9) and studies of Renneville et al. suggested EHMT1/2 as epigenetic regulators of γ-globin. Indeed, biallelic Ehmt2 knockout in mouse erythroleukemia cells led to an increase in the expression of embryonic β-globin genes: Hbb-εy and Hbb-βh1 in murine erythroleukemia (MEL) cells (Renneville et al. 2015). It is important to note that Renneville and colleagues were unsuccessful in creating biallelic deletions of Ehmt1 in MEL cells which indicates that Ehmt1 plays an essential role in these cells. These observations further reinforce the need for robust validation of the essential functions of the aforementioned therapeutic targets in treatment of SCD.
Several key regulators of γ-globin such as BCL11A, KLF1 and MYB have been identified and corroborated both by genetic and functional data, whereas other factors such as Ehmt1/2, SOX6 and NF-E4 have relatively unclear roles both functionally and quantitatively. To fully understand the emerging network of fetal hemoglobin regulation, novel regulatory factors and their mechanism of action remain to be uncovered in future high-throughput screens. Thereafter, therapeutic modality design and validation are required before proceeding to clinical trials to demonstrate the safety and efficacy in humans. A major impediment for the model systems described above is that it is difficult to directly extrapolate the percentage HbF induction expected in humans based on experiments in cells or animals. For better efficacy, one could envision combinatorial therapies focusing on HbF induction and gene addition or correction methods as the future treatment regimen for SCD and other β-globin disorders.
Major obstacles in translation of gene-editing tools for clinical applications
Programmable nucleases have been widely used for gene-editing purposes in various disease cell and animal models in vitro as well as in vivo, although there are still important concerns to be addressed before their use in humans for therapeutic applications. In this section, we will focus on concerns raised due to the use of programmable nucleases. We will discuss three important issues: efficiency, off-target effects and safe delivery methods. As mentioned in previous sections, issues with stem cell transplantation that are not directly related to the use of programmable nucleases are beyond the scope of this paper and can be found in other reviews (Lengerke and Daley 2010; Arora and Daley 2012; Slukvin 2013; Kim 2014; Focosi et al. 2014; Singh et al. 2015; Ackermann et al. 2015).
Efficiency
As previously noted, DSBs generated by programmable nucleases are repaired mainly by NHEJ or HDR. Efficiencies of these pathways vary significantly among different cell types and cell cycle stages, but generally NHEJ is more active than HDR in most of the mammalian cell types due to its versatile activity in all stages of cell cycle. As mutation correction in diseased HSCs rely on homology-dependent repair mechanism, genome editing based on HDR repair template proves to be very challenging for applications in SCD as HDR is restricted to actively dividing cells, whereas HSCs are mostly quiescent. Indeed, low HDR efficiency has been a severe limitation of the Hoban study in which the mutation correction efficiency in HSCs was significantly low as compared to the mixed population of CD34+ cells (Hoban et al. 2015).
Regulation of cell cycle can be altered in slowly cycling cell types through stimulation of mitosis by ex vivo use of small molecules (Genovese et al. 2014). A drawback of this method is that true post-mitotic cells are usually not amenable to such perturbation, limiting the applicability of this methodology. Suppressing competing NHEJ repair pathway has been shown to increase HDR rates up to 19-fold in mouse and human cells (Chu et al. 2015; Maruyama et al. 2015), although the implementation and safety of this strategy in disease models of SCD are not known and should be carefully assessed. The use of HR-stimulating molecules was also shown to increase HR rates (Song et al. 2016). Simultaneous application of the above-mentioned approaches could result in increased HDR rates that are capable of bypassing the therapeutic threshold of SCD mutation correction.
Considering the preference of NHEJ over HR in mammalian cells, gene knockout strategies can be more useful. A successful clinical precedence can be seen in the studies of Tebas et al. where ZFNs were used to disrupt CCR5 gene ex vivo for an anti-HIV therapy (Tebas et al. 2014). The therapeutic approach that was based on gene disruption by NHEJ led to an efficiency of 11–28 % gene modification in CD4 T cells, which was suitable for clinical trials. In case of SCD, an efficiency of as low as 2 % can be enough to get significant clinical benefit, as demonstrated in previous studies (Walters et al. 2001; Iannone et al. 2003; Wu et al. 2007). To obtain clinically relevant gene correction efficiencies in HSCs, recent novel strategies utilizing NHEJ-based ligation of the insert to the ends of the DSB can be useful (Maresca et al. 2013; Geisinger et al. 2016). However, the specificity of such methods needs further evaluation. Another emerging strategy to insert an exogenous DNA into the target locus independent of HDR is microhomology-mediated end joining (MMEJ) (Sakuma et al. 2016); although, it is not known whether MMEJ is efficient in HSCs or iPSCs. A systematic analysis of the suggested technologies can help in determining the most efficient strategy for correction of the SCD.
In addition to gene targeting efficiency, another issue in the field that needs further improvement is the correct quantification of the gene modification in HSCs. CD34+ cells, which have been used in many studies, are a mixture of many progenitors, containing only few true HSCs. Currently, the true assessment of gene modified HSCs within this population is done by engraftment studies, which is time consuming and also may not correctly represent human hematopoiesis. To overcome this limitation, improved methods to enrich and expand HSCs in vitro will be necessary. Studies of Hoban et al. show that long-term repopulating HSCs may have significantly low gene-targeting efficiency (Hoban et al. 2015). As can be seen in the studies described in “Gene targeting as a strategy for sickle cell therapeutics”, iPSCs can have higher gene-editing efficiency compared to HSCs, but a challenge of using iPSCs for the treatment of hemaglobinopathies is the lack of efficient methods to derive HSCs from iPSCs, and the current progress in this field is discussed in more detail in a recent review (Ackermann et al. 2015).
Off-target effects and toxicity
Programmable nucleases are designed to recognize a specific site only; however, sometimes they can recognize some additional unintended “off-target” sites as well. This off-target recognition could be especially problematic while targeting sequences with high homology to other regions in the genome. Off-target mutagenesis could be a big problem in human applications, since even one rare mutation could have unexpected consequences and even remote risks need to be anticipated. For instance, although it has been used in many studies, ZFNs can have high off-target rates and could be cytotoxic (Cornu et al. 2007). Development of obligate heterodimer ZFNs and using an optimum spacer length between the monomers helped improve specificity (Miller et al. 2007; Szczepek et al. 2007; Händel et al. 2009; Doyon et al. 2011). However, another problem that makes it hard to design specific ZFNs is the lack of zinc finger proteins efficiently recognizing all 64 combinations of 3 bp.
One study suggested TALENs as a better option due to reduced off-target effects and cytotoxicity (Mussolino et al. 2011). One reason could be that TALENs are typically designed to have longer recognition sites. Similar to ZFNs, optimal spacer length between monomers is important for TALEN specificity (Sun and Zhao 2013). Although TALENs can still induce off-target mutations, off-targets can be minimized by their careful design. Preference of NH or NK instead of NN for guanine recognition could be another way of improving specificity (Sun and Zhao 2013). In a different approach to increase specificity of TALENs, Lin et al. fused a TALEN monomer to an engineered homing endonuclease I-SceI (Lin et al. 2015). Although they have very high specificity, homing endonucleases are not modular as programmable nucleases and it is difficult to change their targeting specificity. However, Lin et al. was able to engineer I-SceI to recognize an 18 bp sequence in the HBB locus, albeit that chimeric proteins were designed to target close to β-thalassemia mutations. Still, it suggests an approach to increase the specificity of TALENs in the HBB locus.
CRISPRs cause the biggest concern due to lack of requirement for obligate heterodimers and their shorter recognition site. Finding specific targets recognized by CRISPR gRNA can be hard, since its recognition site is only 20 bp and only the seed sequence of 8–12 bp proximal to PAM is critical for specificity (Cong et al. 2013). Another reason for unwanted mutations could be that although Cas9 from S. pyogenes is assumed to recognize a PAM sequence of 5′-NGG-3′, it can also recognize 5′-NAG-3′ (Hsu et al. 2013). In a systematic analysis, it was indeed suggested that CRISPR/Cas9 system has higher off-target effects compared to TALENs (Wang et al. 2015). However, it was shown that optimization of gRNAs could increase specificity by up to 5000-fold or more (Fu et al. 2014). Another attempt to increase specificity was the use of nickases or Cas9–FokI fusions to require binding of a pair of CRISPRs rather than one for DSB (Mali et al. 2013; Ran et al. 2013a; Tsai et al. 2014). Two recently engineered Cas9 variants eSpCas9 and SpCas9-HF1 were also highly successful at improving specificity (Slaymaker et al. 2016; Kleinstiver et al. 2016).
Another challenge in the genome-editing field is the sensitive detection of off-target events. Although there are tools to predict off-target sites for these three nucleases, experimental evidence shows that the predicted and the actual off-targets could be different (Tsai et al. 2015). Thus, an analysis of predicted off-target sites would not be enough to claim lack of off-target effects. Recently, several methods were developed for a more comprehensive analysis of off-targets (Frock et al. 2015; Tsai et al. 2015; Wang et al. 2015; Kim et al. 2015; O’Geen et al. 2015). However, they are still not sensitive enough to detect very rare mutations. Thus, further improvements in off-target detection methods would be crucial.
In addition to the activity of nucleases, another reason for off-target modifications could be the donor DNA itself. It is well known that linear or plasmid DNA can randomly integrate into the mammalian genome (Vasquez et al. 2001). Thus, once the cells are transfected with such exogenous DNA, the DNA can be inserted into the genome by random integration in addition to by HR. Actually, random integration is more frequent than HR; it happens in one per 102–104 treated cells (Vasquez et al. 2001). A recent study suggested that the use of adenoviral donor DNA could minimize random integration compared to plasmid or IDLV donors (Holkers et al. 2014). The presence of a protein cap in AdVs was shown to be the main reason for increased specificity by potentially blocking ligation of the donor to off-target sites.
Safe delivery methods
It is known that the delivery method of programmable nucleases has an effect on efficiency, off-target effects and toxicity (Maggio and Gonçalves 2015). Thus, finding an optimal method is essential. Viral delivery methods such as lentiviral or adenoviral transduction are commonly used for exogenous DNA delivery due to their high transduction efficiency even for hard-to-transfect cells. However, due to the serious side effects of viral vectors as mentioned in “Introduction”, it would be crucial to deliver the programmable nucleases by non-viral methods for clinical use. Although it is still a viral delivery method, IDLVs can be safer than lentiviruses since they cannot integrate their genome into the host cell. So far, IDLV-mediated delivery of nucleases was shown for ZFNs and TALENs (Lombardo et al. 2007; Mock et al. 2014), although IDLV delivery of CRISPR/Cas9 could also be feasible.
Cationic liposome or cationic polymer-based transfection methods are among the commonly used non-viral DNA delivery methods in mammalian cells. However, those methods are not very efficient in some cell types, especially in stem cells. Thus, electroporation could be a better option in such cases. Considering the fact that off-target effects can be dependent on nuclease levels, microinjection of TALEN or CRISPR/Cas9 coding plasmids was suggested as a method for better control of the amount of nucleases delivered into the cells (Cottle et al. 2015).
Another factor that can contribute to off-target effects is long-term persistence of programmable nucleases in the cells. Delivering programmable nucleases as a protein or mRNA instead of expressing them from an exogenous DNA could be a better therapeutic approach, since there is no risk of persistence of a transgene in the cell. Shorter life of proteins and mRNAs within the cell compared to transfected DNA can lead to less off-target effects, since the cells will be exposed less to the nucleases. Direct protein delivery was easier to utilize for ZFNs due to their inherent ability to penetrate the cell membrane (Gaj et al. 2012). For TALENs, fusion of the protein with cell-penetrating peptides was necessary, and the same requirement exists for the Cas9 protein (Ru et al. 2013; Liu et al. 2014; Ramakrishna et al. 2014). The need for RNA delivery in addition to protein delivery made this kind of approach more complex for CRISPR/Cas9, although forming a complex between gRNAs and cell-penetrating peptides solved this issue (Ramakrishna et al. 2014). Another emerging method for delivery of Cas9 and gRNA is the delivery of pre-formed ribonucleoprotein (RNP) complexes via electroporation (Kim et al. 2014; Lin et al. 2014). Considering the significant improvement in HSC targeting after RNP delivery of CRISPR/Cas9 (DeWitt et al. 2016), this approach could be revolutionary in the field. In case the direct protein delivery route is chosen, lentiviral vectors can also be used for protein delivery. Lentiviral protein delivery of ZFNs was successful; however, TALEN subdomains were cleaved by HIV-1 proteases (Cai et al. 2014). mRNA delivery could be another transient delivery method that is used in various cell types including CD34+ cells (Genovese et al. 2014; Maggio and Gonçalves 2015; Cox et al. 2015; Mock et al. 2015; Liang et al. 2015a).
Conclusions and future prospects
In recent years, programmable nucleases, especially CRISPR/Cas9, have been the center of attention among the scientific community due to their numerous applications in versatile model systems. Insertion of virtually any desired artificial sequence into the genome is one such application that is relevant for the treatment of genetic disorders. Being a monogenic disease, SCD has a high potential to be treated by programmable nucleases. So far, ZFNs, TALENs and CRISPR/Cas9 were demonstrated to be successful in the correction of sickle mutation by gene targeting in human cells. Novel uses of zinc fingers, TALEs and CRISPRs can also induce HbF levels in clinically relevant cells. HbF induction approach, together with correction of the sickle mutation, can be a beneficial combinatorial therapy to fully alleviate disease complications. In addition to correction of the SCD for therapeutic applications, another beneficial utilization of gene targeting could be creating disease models to help study the disease state and design better treatment strategies.
Genome editing is a rapidly developing technology for which several parameters are yet to be fully analyzed. For instance, modification of HSCs by genome-editing techniques has been very inefficient. DeWitt et al.’s study (DeWitt et al. 2016) is the only example in which HSCs were modified at clinically relevant levels. Further analysis of their approach can help overcome issues related to modification of HSCs.
Although genome editing is widely used in various cell lines and model organisms, a superior understanding of the specificity and mechanism of these tools is also necessary before their use for treatment of genetic disorders like SCD. Off-target effects, which create additional mutations in undesired genomic locations, could impose a serious risk for the applications of genome-editing tools in humans. Moreover, finding safer delivery methods is of utmost importance to reduce toxicities and side effects associated with current modes of delivery. Another issue that needs to be addressed is the nature of donor DNA itself and it is necessary to find ways of decreasing random integration of the donor DNA into the genome. Although discoveries in mouse models are promising, long-term effects of programmable nucleases are yet to be determined. Tremendous efforts of the researchers have enabled genome-editing therapies to be of immense value for treatment of patients with SCD and the future holds further promise.
References
Abil Z, Xiong X, Zhao H (2015) Synthetic biology for therapeutic applications. Mol Pharm 12:322–331
Ackermann M, Liebhaber S, Klusmann JH, Lachmann N (2015) Lost in translation: pluripotent stem cell-derived hematopoiesis. EMBO Mol Med 7:1388–1402
Aliyu ZY, Tumblin AR, Kato GJ (2006) Current therapy of sickle cell disease. Haematologica 91:7–10
Arora N, Daley GQ (2012) Pluripotent stem cells in research and treatment of hemoglobinopathies. Cold Spring Harb Perspect Med 2(4):a011841
Ashley-Koch A, Yang Q, Olney RS (2000) Sickle hemoglobin (Hb S) allele and sickle cell disease: a HuGE review. Am J Epidemiol 151:839–845
Ballas SK (2015) Pathophysiology and principles of management of the many faces of the acute vaso-occlusive crisis in patients with sickle cell disease. Eur J Haematol 95:113–123
Barrangou R, Fremaux C, Deveau H et al (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712
Bauer DE, Kamran SC, Orkin SH (2012) Reawakening fetal hemoglobin: prospects for new therapies for the β-globin disorders. Blood 120:2945–2953
Baum C, Düllmann J, Li Z et al (2003) Side effects of retroviral gene transfer into hematopoietic stem cells. Blood 101:2099–2113
Berry M, Grosveld F, Dillon N (1992) A single point mutation is the cause of the Greek form of hereditary persistence of fetal haemoglobin. Nature 358:499–502
Bianchi E, Zini R, Salati S et al (2010) c-myb supports erythropoiesis through the transactivation of KLF1 and LMO2 expression. Blood 116:e99–110
Bibikova M, Carroll D, Segal DJ et al (2001) Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol 21:289–297
Bibikova M, Golic M, Golic KG, Carroll D (2002) Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161:1169–1175
Boch J, Bonas U (2010) Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol 48:419–436
Boch J, Scholze H, Schornack S et al (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512
Bollag RJ, Waldman AS, Liskay RM (1989) Homologous recombination in mammalian cells. Annu Rev Genet 23:199–225
Borg J, Papadopoulos P, Georgitsi M et al (2010) Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet 42:801–805
Cai Y, Bak RO, Mikkelsen JG (2014) Targeted genome editing by lentiviral protein transduction of zinc-finger and TAL-effector nucleases. eLife 3:e01911
Canver MC, Smith EC, Sher F et al (2015) BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527:192–197
Carlson DF, Fahrenkrug SC, Hackett PB (2012) Targeting DNA with fingers and TALENs. Mol Ther Nucleic Acids 1:e3. doi:10.1038/mtna.2011.5
Carroll D (2011) Genome engineering with zinc-finger nucleases. Genetics 188:773–782
Cathomen T, Joung J (2008) Zinc-finger nucleases: the next generation emerges. Mol Ther 16:1200–1207
Chandrakasan S, Malik P (2014) Gene therapy for hemoglobinopathies. Hematol Oncol Clin North Am 28:199–216
Chang JC, Ye L, Kan YW (2006) Correction of the sickle cell mutation in embryonic stem cells. Proc Natl Acad Sci USA 103:1036–1040
Check E (2002) Gene therapy: a tragic setback. Nature 420:116–118
Choulika A, Perrin A, Dujon B, Nicolas JF (1995) Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol Cell Biol 15:1968–1973
Christian M, Cermak T, Doyle EL et al (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186:757–761
Chu VT, Weber T, Wefers B et al (2015) Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33:543–548
Collins FS, Metherall JE, Yamakawa M et al (1985) A point mutation in the A gamma-globin gene promoter in Greek hereditary persistence of fetal haemoglobin. Nature 313:325–326
Cong L, Ran FA, Cox D et al (2013) Multiplex genome engineering using CRISPR/Cas Systems. Science 339:819–823
Cornu TI, Thibodeau-Beganny S, Guhl E et al (2007) DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol Ther 16:352–358
Costa FC, Fedosyuk H, Neades R et al (2012) Induction of fetal hemoglobin in vivo mediated by a synthetic γ-globin zinc finger activator. Anemia 2012:e507894
Cottle RN, Lee CM, Archer D, Bao G (2015) Controlled delivery of β-globin-targeting TALENs and CRISPR/Cas9 into mammalian cells for genome editing using microinjection. Sci Rep 5:16031. doi:10.1038/srep16031
Cox DBT, Platt RJ, Zhang F (2015) Therapeutic genome editing: prospects and challenges. Nat Med 21:121–131
Cradick TJ, Fine EJ, Antico CJ, Bao G (2013) CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 41:9584–9592
Deng W, Rupon JW, Krivega I et al (2014) Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell 158:849–860
Deng C, Capecchi MR (1992) Reexamination of gene targeting frequency as a function of the extent of homology between the targeting vector and the target locus. Mol Cell Biol 12:3365–3371
DeWitt M, Magis W, Bray NL et al (2016) Efficient correction of the sickle mutation in human hematopoietic stem cells using a Cas9 ribonucleoprotein complex. bioRxiv. doi:10.1101/036236
Dong A, Rivella S, Breda L (2013) Gene therapy for hemoglobinopathies: progress and challenges. Transl Res 161:293–306
Doyon Y, Vo TD, Mendel MC et al (2011) Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods 8:74–79
Filipe A, Li Q, Deveaux S et al (1999) Regulation of embryonic/fetal globin genes by nuclear hormone receptors: a novel perspective on hemoglobin switching. EMBO J 18:687–697
Focosi D, Amabile G, Di Ruscio A et al (2014) Induced pluripotent stem cells in hematology: current and future applications. Blood Cancer J 4:e211
Frenette PS, Atweh GF (2007) Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest 117:850–858
Frock RL, Hu J, Meyers RM et al (2015) Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33:179–186
Fu Y, Sander JD, Reyon D et al (2014) Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32:279–284
Gaj T, Guo J, Kato Y et al (2012) Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods 9:805–807
Galarneau G, Palmer CD, Sankaran VG et al (2010) Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 42:1049–1051
Geisinger JM, Turan S, Hernandez S et al (2016) In vivo blunt-end cloning through CRISPR/Cas9-facilitated non-homologous end-joining. Nucleic Acids Res. doi:10.1093/nar/gkv1542
Genovese P, Schiroli G, Escobar G et al (2014) Targeted genome editing in human repopulating hematopoietic stem cells. Nature 510:235–240
Goncz KK, Prokopishyn NL, Chow BL et al (2002) Application of SFHR to gene therapy of monogenic disorders. Gene Ther 9:691–694
Gräslund T, Li X, Magnenat L et al (2005) Exploring strategies for the design of artificial transcription factors: targeting sites proximal to known regulatory regions for the induction of γ-globin expression and the treatment of sickle cell disease. J Biol Chem 280:3707–3714
Hacein-Bey-Abina S, Garrigue A, Wang GP et al (2008) Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest 118:3132–3142
Händel EM, Alwin S, Cathomen T (2009) Expanding or restricting the target site repertoire of zinc-finger nucleases: the inter-domain linker as a major determinant of target site selectivity. Mol Ther 17:104–111
Hanna J, Wernig M, Markoulaki S et al (2007) Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 318:1920–1923
Hassell KL (2010) Population estimates of sickle cell disease in the U.S. Am J Prev Med 38:S512–521
Hendrie PC, Russell DW (2005) Gene targeting with viral vectors. Mol Ther 12:9–17
Hoban MD, Cost GJ, Mendel MC et al (2015) Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 125:2597–2604
Hockemeyer D, Soldner F, Beard C et al (2009) Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 27:851–857
Holkers M, Maggio I, Henriques SFD et al (2014) Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat Methods 11:1051–1057
Hsu PD, Scott DA, Weinstein JA et al (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832
Huang X, Wang Y, Yan W et al (2015) Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells 33:1470–1479
Iannone R, Casella JF, Fuchs EJ et al (2003) Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant 9:519–528
Ingram VM (1956) A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature 178:792–794
Isalan M (2012) Zinc-finger nucleases: how to play two good hands. Nat Methods 9:32–34
Jane SM, Nienhuis AW, Cunningham JM (1995) Hemoglobin switching in man and chicken is mediated by a heteromeric complex between the ubiquitous transcription factor CP2 and a developmentally specific protein. EMBO J 14:97–105
John A, Brylka H, Wiegreffe C et al (2012) Bcl11a is required for neuronal morphogenesis and sensory circuit formation in dorsal spinal cord development. Development 139:1831–1841
Johnson RD, Jasin M (2001) Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans 29:196–201
Khan IF, Hirata RK, Russell DW (2011) AAV-mediated gene targeting methods for human cells. Nat Protoc 6:482–501
Kim C (2014) Disease modeling and cell based therapy with iPSC: future therapeutic option with fast and safe application. Blood Res 49:7–14
Kim S, Kim D, Cho SW et al (2014) Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24(6):1012–1019
Kim Y, Kweon J, Kim A et al (2013) A library of TAL effector nucleases spanning the human genome. Nat Biotechnol 31:251–258
Kim D, Bae S, Park J et al (2015) Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 12:237–243
Kleinstiver BP, Pattanayak V, Prew MS et al (2016) High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529:490–495
Kuo TY, Chen CY, Hsueh YP (2010) Bcl11A/CTIP1 mediates the effect of the glutamate receptor on axon branching and dendrite outgrowth. J Neurochem 114:1381–1392
LaFountaine JS, Fathe K, Smyth HDC (2015) Delivery and therapeutic applications of gene editing technologies ZFNs, TALENs, and CRISPR/Cas9. Int J Pharm 494:180–194
Lanzkron S, Strouse JJ, Wilson R et al (2008) Systematic review: hydroxyurea for the treatment of adults with sickle cell disease. Ann Intern Med 148:939–955
Lanzkron S, Carroll CP, Haywood C (2013) Mortality rates and age at death from sickle cell disease: US, 1979–2005. Public Health Rep 128:110–116
Larochelle A, Dunbar CE (2008) HOXB4 and retroviral vectors: adding fuel to the fire. J Clin Invest 118:1350–1353
Lengerke C, Daley GQ (2010) Autologous blood cell therapies from pluripotent stem cells. Blood Rev 24:27–37
Levasseur DN, Ryan TM, Pawlik KM, Townes TM (2003) Correction of a mouse model of sickle cell disease: lentiviral/antisickling β-globin gene transduction of unmobilized, purified hematopoietic stem cells. Blood 102:4312–4319
Li T, Huang S, Jiang WZ et al (2011a) TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res 39:359–372
Li M, Suzuki K, Qu J et al (2011b) Efficient correction of hemoglobinopathy-causing mutations by homologous recombination in integration-free patient iPSCs. Cell Res 21:1740–1744
Liang J, Chao R, Abil Z et al (2014) FairyTALE: a high-throughput TAL effector synthesis platform. ACS Synth Biol 3:67–73
Liang X, Potter J, Kumar S et al (2015a) Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol 208:44–53
Liang P, Xu Y, Zhang X et al (2015b) CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 6:363–372
Lin S, Staahl BT, Alla RK, Doudna JA (2014) Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 3:e04766
Lin J, Chen H, Luo L et al (2015) Creating a monomeric endonuclease TALE-I-SceI with high specificity and low genotoxicity in human cells. Nucleic Acids Res 43:1112–1122
Liu P, Keller JR, Ortiz M et al (2003) Bcl11a is essential for normal lymphoid development. Nat Immunol 4:525–532
Liu J, Gaj T, Patterson JT et al (2014) Cell-penetrating peptide-mediated delivery of TALEN proteins via bioconjugation for genome engineering. PLoS ONE 9:e85755
Locatelli F, Pagliara D (2012) Allogeneic hematopoietic stem cell transplantation in children with sickle cell disease. Pediatr Blood Cancer 59:372–376
Lombardo A, Genovese P, Beausejour CM et al (2007) Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol 25:1298–1306
Luo Y, Zhu D, Zhang Z et al (2015) Integrative analysis of CRISPR/Cas9 target sites in the human HBB gene. BioMed Res Int 2015:514709. doi:10.1155/2015/514709
Maggio I, Gonçalves MAFV (2015) Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol 33:280–291
Mali P, Yang L, Esvelt KM et al (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Maresca M, Lin VG, Guo N, Yang Y (2013) Obligate Ligation-Gated Recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res 23:539–546
Maruyama T, Dougan SK, Truttmann MC et al (2015) Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol 33:538–542
Menzel S, Garner C, Gut I et al (2007) A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet 39:1197–1199
Miller JC, Holmes MC, Wang J et al (2007) An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25:778–785
Mock U, Riecken K, Berdien B et al (2014) Novel lentiviral vectors with mutated reverse transcriptase for mRNA delivery of TALE nucleases. Sci Rep 4:6409
Mock U, Machowicz R, Hauber I et al (2015) mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Res 43:5560–5571
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326:1501
Mussolino C, Morbitzer R, Lütge F et al (2011) A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res 39:9283–9293
Noordermeer D, de Laat W (2008) Joining the loops: beta-globin gene regulation. IUBMB Life 60:824–833
O’Geen H, Henry IM, Bhakta MS et al (2015) A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res 43:3389–3404
Ochiai H (2015) Single-base pair genome editing in human cells by using site-specific endonucleases. Int J Mol Sci 16:21128–21137
Paradowski K (2015) Pathophysiology and perioperative management of sickle cell disease. J Perioper Pract 25:101–104
Pawliuk R, Westerman KA, Fabry ME et al (2001) Correction of sickle cell disease in transgenic mouse models by gene therapy. Science 294:2368–2371
Pestina TI, Hargrove PW, Jay D et al (2009) Correction of murine sickle cell disease using γ-globin lentiviral vectors to mediate high-level expression of fetal hemoglobin. Mol Ther 17:245–252
Pfeiffer P, Goedecke W, Obe G (2000) Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis 15:289–302
Piel FB (2016) The present and future global burden of the inherited disorders of hemoglobin. Hematol Oncol Clin North Am 30:327–341
Porteus MH (2006) Mammalian gene targeting with designed zinc finger nucleases. Mol Ther 13:438–446
Ramakrishna S, Kwaku Dad AB, Beloor J et al (2014) Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res 24:1020–1027
Ramalingam S, Annaluru N, Kandavelou K, Chandrasegaran S (2014) TALEN-mediated generation and genetic correction of disease-specific human induced pluripotent stem cells. Curr Gene Ther 14:461–472
Ran FA, Hsu PD, Lin CY et al (2013a) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154:1380–1389
Ran FA, Hsu PD, Wright J et al (2013b) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308
Renneville A, Galen PV, Canver MC et al (2015) EHMT1 and EHMT2 inhibition induce fetal hemoglobin expression. Blood 126:1930–1939
Ru R, Yao Y, Yu S et al (2013) Targeted genome engineering in human induced pluripotent stem cells by penetrating TALENs. Cell Regen 2:5
Sadelain M, Rivella S, Lisowski L et al (2004) Globin gene transfer for treatment of the β-thalassemias and sickle cell disease. Best Pract Res Clin Haematol 17:517–534
Sakuma T, Nakade S, Sakane Y et al (2016) MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat Protoc 11:118–133
Sebastiano V, Maeder ML, Angstman JF et al (2011) In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 29:1717–1726
Sedgewick AE, Timofeev N, Sebastiani P et al (2008) BCL11A is a major HbF quantitative trait locus in three different populations with β-hemoglobinopathies. Blood Cells Mol Dis 41:255–258
Serjeant GR (2013) The natural history of sickle cell disease. Cold Spring Harb Perspect Med 3:a011783
Shenoy S (2011) Hematopoietic stem cell transplantation for sickle cell disease: current practice and emerging trends. Hematol Am Soc Hematol Educ Program 2011:273–279
Singh VK, Kalsan M, Kumar N et al (2015) Induced pluripotent stem cells: applications in regenerative medicine, disease modeling, and drug discovery. Front Cell Dev Biol 3:2. doi:10.3389/fcell.2015.00002
Slaymaker IM, Gao L, Zetsche B et al (2016) Rationally engineered Cas9 nucleases with improved specificity. Science 351:84–88
Slukvin II (2013) Hematopoietic specification from human pluripotent stem cells: current advances and challenges toward de novo generation of hematopoietic stem cells. Blood 122:4035–4046
Smithies O, Gregg RG, Boggs SS et al (1985) Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature 317:230–234
Song J, Yang D, Xu J et al (2016) RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat Commun 7:10548. doi:10.1038/ncomms10548
Steinberg MH, Sebastiani P (2012) Genetic modifiers of sickle cell disease. Am J Hematol 87:795–803
Steinberg MH, Lu ZH, Barton FB et al (1997) Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Blood 89:1078–1088
Sun N, Zhao H (2013) Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol Bioeng 110:1811–1821
Sun N, Zhao H (2014) Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol Bioeng 111:1048–1053
Sun N, Liang J, Abil Z, Zhao H (2012) Optimized TAL effector nucleases (TALENs) for use in treatment of sickle cell disease. Mol BioSyst 8:1255–1263
Sunshine HR, Hofrichter J, Eaton WA (1978) Requirement for therapeutic inhibition of sickle haemoglobin gelation. Nature 275:238–240
Suzuki K, Yu C, Qu J et al (2014) Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell 15:31–36
Szczepek M, Brondani V, Büchel J et al (2007) Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol 25:786–793
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676
Tebas P, Stein D, Tang WW et al (2014) Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370:901–910
Thein SL, Menzel S, Peng X et al (2007) Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci USA 104:11346–11351
Thomas KR, Capecchi MR (1987) Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51:503–512
Thomas CE, Ehrhardt A, Kay MA (2003) Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet 4:346–358
Tsai SQ, Wyvekens N, Khayter C et al (2014) Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32:569–576
Tsai SQ, Zheng Z, Nguyen NT et al (2015) GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33:187–197
Tsang JCH, Yu Y, Burke S et al (2015) Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells. Genome Biol 16:178. doi:10.1186/s13059-015-0739-5
Uda M, Galanello R, Sanna S et al (2008) Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc Natl Acad Sci USA 105:1620–1625
Urnov FD, Rebar EJ, Holmes MC et al (2010) Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11:636–646
van Dijk TB, Gillemans N, Pourfarzad F et al (2010) Fetal globin expression is regulated by Friend of Prmt1. Blood 116:4349–4352
Vasquez KM, Marburger K, Intody Z, Wilson JH (2001) Manipulating the mammalian genome by homologous recombination. Proc Natl Acad Sci USA 98:8403–8410
Voit RA, Hendel A, Pruett-Miller SM, Porteus MH (2014) Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res 42:1365–1378
Wah DA, Bitinaite J, Schildkraut I, Aggarwal AK (1998) Structure of FokI has implications for DNA cleavage. Proc Natl Acad Sci USA 95:10564–10569
Walters MC, Patience M, Leisenring W et al (2001) Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant 7:665–673
Wang L, Menendez P, Shojaei F et al (2005) Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J Exp Med 201:1603–1614
Wang X, Wang Y, Wu X et al (2015) Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol 33:175–178
Weatherall DJ, Clegg JB (2001) Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ 79:704–712
Wilber A, Tschulena U, Hargrove PW et al (2010) A zinc-finger transcriptional activator designed to interact with the gamma-globin gene promoters enhances fetal hemoglobin production in primary human adult erythroblasts. Blood 115:3033–3041
Wilber A, Hargrove PW, Kim YS et al (2011a) Therapeutic levels of fetal hemoglobin in erythroid progeny of β-thalassemic CD34+ cells after lentiviral vector-mediated gene transfer. Blood 117:2817–2826
Wilber A, Nienhuis AW, Persons DA (2011b) Transcriptional regulation of fetal to adult hemoglobin switching: new therapeutic opportunities. Blood 117:3945–3953
Woods NB, Bottero V, Schmidt M et al (2006) Gene therapy: therapeutic gene causing lymphoma. Nature 440:1123
Wright AV, Nuñez JK, Doudna JA (2016) Biology and applications of CRISPR systems: harnessing nature’s toolbox for genome engineering. Cell 164:29–44
Wu LC, Sun CW, Ryan TM et al (2006) Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood 108:1183–1188
Wu CJ, Gladwin M, Tisdale J et al (2007) Mixed haematopoietic chimerism for sickle cell disease prevents intravascular haemolysis. Br J Haematol 139:504–507
Xiao-Jie L, Hui-Ying X, Zun-Ping K et al (2015) CRISPR-Cas9: a new and promising player in gene therapy. J Med Genet 52:289–296
Xu J, Sankaran VG, Ni M et al (2010) Transcriptional silencing of {gamma}-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev 24:783–798
Zhang X-B, Beard BC, Trobridge GD et al (2008) High incidence of leukemia in large animals after stem cell gene therapy with a HOXB4-expressing retroviral vector. J Clin Invest 118:1502–1510
Zhou W, Zhao Q, Sutton R et al (2004) The role of p22 NF-E4 in human globin gene switching. J Biol Chem 279:26227–26232
Zhou D, Liu K, Sun CW et al (2010) KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat Genet 42:742–744
Zou J, Mali P, Huang X et al (2011) Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 118:4599–4608
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (1U54DK107965) and Centennial Chair Professorship (HZ) in the Department of Chemical and Biomolecular Engineering at the University of Illinois at Urbana-Champaign.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Tasan, I., Jain, S. & Zhao, H. Use of genome-editing tools to treat sickle cell disease. Hum Genet 135, 1011–1028 (2016). https://doi.org/10.1007/s00439-016-1688-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-016-1688-0