
Abstract
Sickle cell disease (SCD) is an inherited monogenic disorder resulting in serious mortality and morbidity worldwide. Although the disease was characterized more than a century ago, there are only two FDA approved medications to lessen disease severity, and a definitive cure available to all patients with SCD is lacking. Rapid and substantial progress in genome editing approaches have proven valuable as a curative option given plausibility to either correct the underlying mutation in patient-derived hematopoietic stem/progenitor cells (HSPCs), induce fetal hemoglobin expression to circumvent sickling of red blood cells (RBCs), or create corrected induced pluripotent stem cells (iPSCs) among other approaches. Recent discovery of CRISPR/Cas9 has not only revolutionized genome engineering but has also brought the possibility of translating these concepts into a clinically meaningful reality. Here we summarize genome engineering applications using CRISPR/Cas9, addressing challenges and future perspectives of CRISPR/Cas9 as a curative option for SCD.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Gene editing
- Gene therapy
- Hematopoietic stem cell transplantation
- Hemoglobinopathies
- Programmable endonucleases
1 Introduction
Sickle cell disease (SCD) is an inherited monogenic disorder characterized by a single substitution on chromosome 11 where glutamic acid is replaced by valine in the sixth codon of the β-globin gene. Whether inherited either in a homozygous state or with another abnormal β-globin gene, SCD encompasses a group of disorders with variable clinical phenotypes yet share a common pathophysiologic consequence derived from a single monogenic change. The modified β-globin gene produces an abnormal hemoglobin S (HbS) which rapidly polymerizes in the deoxygenated state altering red blood cell (RBC) rheology and lifespan. This single substitution leads to multiple downstream effects and devastating clinical complications including chronic anemia, chronic inflammation, recurrent vaso-occlusion, acute and chronic pain, stroke, organ failure, and early mortality (Paulukonis et al. 2016).
SCD is the most common inherited hemoglobinopathy worldwide, and despite knowledge of the disorder for over 100 years, it remains a life-limiting disease with few therapeutic options to reduce disease severity. Unlike other more recently identified molecular disorders that have benefited from higher federal, foundational, and per person funding (Smith et al. 2006; Lobner et al. 2013), there are only two FDA approved medications to lessen disease severity, hydroxyurea (HU) (approved for adults in 1998; children in 2017) and L-glutamine (approved in 2018). There remains misinformation, poor adherence, and a reluctance to prescribe HU despite benefit (Wang et al. 2011; Zimmerman et al. 2007; Steinberg et al. 2003; Ware 2010), while insurance companies will often not cover the cost of the highly purified form of L-glutamine approved by the FDA. Whereas the two mainstay treatments for SCD, blood transfusions and HU do not fully eliminate the consequences of the disease, simple public health measures such as newborn screening, penicillin prophylaxis, and vaccinations have significantly reduced early childhood mortality. Between 1979 and 2005, childhood mortality for children with SCD decreased by 3% per year; however, a 1% per year increase during the same period was observed for adults (Lanzkron et al. 2013). As more than 94% of children with SCD in well-resourced countries now survive until age 18, and with an expected rise in birth rate for babies with severe hemoglobin disorders to be over 400,000 by 2050 (Piel et al. 2013), disease management needs to shift to a two-tiered system addressing acute and chronic disease needs while simultaneously searching for curative options to address the global burden and public health issues of the disease. Hematopoietic stem cell transplantation (HSCT) and gene therapy offer a way to reduce disease burden, improve outcomes and quality of life for patients with SCD, and potentially reduce health care costs over the long term (Ballas 2009; Arnold et al. 2017; Saenz and Tisdale 2015; Bhatia et al. 2015).
Since the first HSCT in 1984 for a pediatric patient with SCD and acute myelogenous leukemia, numerous patients have successfully undergone bone marrow (BM) HSCT with a human leukocyte antigen (HLA)-matched sibling donor. Whether using a myeloablative or non-myeloablative preparative regimen, greater than 90% of all patients are cured of SCD with a BM HSCT (Walters et al. 1996; Hsieh et al. 2014; Walters et al. 2001; Gluckman et al. 2017). Between 1986 and 2013, over 1,000 patients have received an HLA-matched sibling HSCT with a 5-year event free survival and overall survival of 91.4% and 92.9%, respectively (Gluckman et al. 2017). HSCT should be considered standard of care when a patient has a clinical indication and an HLA-matched sibling donor, yet less than 15% of patients with SCD have an appropriately matched donor (Walters et al. 2001). Furthermore, only 10% of eligible patients have undergone curative HSCT despite patient willingness to consider HSCT morbidity and mortality at the chance for cure (Chakrabarti and Bareford 2007). HLA-matched unrelated donor (MUD) transplantation, umbilical cord blood transplantation (UCBT), and haploidentical transplantation offer more patients the chance for cure, though high rates of complications currently limit the broad use of these therapies. Such complications, including graft rejection and graft-vs-host disease (GVHD), are addressed in gene therapy models where a patient’s autologous hematopoietic stem and progenitor cells (HSPCs) are modified thereby eliminating such complications.
The premise of gene therapy either by gene editing or insertion into autologous HSPCs raises the promise of a safer cure for SCD that is available to all patients. Such methodology eliminates two major barriers in the cure of SCD: the lack of suitable donors, and the morbidity and mortality associated with GVHD. After decades of scientific progress, gene therapy for the cure of SCD is currently in multiple clinical trials with promising initial results. Potential methods for gene therapy in SCD are multiple: (i) addition of therapeutic globin such as β-globin or βT87Q-globin to make adult hemoglobin (HbA), or γ-globin to enhance fetal hemoglobin (HbF) levels, (ii) HbF induction by editing of globin regulatory elements or knockdown of HbF repressors, or (iii) direct gene correction of the SCD mutation with programmable nucleases. Here we focus on the challenges of CRISPR-Cas9 editing, it’s implications, and future possibilities as a curative option for SCD.
2 Genome Editing in SCD
Given the prospect for genotypic and therefore phenotypic correction in a monogenic disorder like SCD, significant effort has been devoted to find critical genes/chromosomal areas contributing to the pathophysiology of the disease. Antisickling genes such as wild type β-globin, modified β-globin (T87Q) which confers additional antisickling properties, γ-globin or β/γ hybrids have been transferred to sickle HSPCs using various viral constructs; of those, some are currently being tested in clinical trials for both safety and efficacy (reviewed in (Demirci et al. 2018)).
Genome editing is desirable as it leads to permeant removal or correction of a detrimental mutation, or by the creation of protective insertions or deletions. Theoretically, programmable nucleases create double strand breaks (DSB) at a specific genomic locus followed by recruitment of DNA repair mechanism through either non-homologous end-joining (NHEJ) or homology directed repair (HDR) (using homologous sequences found in sister chromatids, homologous chromosomes or extrachromosomal donor DNA sequence provided for correction purposes) to the DSB site. Until 5 years ago, three major nucleases including meganucleases also known as homing endonucleases (reviewed in (Stoddard 2011)), zinc finger nucleases (ZFNs, reviewed in (Urnov et al. 2010)), and TAL-effector nucleases (TALENs, reviewed in (M Scharenberg et al. 2013)) were introduced for various genome editing purposes. These tools have been successfully used ex vivo to correct the SCD mutation and induce fetal globin by editing regulatory sequences such as promoters or other regulatory sequences including BCL11A, KLF1 and MYB to circumvent the severity of the mutation in sickle HSPCs (reviewed in (Tasan et al. 2016)). While these nucleases are highly specific thereby diminishing off-target effects (OTEs), programming of these enzymes is difficult, time consuming, and requires significant expertise.
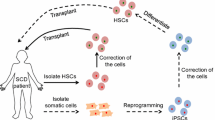
In 2012, Doudna et al. presented a new genome editing technology (Wiedenheft et al. 2012; Jinek et al. 2012), referred to as Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated protein 9 (Cas9), in which a specific RNA (guide RNA) sequence recognizes the target DNA region of interest and directs the effector Cas protein there for editing. This strategy not only revolutionized genome editing strategies but also brought forth the improved possibility of translation of genome editing approaches to the clinical setting due to its advantages: easy to design, highly efficient, and inexpensive. Once introduced into target cells, CRISPR/Cas9 directed DSBs result in activation of DNA repair mechanisms. This machinery would lead to either some insertions/deletions (INDELs), which ideally results in loss-of-function for a given gene, or would repair the DNA break using homology strands if HDR is activated. In this manner, CRISPR/Cas9 technology can target correction of the SCD mutation or induce fetal hemoglobin expression by editing chromosomal areas controlling its expression (Fig. 1), yet challenges in the use of this technology remain surrounding efficiency, safety, and delivery.

Potential CRISPR/Cas9 applications for sickle cell disease (SCD). The proof-of-principle experiments have proven the possibility of SCD mutation correction and fetal hemoglobin (HbF) induction in SCD derived HSCs and iPSCs, and subsequent normal red blood cell derivation for transfusion purposes. However, these advances are waiting to be addressed by clinical trials to explore the full potential
2.1 HbF Induction
HbF is the predominant globin type after the first trimester of gestation and is replaced by HbA by 6 months after birth. Both HbA and HbF are maintained on chromosome 11, with switching from HbF to adult globin mainly controlled by a powerful upstream enhancer known as the locus control region (LCR) that loops to each globin promoter to activate their expression (Li et al. 2002). After the switch to HbA, HbF is not entirely suppressed, though it is not evenly distributed among RBCs. When there is not a genotypic cause for persistence of HbF in all RBCs, HbF can be minimal in some cells or concentrated in specific cells referred to as F-cells (Demirci et al. 2018).
After the initial observation by Janet Watson and colleagues that newborn babies do not show SCD complications for a certain period due to high levels of HbF in the infant’s blood (Watson et al. 1948), more work has been devoted to increase HbF levels in the adult body. The important role of elevated HbF for SCD protection was further confirmed with the reports showing asymptomatic patients with SCD with elevated HbF as a result of coinheriting hereditary persistence of fetal globin (HPFH) mutations (Forget 1998; Stamatoyannopoulos et al. 1975). Such mutations occur either in the form of large deletions in the β-globin gene, or smaller deletions/single nucleotide polymorphisms (SNPs) in γ-globin promoter or HbF regulating quantitative trait loci (QTL) (Paikari and Sheehan 2018). In line with these reports, deletion or inversion of 13.6 kb chromosomal region to obtain a HPFH-like phenotype in SCD patient derived HSPCs resulted in elevated levels of HbF in erythroblast and ameliorated the ex vivo sickling (Antoniani et al. 2018). Similarly, point mutations created by CRISPR/Cas9 approach in the −115 and − 200 clusters of the γ-globin promoter, inhibiting the binding of validated HbF transcriptional repressors BCL11A and LRF (also known as ZBTB7A), respectively (Wang and Thein 2018), de-repressed the expression of HbF (Martyn et al. 2018; Liu et al. 2018). To show the applicability of these approaches, animal models are required for in vivo evaluations prior to human studies. Immunodeficient mice are generally used for human cell engraftment studies but are not proper for in vivo erythropoiesis. To overcome this problem, Li et al. used a human β-globin locus transgenic (β-YAC) mice model to study the in vivo effect of disruption of the repressor binding region within the γ-globin promoter (Li et al. 2018). Along with significant target site distribution which was sustained in the secondary transplantation experiments, no hematological abnormality was seen and pronounced switch from human β to γ globin expression in RBCs of adult mice was noted.
Gene edition of transcriptional regulators is an alternative methodology to stimulate rare naturally occurring HPFH mutations to control HbF expression. Several transcription factors including SCA/TAL1, GATA1 and KLF1 are reported to be involved in HbF regulation (Sankaran and Orkin 2013). While all of them could be considered potential candidates, direct targeting of these factors for HbF induction is challenging as all of them have either broader roles in non-erythroid linages or have significant roles in normal erythropoiesis. Significant candidates, LFR and BCL11A, are validated HbF silencers (Uda et al. 2008; Menzel et al. 2007), and have been edited in the erythrocyte progenitor cell line (HUDEP-2) leading to robust HbF expression (Masuda et al. 2016). BCL11A is important for HSPC function (Tsang et al. 2015) and normal lymphoid development (Liu et al. 2003), with only one paper demonstrating very low level indels and a slight increase in γ-globin expression in a non-human primate model using TALE nuclease mRNA targeting the BCL11A coding sequence with respect to control transplants (Humbert et al. 2018). The safety and feasibility of the BCL11A knockdown is still awaiting to be addressed by large animal models with high indel ratios and subsequent clinical trials with large patients cohorts. The first clinical trial launched in February 2018 uses a lentiviral gene transfer vector encoding a microRNA-adapted small hairpin (sh) RNAs (shRNAmiR) targeting BCL11A in patients with severe SCD is currently ongoing with the first patient demonstrating 23% HbF (NCT03282656, Shim et al. 2017; Esrick et al. 2018). Recently, Daniel Bauer and colleagues have presented a different approach in which they induce comparable levels of HbF in CD34+ cells by targeting the +58 intronic site of the BCL11A gene that acts as an erythroid specific enhancer (Bauer et al. 2013; Canver et al. 2015). They were able to show that while guide RNA directed disruption of the enhancer site provided substantial reduction in Bcl11a expression in erythrocyte cells leading to elevated HbF expression in mice, it did not affect the expression in non-erythroid lineages (Smith et al. 2016). The results were extended to erythroid cells derived from progenitor cells of patients with β-Thalassemia major (Psatha et al. 2018), supporting that this enhancer disruption strategy would be favorable for clinical use if it is proven safe with preclinical and clinical studies.
With the establishment of guide RNA screening models, it has become possible to discover novel genomic sites/genes controlling HbF expression. In a recent paper, protein kinase domain–focused CRISPR/Cas9–based genetic screening revealed that heme-regulated inhibitor HRI (also known as EIF2AK1), an erythroid-specific kinase that controls protein translation as an HbF repressor, could be used as a potential candidate for the treatment of hemoglobinopathies (Grevet et al. 2018). Using similar methodology, the same group also identified that SPOP, a substrate adaptor of the CUL3 ubiquitin ligase complex, as a HbF repressor in both HUDEP-2 and CD34+ cells (Lan et al. 2018). Extending these guide RNA screening strategies to non-coding regions and epigenetics would allow identification of stronger candidates or gene combinations to enhance HbF expression to clinically meaningful levels that reverse the sickling of RBCs and reverse the disease phenotype as seen in patients with HPFH.
2.2 SCD Mutation Correction
As the pathologic mutation for SCD is already clearly identified, correction of the SCD mutation seems the most difficult but potentially the most feasible and promising approach as Cas9 cuts sickle β-globin and this break can be repaired if a normal β-globin sequence flanked with homology arms to the DSB is supplied. Genotypic correction appears possible by targeting the specific locus at the genome and providing the correct sequence for β-globin without the necessity of exogenous transgene activation.
To ensure proper correction, an increasing number of researchers are using gene editing technologies for correcting the SCD mutation in different cell types (Table 1). Most of these works use the CRISPR/Cas9 system as it has shown better correction efficiency and lower OTEs than other gene editing tools such as TALENs (Bak et al. 2018; Hoban et al. 2016a). The HSPC source is historically bone marrow derived CD34+ HSPCs, currently used in the majority of genome editing studies, though recently peripherally mobilized CD34+ HSPCs using plerixafor has shown promise in patients with SCD given safety concerns regarding granulocyte colony stimulating factor use in these patients. These CD34+ cells can be modified to be infused back into the patient. However, differences in the cell cycle or the presence of specific nucleases that might disrupt the correction pathways used by the cells after the DSBs offer overall resistance to successful gene editing (Lomova et al. 2018). In order to maximize success, the preferred delivery method in these studies is electroporation with an Adeno-associated virus (AAV)-6 viral vector for the delivery of the CRISPR/Cas9 system with the donor DNA. For evaluating the correction of the SCD mutation, several studies analyzed gene editing at the DNA level using either targeted deep sequencing (Lomova et al. 2018; Wen et al. 2017) or nested droplet digital (dd)PCR (Vakulskas et al. 2018), while others used more functional studies like RNAseq or RNA expression levels (Dever et al. 2016; DeWitt et al. 2016; Chung et al. 2018; Magis et al. 2018) with only three studies using High performance liquid chromatography (HPLC) for measuring protein levels after the correction of the SCD mutation in the β-globin gene (Hoban et al. 2016a; Vakulskas et al. 2018; DeWitt et al. 2016).
Since the publication in 2008 of a protocol for generating human iPSCs from somatic cells, many groups have developed protocols for the differentiation of iPSCs into different cell lineages such as hematopoietic cells to become another viable source of autologous HSPCs (Fujita et al. 2016; Ferreira et al. 2018; Sugimura et al. 2017). Currently however, the hematopoietic cells derived from iPSCs are primitive rather than definitive hematopoietic cells and are therefore unable to engraft in a xenograft mouse model. The available protocols for the differentiation of iPSCs towards HSPCs mainly mimic primitive hematopoiesis, which can be noticed when the generated HSPCs are differentiated into erythroid cells containing mainly ε-globin and γ-globin, with very low amounts of β-globin if present at all. In order to realize the available gene editing tools to correct the SCD mutation in iPSCs for therapeutic purpose, a proper differentiation protocol is needed to produce engraftable HSPCs from iPSCs. Such therapy, as with other autologous modification strategies, would ultimately eliminate two major hurdles in allogeneic transplantation; rejection and GVHD in transplantation therapies. In addition, as low efficiency of correction is a problem for HSPC studies, cloning corrected cells from a bulk iPSC population would allow derivation of a population with 100% of cells corrected.
In addition to the difficulties differentiating iPSCs towards HSPCs, only one study has presented the correction of the SCD mutation in SCD-derived iPSCs at the RNA and protein levels by qPCR and Western blot analyses, respectively (Huang et al. 2015), though several groups have shown the correction of the SCD mutation at the DNA level using nested ddPCR or DNA sequencing (Park et al. 2017; Li et al. 2016; Martin et al. 2018). Correction of underlying mutation in both CD34+ and iPSCs seems promising, yet while significant correction rates are reported in ex vivo conditions, limited corrected human cell engraftment are reported in immunodeficient mouse models (Table 1). While immunodeficient mice transplantation models for human cell engraftment studies are being widely accepted, it is not clear that whether these results completely reflect the clinical outcome of these approaches. After optimization of the correction methodologies, larger animal models are necessary to explore the potential of the application.
Though editing of CD34+ cells is possible, multiple genotypic outcomes are possible and editing of long-term engrafting HSPCs are not yet fully explored. Treating cells with CRISPR/Cas9 and a β-globin donor might result with cells in their native state (uncorrected), as sickle trait (one allele corrected), as healthy (both alleles corrected), as β-thalassemia major (both alleles disrupted), as β-thalassemia trait (one allele corrected and other disrupted), and/or sickle/β-thalassemia (one allele disrupted) due to NHEJ/HDR machinery of the cells (Esrick and Bauer 2018). As precise correction in long-term HSPCs is not yet efficient and editing results in reduction in engrafting HSPCs (Hoban et al. 2016a; Dever et al. 2016), transplantation of mixed culture could be clinically problematic and possible unintended consequences should be addressed before clinical trials.
3 Challenges
Genome editing has been the most attractive tool for scientists seeking to correct genetic mutations either as gene knockout or knock-in. Conventional methods for genome engineering, however, are costly, time-consuming, labor-intensive, and require expertise in protein engineering to design specific nucleases (Roy et al. 2018). On the contrary, CRISPR/Cas9 genome editing is a system that is relatively easier, cheaper and more efficient, and is being used in a large variety of model cells and species. It has not only led to easier and cheaper development of knock-out animal models but has also contributed to the establishment of whole-genome screening libraries that identify therapeutic genes/chromosomal regions that may directly affect a targeted phenotype. While there is a huge international interest in CRISPR/Cas9-based editing approaches, there is still much to improve upon such as the efficiency of cutting and editing (both NHEJ and HDR), improving specificity, and improving delivery methods. Lastly, there is a world-wide concern about safety, particularly as it relates to OTEs, that needs to be clarified and addressed before transferring this approach into routine clinical care.
3.1 Efficiency of Editing
The limiting factor for diverse application of a given CRISPR/Cas9 system has been the dependency on a protospacer-adjacent motif (PAM) sequence flanking the target. For instance, as SCD mutation correction studies need to target a specific chromosomal area, there are not many guide RNA options for different Cas proteins. Therefore, substantial effort has been made to engineer various Cas effector proteins for the recognition of different PAM sequence (Kleinstiver et al. 2015a; Nishimasu et al. 2018; Kim et al. 2017). While the introduction of 19 subtypes of CRISPR systems with various Cas effector proteins recognizing different PAM sites have extended targetable genomic loci (Leenay and Beisel 2017), not all of them have been widely studied in terms of efficacy and safety. Therefore, scientists still tend to use well-established Cas types (i.e Streptococcus pyogenes Cas9-SpCas9 or Cpf1-Cas12a) in their research. SpCas9 has a PAM recognition of 5’ NGG 3′, while some other Cas9 orthologs have been reported to require longer PAM sites (Fonfara et al. 2013; Ran et al. 2015). While these have some advantages over classical SpCas9, their longer PAM sites restrict their use despite potentially more efficient delivery. For example, smaller Cas effector proteins such as Staphylococcus aureus derived Cas9 (SaCas9) with a PAM site of NNGRRT, are more efficient for viral delivery systems (Kleinstiver et al. 2015b). To extend the boundaries of targeting range for Cas9 proteins, PAM preference can successfully be altered by targeted mutations to residues near the PAM DNA duplex (Anders et al. 2016; Hirano et al. 2016).
Understanding the subunits of Cas effector proteins have allowed the modification of PAM specificity. In a recent report, Chatterjee et al. characterized Streptococcus canis Cas9 (ScCas9) displaying 5′-NNG-3′ PAM, reporting an 89.2% sequence similarity to SpCas9 (Chatterjee et al. 2018). Structural analysis showed that two distinct mutational areas [a positive-charged insertion in the REC domain (at 367–376) and a KQ insertion in the PAM-interacting domain (at 1337 and 1338)] are responsible for having the specificity for a minimal PAM sequence. Another group has recently generated Cas9 variants with various PAM compatibilities (including NG, GAA and GAT) using phage-assisted continuous evolution (PACE) approach (Hu et al. 2018). But more intriguingly, although extending PAM recognition capacity of Cas9 variants would be assumed to augment OTE (Hu et al. 2018; Tsai et al. 2015), they reported greater DNA specificity for Cas9 variants with respect to canonical SpCas9 along with lower genome-wide off-target. In a different approach, Sniper-Cas9 (F539S/M763I/K890 N variant) was successfully obtained using directed evolution, and characterized with high on-target and reduced OTEs (Lee et al. 2018). These studies illustrate the potential and the need for further improvements in targetable loci on the genome for various Cas effector proteins. While improving the efficiency, safety should also be parallelly taken into account to realize the approaches in routine clinical applications.
3.2 Potential Immunogenicity of Editing Tools or Edited Cells
The ultimate goal of CRISPR technology is to edit mutations related with disorders or control disease associated gene expressions in patient-derived specific stem/progenitor cells. However, in vivo effects of CRISPR/Cas9 systems have a lot of unanswered questions. In 2019, there are open clinical trials in the United States and abroad using CRISPR/Cas9 for a potential treatment of SCD, Thalassemia, HIV-1, and several cancer types (https://clinicaltrials.gov/keyword CRISPR). Though hope remains for these clinical trials, ex vivo work conducted thus far, demonstrate preliminary data pointing toward possible adverse effects of the technology. The first question is whether guide RNAs or Cas9 itself has any effects on the immune system. To partially address this uncertainty, Kim et al. demonstrated that in vitro transcribed guide RNAs with a 5′-triphosphate group (5′-ppp) leads to cytotoxicity due to the activation of innate immune system in human and mouse cells (Kim et al. 2018). The authors also reported that removal of triphosphate resulted in high mutation rate in primary human CD4+ cells thus avoiding the innate immune system. In a recent pre-print article, Charlesworth et al. showed pre-existing antibodies against Cas9 derived from Staphylococcus aureus (79%) or Streptococcus pyogenes (65%) in a small group of healthy volunteers (Charlesworth et al. 2018). In a follow up work performed with 200 blood samples, prevalence of antibodies against SaCas9 and SpCas9 were reported to be 10% and 2.5%, respectively (Simhadri et al. 2018). While these results are not unexpected, triggering of the immune system by CRISPR/Cas9 is potentially problematic and harmful in vivo. While these observations and potential immune response are awaiting to be addressed by large animal models and clinical studies, Cas9 expression levels, delivery methods, vector types in case of transduction routes, and target cells populations should be optimized in any capacity to diminish a severe immune response.
3.3 Specificity of Editing
Other than a potential immune response, OTEs are one of the biggest challenges of CRISPR/Cas9 system. As Cas9-guide RNA complex can recognize sequences with up to 5 mismatched bases (Fu et al. 2013), the possibility of OTE for a given guide RNA cannot be ignored. A number of advances have been taken to increase the specificity of CRISPR/Cas system, but the guide RNA design is the first critical process for reduction of OTEs. There are vast guide RNA design tools available; of those, newer ones include supplementary algorithms evaluating on-target cutting efficiency other than selectivity for the target. During the synthesis of guide RNA, additional modifications on the guide RNA structure including truncation of spacer RNA (Fu et al. 2014) and chemical modifications (Cromwell et al. 2018) have been reported to increase Cas9 endonuclease specificity. In addition, chemical modifications with 2′-O-methyl 3′ phosphorothioate (Hendel et al. 2015) and 2′-fluoro-ribose (Rahdar et al. 2015) improve the editing efficiency via increasing the stability of guide RNAs in cells.
The second important aspect to reduce OTEs is to enhance Cas9 specificity. A mutated variant of Cas9, nickase (Cas9n), can only cut a single DNA strand such that two close recognition sites in the DNA are required for a double strand break and thus OTEs are drastically reduced (50–1500 fold in human cells) (Ran et al. 2013). However, as some single nicks can be converted to double strand breaks, this approach was further improved with introduction of a catalytically inactive Cas9 (dead (d)Cas9) and Fok1 fusion protein (Tsai et al. 2014). In this approach, recognition of guide RNAs by dCas9 brings Fok1 enzyme the close proximity that is required for active dimerized Fok1 nuclease. While these approaches provided significant reduction in the off-target issues, requirement for a double recognition site might result in less editing efficiencies, and the necessity for double guide RNA usage might limit viral delivery approaches. To therefore keep editing efficiency high enough for clinical application, active nucleases are being engineered for higher specificities. The initial idea for high specificity nucleases was to decrease the interactions of Cas9 with its DNA target to lessen OTEs while keeping enough energy for on-target recognition. With the introduction of high fidelity Cas9 (SpCas9-HF1, N497A/R661A/Q695A/Q926A) (Kleinstiver et al. 2016) and enhanced specificity Cas9 (eSpCas9 (1.1), K848A/K1003A/R1060A) (Slaymaker et al. 2016), there are no or significantly reduced OTEs compared to wild type nucleases while maintaining robust on-target activities. Recently, Doudna et al. has published that both SpCas9-HF1 and eSpCas9(1.1) are trapped in an inactive state when bound to mismatched targets and that the non-catalytic domain of Cas9, REC3, is responsible for target recognition and direction of nuclease activity (Chen et al. 2017). Using these observations, they were able to create hyper-accurate Cas9 variant (HypaCas9) with wide-range genome specificity without compromising any detectable OTEs.
Recently, several publications have raised appropriate concern about the CRISPR/Cas system showing unintended consequences such as large deletions, insertions, and rearrangement of the chromosome when used in clinical trials (Kosicki et al. 2018; Shin et al. 2017; Adikusuma et al. 2018). It is not clear that this uncertainty is going to be elucidated, or is clinically relevant, but it is sensible to urge more pre-clinical studies addressing these valid safety concerns.
3.4 Delivery
To apply CRISPR/Cas9 system to a given cell type/organism, the structure and vehicle of the components should be determined based on requirements for protein amount, exposure time, efficiency, and restrictions for OTEs and other safety issues. For the structure of the system, it could be (i) integrating/non-integrating viral vectors/plasmids expressing both mRNAs for guide RNA and Cas9, (ii) Cas9 mRNA and guide RNA, or (iii) ribonucleoprotein complex (RNP) constituting Cas9 protein and guide RNA. A short time after the discovery that CRISPR/Cas9 system could be used in human cells for genome editing purposes, viral constructs providing continuous expression of Cas9 and guide RNAs were used to explore this potential. However, while it might be advantageous for gene editing approaches requiring long-term expression, it was also recognized that sustained expression of guide RNAs and Cas9 augmented the possibility of mismatch bindings and OTEs (Pattanayak et al. 2013). For precise temporal control of expression, several inducible systems have been presented (Nihongaki et al. 2015; Zetsche et al. 2015). Using vectoral delivery in the lab is stable and cheap; however, there is still ongoing debate about the problems of viral systems with the immune system (Yin et al. 2014) and insertional mutagenesis (Hoban et al. 2016b). An alternative method to plasmids/vectors carrying Cas9 sequence is the introduction of mRNA for Cas9 that is translated to active protein once it is transferred to the cell. While this system avoids the time needed for transcription for Cas9 transferred with plasmids, it is also applicable only for genome editing approaches doable with transient Cas9 expressions. In addition, as mRNAs are not as stable as DNAs, delivery time of RNAs for Cas9 and guide RNA would be critical. Jiang et al. showed that Cas9 protein was at maximum level 6 h after delivering Cas9 mRNA and not detectable after 24 h in mice (Jiang et al. 2017). One way to optimize efficiency would be different delivery times or chemical modifications to provide the stability of RNAs as was mentioned earlier (Safety section).
The RNP complex is another alternative in which native Cas9 protein and guide RNA form a single complex that is readily active once it is in the cell. Other than the question of whether native foreign protein to the human cells is significantly immunogenic to hinder the potential of RNP usage, the main drawback of this application is that Cas9-guide RNA structure is a relatively large complex. Non-viral delivery systems including electroporation, encapsulation, and delivery by modification are trending for not only transferring this large cargo but also for other DNA and RNA systems (reviewed in (Glass et al. 2018)). Electroporation, a non-selective delivery method, has been used for a long time for various DNA, RNA, and protein transfers through the cell membrane by enlarging the pores on the cell membrane via a strong electric field. While this method is highly efficient in transferring Cas9 and guide RNA to HSPCs for an aim of correction of SCD mutation (Hoban et al. 2016a; Dever et al. 2016; Magis et al. 2018), toxicity and the long-term viability issue of electroporation for a clinical setting is still being questioned. From a clinical point of view, huge quantities of Cas9 protein might be required for a clinical setting, and purification of endotoxin-free Cas9 protein is not economically feasible at this time. More industrial work is warranted to explore feasible ways for GMP grade Cas9 production in order for this technique to be practical in a clinical setting.
4 Future Perspective and Directions
While SCD was characterized more than century ago, definitive treatment for all patients is not currently available given a lack of suitable donors for curative HSCT. As monogenic disease, SCD is one of the most important candidates for programmable nucleases, particularly CRISPR/Cas9 due to being cost-effective, easily applicable, and highly efficient. Proof-of-principle studies have shown that CRISPR/Cas9 can efficiently be used to correct the SCD mutation or induce HbF expression in ex vivo cell culture conditions and mouse models. However, there is still concerns about the safety due to random off-target effect and subtherapeutic efficiency. More work should be conducted in larger animal models to demonstrate the safety of the approach along with optimization studies in ex vivo conditions.
Clinical trials investigating the prospective of CRISPR/Cas9 for SCD are in progress or are starting soon, which will certainly direct the future of this approach. The application itself is promising but it is not currently feasible for translation into routine use especially for less developed counties such as Africa where prevalence of SCD is high. Additional cost-effective manufacturing processes for clinical grade guide RNAs and Cas9 proteins should be implemented to extend the use, and ensure a safer, more efficient product. The premise of gene therapy for the cure of SCD is moving closer to reality, though questions and challenges remain to ensure this as a feasible, safe, and lifelong curative strategy.
Abbreviations
- AAV:
-
Adeno-associated virus
- BM:
-
Bone marrow
- Cas9:
-
CRISPR associated protein 9
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- DSB:
-
Double strand breaks
- dCas9:
-
Dead Cas9
- ddPCR:
-
Droplet digital PCR
- eSpCas9:
-
Enhanced specificity Streptococcus pyogenes Cas9
- GVHD:
-
Graft-vs-host disease
- HbA:
-
Adult hemoglobin
- HbF:
-
Fetal hemoglobin
- HbS:
-
Hemoglobin S
- HDR:
-
Homology directed repair
- HLA:
-
Human leukocyte antigen
- HPFH:
-
Hereditary persistence of fetal globin
- HPLC:
-
High performance liquid chromatography
- HRI:
-
Heme-regulated inhibitor
- HSCT:
-
Hematopoietic stem cell transplantation
- HSPCs:
-
Hematopoietic stem/progenitor cells
- HU:
-
Hydroxyurea
- INDELs:
-
Insertions/deletions
- iPSCs:
-
Induced pluripotent stem cells
- LCR:
-
Locus control region
- MUD:
-
Matched unrelated donor
- NHEJ:
-
Non-homologous end-joining
- OTEs:
-
Off-target effects
- PACE:
-
Phage-assisted continuous evolution
- PAM:
-
Protospacer-adjacent motif
- QTL:
-
Quantitative trait loci
- RBCs:
-
red blood cells
- ScCas9:
-
Streptococcus canis Cas9
- SCD:
-
Sickle cell disease
- shRNAmiR :
-
MicroRNA-adapted small hairpin (sh) RNAs
- SpCas9-HF1:
-
high fidelity Streptococcus pyogenes Cas9
- TALENs:
-
TAL-effector nucleases
- UCBT:
-
Umbilical cord blood transplantation
- ZFNs:
-
Zinc finger nucleases
References
Adikusuma F, Piltz S, Corbett MA, Turvey M, McColl SR, Helbig KJ, Beard MR, Hughes J, Pomerantz RT, Thomas PQ (2018) Large deletions induced by Cas9 cleavage. Nature 560(7717):E8–E9
Anders C, Bargsten K, Jinek M (2016) Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Mol Cell 61(6):895–902
Antoniani C, Meneghini V, Lattanzi A, Felix T, Romano O, Magrin E, Weber L, Pavani G, El Hoss S, Kurita R (2018) Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood 131(17):1960–1973. https://doi.org/10.1182/blood-2017-10-811505
Arnold SD, Brazauskas R, He N, Li Y, Aplenc R, Jin Z, Hall M, Atsuta Y, Dalal J, Hahn T (2017) Clinical risks and healthcare utilization of haematopoietic cell transplantation for sickle cell disease in the US using merged databases. Haematologica 102(11):1823–1832. https://doi.org/10.3324/haematol.2017.169581
Bak RO, Gomez-Ospina N, Porteus MH (2018) Gene editing on center stage. Trends Genet 34(8):600–611
Ballas SK (2009) The cost of health care for patients with sickle cell disease. Am J Hematol 84(6):320–322
Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L (2013) An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 342(6155):253–257
Bhatia M, Kolva E, Cimini L, Jin Z, Satwani P, Savone M, George D, Garvin J, Paz ML, Briamonte C (2015) Health-related quality of life after allogeneic hematopoietic stem cell transplantation for sickle cell disease. Biol Blood Marrow Transplant 21(4):666–672
Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP (2015) BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527(7577):192–197
Chakrabarti S, Bareford D (2007) A survey on patient perception of reduced-intensity transplantation in adults with sickle cell disease. Bone Marrow Transplant 39(8):447–451
Charlesworth CT, Deshpande PS, Dever DP, Dejene B, Gomez-Ospina N, Mantri S, Pavel-Dinu M, Camarena J, Weinberg KI, Porteus MH (2018) Identification of pre-existing adaptive immunity to Cas9 proteins in humans. BioRxiv:243345. https://doi.org/10.1101/243345
Chatterjee P, Jakimo N, Jacobson JM (2018) Minimal PAM specificity of a highly similarSpCas9 ortholog. Sci Adv 4:eaau0766
Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, Doudna JA (2017) Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 550(7676):407–410
Chung JE, Magis W, Vu J, Heo S-J, Wartiovaara K, Walters MC, Kurita R, Nakamura Y, Boffelli D, Martin DI (2018) CRISPR-Cas9 interrogation of a putative fetal globin repressor in human erythroid cells. BioRxiv:335729. https://doi.org/10.1101/335729
Cromwell CR, Sung K, Park J, Krysler AR, Jovel J, Kim SK, Hubbard BP (2018) Incorporation of bridged nucleic acids into CRISPR RNAs improves Cas9 endonuclease specificity. Nat Commun 9(1):1448
Demirci S, Uchida N, Tisdale JF (2018) Gene therapy for sickle cell disease: an update. Cytotherapy 20(7):899–910
Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB, Mantri S (2016) CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 539(7629):384–389
DeWitt MA, Magis W, Bray NL, Wang T, Berman JR, Urbinati F, Heo S-J, Mitros T, Muñoz DP, Boffelli D (2016) Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Transl Med 8(360):360ra134–360ra134
Esrick EB, Bauer DE (2018) Genetic therapies for sickle cell disease. Semin Hematol 55(8):76–86
Esrick EB, Brendel C, Manis JP, Armant MA, Negre H, Dansereau C, Ciuculescu MF, Patriarca S, Mackinnon B, Daley H (2018) Flipping the switch: initial results of genetic targeting of the fetal to adult globin switch in sickle cell patients. Blood 132:1023
Ferreira AF, Calin GA, Picanço-Castro V, Kashima S, Covas DT, de Castro FA (2018) Hematopoietic stem cells from induced pluripotent stem cells–considering the role of microRNA as a cell differentiation regulator. J Cell Sci 131(4):jcs203018
Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lecrivain A-L, Bzdrenga J, Koonin EV, Charpentier E (2013) Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res 42(4):2577–2590
Forget BG (1998) Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci 850(1):38–44
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31(9):822–826
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK (2014) Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32(3):279–284
Fujita A, Uchida N, Haro-Mora JJ, Winkler T, Tisdale J (2016) β-globin-expressing definitive erythroid progenitor cells generated from embryonic and induced pluripotent stem cell-derived sacs. Stem Cells 34(6):1541–1552
Glass Z, Lee M, Li Y, Xu Q (2018) Engineering the delivery system for CRISPR-based genome editing. Trends Biotechnol 36(2):173–185
Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F, Carreras J, Simões BP, Ferster A, Dupont S, De La Fuente J (2017) Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 129(11):1548–1556
Grevet JD, Lan X, Hamagami N, Edwards CR, Sankaranarayanan L, Ji X, Bhardwaj SK, Face CJ, Posocco DF, Abdulmalik O (2018) Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science 361(6399):285–290
Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB (2015) Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol 33(9):985–989. https://doi.org/10.1038/nbt.3290
Hirano S, Nishimasu H, Ishitani R, Nureki O (2016) Structural basis for the altered PAM specificities of engineered CRISPR-Cas9. Mol Cell 61(6):886–894
Hoban MD, Lumaquin D, Kuo CY, Romero Z, Long J, Ho M, Young CS, Mojadidi M, Fitz-Gibbon S, Cooper AR (2016a) CRISPR/Cas9-mediated correction of the sickle mutation in human CD34+ cells. Mol Ther 24(9):1561–1569
Hoban MD, Orkin SH, Bauer DE (2016b) Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood 127(7):839–848
Hsieh MM, Fitzhugh CD, Weitzel RP, Link ME, Coles WA, Zhao X, Rodgers GP, Powell JD, Tisdale JF (2014) Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 312(1):48–56
Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z (2018) Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556(7699):57–63
Huang X, Wang Y, Yan W, Smith C, Ye Z, Wang J, Gao Y, Mendelsohn L, Cheng L (2015) Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells 33(5):1470–1479
Humbert O, Peterson CW, Norgaard ZK, Radtke S, Kiem H-P (2018) A nonhuman primate transplantation model to evaluate hematopoietic stem cell gene editing strategies for β-hemoglobinopathies. Mol Ther Methods Clin Dev 8:75–86
Jiang C, Mei M, Li B, Zhu X, Zu W, Tian Y, Wang Q, Guo Y, Dong Y, Tan X (2017) A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res 27(3):440–443
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821. https://doi.org/10.1126/science.1225829
Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR (2017) Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol 35(4):371–376
Kim S, Koo T, Jee H-G, Cho H-Y, Lee G, Lim D-G, Shin HS, Kim J-S (2018) CRISPR RNAs trigger innate immune responses in human cells. Genome Res 28:367–373. https://doi.org/10.1101/gr.231936.117
Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales AP, Li Z, Peterson RT, Yeh J-RJ (2015a) Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523(7561):481–485
Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, Joung JK (2015b) Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 33(12):1293–1298
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK (2016) High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529(7587):490–495
Kosicki M, Tomberg K, Bradley A (2018) Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol 36(8):765–771
Lan X, Khandros E, Grevet JD, Peslak SA, Bhardwaj S, Keller CA, Giardine B, Garcia BA, Hardison RC, Shi J (2018) Domain-focused CRISPR-Cas9 screen identifies the E3 ubiquitin ligase substrate adaptor protein SPOP as a novel repressor of fetal hemoglobin. Blood 132:414
Lanzkron S, Carroll CP, Haywood C Jr (2013) Mortality rates and age at death from sickle cell disease: US, 1979–2005. Public Health Rep 128(2):110–116
Lee JK, Jeong E, Lee J, Jung M, Shin E, Kim Y-h, Lee K, Jung I, Kim D, Kim S (2018) Directed evolution of CRISPR-Cas9 to increase its specificity. Nat Commun 9(1):3048
Leenay RT, Beisel CL (2017) Deciphering, communicating, and engineering the CRISPR PAM. J Mol Biol 429(2):177–191
Li Q, Peterson KR, Fang X, Stamatoyannopoulos G (2002) Locus control regions. Blood 100(9):3077–3086
Li C, Ding L, Sun C-W, Wu L-C, Zhou D, Pawlik KM, Khodadadi-Jamayran A, Westin E, Goldman FD, Townes TM (2016) Novel HDAd/EBV reprogramming vector and highly efficient Ad/CRISPR-Cas sickle cell disease gene correction. Sci Rep 6:30422
Li C, Psatha N, Sova P, Gil S, Wang H, Kim J, Kulkarni C, Valensisi C, Hawkins RD, Stamatoyannopoulos G (2018) Reactivation of γ-globin in adult β-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing. Blood 131:2915–2928. https://doi.org/10.1182/blood-2018-03-838540
Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, Jenkins NA, Copeland NG (2003) Bcl11a is essential for normal lymphoid development. Nat Immunol 4(6):525–532
Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R (2018) Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 173(2):430–442. e417
Lobner K, Lanzkron S, Haywood C (2013) NIH and National Foundation Expenditures for sickle cell disease and cystic fibrosis are associated with Pubmed publications and FDA approvals. Blood 122:1739
Lomova A, Clark DN, Campo-Fernandez B, Flores-Bjurström C, Kaufman ML, Fitz-Gibbon S, Wang X, Miyahira EY, Brown D, DeWitt MA (2018) Improving gene editing outcomes in human hematopoietic stem and progenitor cells by temporal control of DNA repair. Stem Cells:1–11. https://doi.org/10.1002/stem.2935
M Scharenberg A, Duchateau P, Smith J (2013) Genome engineering with TAL-effector nucleases and alternative modular nuclease technologies. Curr Gene Ther 13(4):291–303
Magis W, DeWitt MA, Wyman SK, Vu JT, Heo S-J, Shao SJ, Hennig F, Romero ZG, Campo-Fernandez B, McNeill M (2018) In vivo selection for corrected β-globin alleles after CRISPR/Cas9 editing in human sickle hematopoietic stem cells enhances therapeutic potential. BioRxiv:432716. https://doi.org/10.1101/432716
Martin R, Ikeda K, Uchida N, Cromer MK, Nishimura T, Dever DP, Camarena J, Bak R, Lausten A, Jakobsen MR (2018) Selection-free, high frequency genome editing by homologous recombination of human pluripotent stem cells using Cas9 RNP and AAV6. BioRxiv:252163. https://doi.org/10.1101/252163
Martyn GE, Wienert B, Yang L, Shah M, Norton LJ, Burdach J, Kurita R, Nakamura Y, Pearson RC, Funnell AP (2018) Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet 50(4):498–503
Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell AP, Fisher C, Suciu M, Martyn GE, Norton LJ (2016) Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 351(6270):285–289
Menzel S, Garner C, Gut I, Matsuda F, Yamaguchi M, Heath S, Foglio M, Zelenika D, Boland A, Rooks H (2007) A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet 39(10):1197–1199
Nihongaki Y, Kawano F, Nakajima T, Sato M (2015) Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat Biotechnol 33(7):755–760
Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, Noda T, Abudayyeh OO, Gootenberg JS, Mori H (2018) Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361(6408):1259–1262
Paikari A, Sheehan VA (2018) Fetal haemoglobin induction in sickle cell disease. Br J Haematol 180(2):189–200
Park S, Gianotti-Sommer A, Molina-Estevez FJ, Vanuytsel K, Skvir N, Leung A, Rozelle SS, Shaikho EM, Weir I, Jiang Z (2017) A comprehensive, ethnically diverse library of sickle cell disease-specific induced Pluripotent stem cells. Stem Cell Rep 8(4):1076–1085
Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR (2013) High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31(9):839–843
Paulukonis ST, Eckman JR, Snyder AB, Hagar W, Feuchtbaum LB, Zhou M, Grant AM, Hulihan MM (2016) Defining sickle cell disease mortality using a population-based surveillance system, 2004 through 2008. Public Health Rep 131(2):367–375
Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN (2013) Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med 10(7):e1001484
Psatha N, Reik A, Phelps S, Zhou Y, Dalas D, Yannaki E, Levasseur DN, Urnov FD, Holmes MC, Papayannopoulou T (2018) Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in erythroid cells of patients with β-thalassemia major. Mol Ther Methods Clin Dev 10:313–326
Rahdar M, McMahon MA, Prakash TP, Swayze EE, Bennett CF, Cleveland DW (2015) Synthetic CRISPR RNA-Cas9–guided genome editing in human cells. Proc Natl Acad Sci 112(51):E7110–E7117
Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154(6):1380–1389
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature 520(7546):186–191
Roy B, Zhao J, Yang C, Luo W, Xiong T, Li Y, Fang X, Gao G, Singh CO, Madsen L (2018) CRISPR/cascade 9-mediated genome editing-challenges and opportunities. Front Genet 9:240–252
Saenz C, Tisdale JF (2015) Assessing costs, benefits, and risks in chronic disease: taking the long view. Biol Blood Marrow Transplant 21(7):1149–1150
Sankaran VG, Orkin SH (2013) The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med 3(1):a011643
Shim G, Kim D, Park GT, Jin H, Suh S-K, Oh Y-K (2017) Therapeutic gene editing: delivery and regulatory perspectives. Acta Pharmacol Sin 38(6):738–753
Shin HY, Wang C, Lee HK, Yoo KH, Zeng X, Kuhns T, Yang CM, Mohr T, Liu C, Hennighausen L (2017) CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat Commun 8:15464
Simhadri VL, McGill J, McMahon S, Wang J, Jiang H, Sauna ZE (2018) Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the US population. Mol Ther Methods Clin Dev 10:105–112
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F (2016) Rationally engineered Cas9 nucleases with improved specificity. Science 351(6268):84–88
Smith LA, Oyeku SO, Homer C, Zuckerman B (2006) Sickle cell disease: a question of equity and quality. Pediatrics 117(5):1763–1770
Smith EC, Luc S, Croney DM, Woodworth MB, Greig LC, Fujiwara Y, Nguyen M, Sher F, Macklis JD, Bauer DE (2016) Strict in vivo specificity of the Bcl11a erythroid enhancer. Blood 128(19):2338–2342. https://doi.org/10.1182/blood-2016-08-736249
Stamatoyannopoulos G, Wood W, Papayannopoulou T, Nute P (1975) A new form of hereditary persistence of fetal hemoglobin in blacks and its association with sickle cell trait. Blood 46(5):683–692
Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, Orringer E, Bellevue R, Olivieri N, Eckman J (2003) Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA 289(13):1645–1651
Stoddard BL (2011) Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure 19(1):7–15
Sugimura R, Jha DK, Han A, Soria-Valles C, da Rocha EL, Lu Y-F, Goettel JA, Serrao E, Rowe RG, Malleshaiah M (2017) Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 545(7655):432–438
Tasan I, Jain S, Zhao H (2016) Use of genome-editing tools to treat sickle cell disease. Hum Genet 135(9):1011–1028
Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK (2014) Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32(6):569–576
Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP (2015) GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33(2):187–197
Tsang JC, Yu Y, Burke S, Buettner F, Wang C, Kolodziejczyk AA, Teichmann SA, Lu L, Liu P (2015) Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells. Genome Biol 16(1):178
Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, Usala G, Busonero F, Maschio A, Albai G (2008) Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc Natl Acad Sci 105(5):1620–1625
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD (2010) Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11(9):636–646
Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, Bode NM, McNeill MS, Yan S, Camarena J (2018) A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med 24(8):1216–1224
Walters MC, Patience M, Leisenring W, Eckman JR, Scott JP, Mentzer WC, Davies SC, Ohene-Frempong K, Bernaudin F, Matthews DC (1996) Bone marrow transplantation for sickle cell disease. N Engl J Med 335(6):369–376
Walters M, Patience M, Leisenring W, Rogers Z, Aquino V, Buchanan G, Roberts I, Yeager A, Hsu L, Adamkiewicz T (2001) Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant 7(12):665–673
Wang X, Thein SL (2018) Switching from fetal to adult hemoglobin. Nat Genet 50(4):478–480
Wang WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, Rana S, Thornburg CD, Rogers ZR, Kalpatthi RV (2011) Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 377(9778):1663–1672
Ware RE (2010) How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 115(26):5300–5311. https://doi.org/10.1182/blood-2009-Blood
Watson J, Stahman AW, Bilello FP (1948) The significance of the paucity of sickle cells in newborn Negro infants. Obstet Gynecol Surv 3(6):819–820
Wen J, Tao W, Hao S, Zu Y (2017) Cellular function reinstitution of offspring red blood cells cloned from the sickle cell disease patient blood post CRISPR genome editing. J Hematol Oncol 10(1):119
Wiedenheft B, Sternberg SH, Doudna JA (2012) RNA-guided genetic silencing systems in bacteria and archaea. Nature 482(7385):331–338
Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG (2014) Non-viral vectors for gene-based therapy. Nat Rev Genet 15(8):541–555
Zetsche B, Volz SE, Zhang F (2015) A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat Biotechnol 33(2):139–142
Zimmerman SA, Schultz WH, Burgett S, Mortier NA, Ware RE (2007) Hydroxyurea therapy lowers transcranial Doppler flow velocities in children with sickle cell anemia. Blood 110(3):1043–1047
Conflicts of Interest
The authors have no commercial, proprietary, or financial interest in the products described in this article.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Demirci, S., Leonard, A., Haro-Mora, J.J., Uchida, N., Tisdale, J.F. (2019). CRISPR/Cas9 for Sickle Cell Disease: Applications, Future Possibilities, and Challenges. In: Turksen, K. (eds) Cell Biology and Translational Medicine, Volume 5. Advances in Experimental Medicine and Biology(), vol 1144. Springer, Cham. https://doi.org/10.1007/5584_2018_331
Download citation
DOI: https://doi.org/10.1007/5584_2018_331
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-17588-7
Online ISBN: 978-3-030-17589-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)