
Abstract
Endometrial cancer is the most common invasive gynaecological cancer in women, and relatively little is known about inherited risk factors for this disease. This is the first genome-wide study to explore the role of common and rare germline copy number variants (CNVs) in predisposition to endometrial cancer. CNVs were called from germline DNA of 1,209 endometrioid endometrial cancer cases and 528 cancer-unaffected female controls. Overall CNV load of deletions or DNA gains did not differ significantly between cases and controls (P > 0.05), but cases presented with an excess of rare germline deletions overlapping likely functional genomic regions including genes (P = 8 × 10−10), CpG islands (P = 1 × 10−7) and sno/miRNAs regions (P = 3 × 10−9). On average, at least one additional gene and two additional CpG islands were disrupted by rare deletions in cases compared to controls. The most pronounced difference was that over 30 sno/miRNAs were disrupted by rare deletions in cases for every single disruption event in controls. A total of 13 DNA repair genes were disrupted by rare deletions in 19/1,209 cases (1.6 %) compared to one gene in 1/528 controls (0.2 %; P = 0.007), and this increased DNA repair gene loss in cases persisted after excluding five individuals carrying CNVs disrupting mismatch repair genes MLH1, MSH2 and MSH6 (P = 0.03). There were 34 miRNA regions deleted in at least one case but not in controls, the most frequent of which encompassed hsa-mir-661 and hsa-mir-203. Our study implicates rare germline deletions of functional and regulatory regions as possible mechanisms conferring endometrial cancer risk, and has identified specific regulatory elements as candidates for further investigation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Endometrial cancer is the most common invasive gynaecological cancer in women with more than a quarter of a million new cases each year (Ferlay et al. 2010). Moreover, the incidence of endometrial cancer has increased significantly in western countries over the past 20 years (Evans et al. 2011). The majority of endometrial cancers (80–90 %) are histologically classified as endometrioid subtype at the time of presentation (Clement and Young 2002) and are primarily treated by surgical resection. Patients are vulnerable to treatment-related morbidity due to the high prevalence of comorbidities, such as obesity (Dowdy et al. 2012).
A small proportion of endometrial cancer cases (5–25 %, depending on selection criteria) are due to high-risk germline mutations in one of the mismatch repair genes MLH1, MSH2, MSH6, and PMS2 (Buchanan et al. 2014), often presenting with early age of cancer onset, and strong family history of cancer as part of Lynch Syndrome. Evidence also exists for the contribution of other DNA repair-related genes to endometrial cancer risk, including POLD1 (Palles et al. 2013). Candidate gene and genome-wide searches for single-nucleotide polymorphisms (SNPs) associated with modest risk of endometrial cancer have to date convincingly identified one locus at 17q12 (HNF1B) (Spurdle et al. 2011), with evidence for risk loci at 15q21 (CYP19A1) (Setiawan et al. 2009) and 1q42 (near CAPN9) (Long et al. 2012). Thus, the genetic changes underlying the disease remain unknown for most endometrial cancer affected women.
DNA copy number variation (CNV) in the human genome is increasingly recognised as a major source of genetic variation that may influence cancer risk (Krepischi et al. 2012b; Kuiper et al. 2010). Indeed, cancer-predisposing CNVs are known to occur in important cancer-associated genes and/or pathways in numerous cancers, including the mismatch repair genes, MLH1, MSH2, MSH6 and PMS2 in Lynch Syndrome affected families (Thompson et al. 2014). There is also evidence that CNV frequency and/or size across the genome may play an important role in disease development. Increased germline CNV load has been shown to be associated with earlier age of cancer onset in TP53 mutation carriers (Shlien et al. 2008), and also with predisposition to colorectal cancer in high-risk families with or without mutations in mismatch repair genes (Talseth-Palmer et al. 2013; Yang et al. 2014), breast cancer in families that do not carry BRCA1 or BRCA2 mutations (Pylkas et al. 2012) and germline deletions were associated with ovarian cancer in women carrying a mutation in BRCA1 (Yoshihara et al. 2011). However, these associations have only been found in patients from high-risk families. Despite a number of reported population-based genome-wide association studies of individual CNV genotypes across various cancers (Craddock et al. 2010; Krepischi et al. 2012a; Long et al. 2013), we are unaware of any study assessing role of CNV load in cancer predisposition at the population level. Further, many CNV studies published to date have utilised convenience “control” groups with no or little demographic and clinical annotation, limiting interpretation of the results.
It has been shown that the extent of copy number change can correlate with the level of gene expression of the variant (Stranger et al. 2007). Similarly, copy number changes may disrupt expression of small non-coding RNA regions, namely microRNAs (miRNAs) which have a role in key cellular processes including development, cellular proliferation and apoptosis, and small nucleolar RNAs (snoRNAs) and Cajal body-specific RNAs (scaRNA), which are involved in post-transcriptional processing of other non-coding RNAs (Marcinkowska et al. 2011). Finally, alterations to CpG islands by CNVs have been shown to directly influence gene expression levels by changing methylation patterns (Robinson et al. 2010) and modification of transcription factor binding sites (Elango and Yi 2011; Zhang et al. 2009). Thus, consideration of the functional consequences of CNVs is an important aspect to assess in disease risk analyses.
In this study, we utilised SNP array data from a previously reported genome-wide association study of endometrioid endometrial cancer cases (Spurdle et al. 2011) and compared results to those from a well-annotated comparable control group, to assess whether CNVs across the genome show an altered frequency in endometrial cancer cases compared with healthy controls. Our analysis considered the likely functional importance of CNVs by assessing their location relative to genes and gene regulatory elements, and also CNV frequency in the cohort.
Materials and methods
Study cohorts
Detailed descriptions of the case and control sample sets utilised in this study have been previously reported (McEvoy et al. 2010; Spurdle et al. 2011). Briefly, 1,343 endometrioid endometrial cancer cases with self-reported European ancestry were selected for genome-wide genotyping from the Australian National Endometrial Cancer Study (ANECS) or the Studies of Epidemiology and Risk factors in Cancer Heredity study (SEARCH) in the UK (Spurdle et al. 2011). Control samples (n = 528) were female participants in the Hunter Community Study (HCS), a population-based cohort aged 55–86 years, predominantly of European ancestry and residing in the Hunter Region in New South Wales, Australia (McEvoy et al. 2010). Control individuals with report of any cancer were excluded from the analysis.
Genotyping, identification of CNVs and quality control
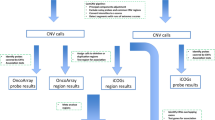
All DNA samples were genotyped with the Human610-Quad BeadChip (Illumina, Inc, San Diego, CA, USA) with ~610,000 markers, as described previously (Spurdle et al. 2011; Talseth-Palmer et al. 2013). Data for each array were normalised using GenomeStudio 2011.1 software (Illumina). Probe information including, genomic location, signal intensity (Norm R), allele frequency (Norm θ), log R Ratios (LRRs), and B Allele frequencies (BAF) for each sample was calculated and exported from GenomeStudio. All samples had a call rate >95 %. Results from parallel quantitative PCR validation studies (see below: Quantitative PCR CNV validation section, Online Resource 1) demonstrated that PennCNV (Wang et al. 2007) had the highest accuracy in identifying Multiplex ligation-dependent probe amplification (MLPA)- and/or quantitative PCR (qPCR)-detected gene deletions of MMR and other selected genes identified in this cohort, with 13/13 copy number variants called by PennCNV confirmed using these technologies, but lower rates of validation for CNVs called using other algorithms [QuantiSNP (Colella et al. 2007), CNV Partition (http://www.illumina.com/software/illumina_connect.ilmn), Gnosis (http://www.cnvision.org)] (Online Resource 1A). Based on this observation, CNV calls were then generated using the PennCNV program (version 2009 Aug 27), using the default program parameters, library files and genomic wave adjustment.
Case and control individuals were subject to quality control measures as previously defined, including measures of heterozygosity, relatedness, and non-European ancestry (Spurdle et al. 2011; Talseth-Palmer et al. 2013). Additional quality control procedures were performed for this copy number analysis to remove poor-quality array data (Online Resource 2) using the following exclusion criteria: log R ratio standard deviation >0.28, B allele frequency drift >0.01, waviness factor deviating from 0 by >0.04 and/or with the number of CNV calls exceeding 70. To reduce false positives, CNV calls were excluded if they contained <5 probes and/or were ≥1,000 kb in size. A total of 1,209 cases and 528 female controls passed quality control and were included in the analysis.
Quantitative PCR- and multiplex ligation-dependent probe amplification-CNV validations
Experimental validation of predicted CNVs using MLPA and/or qPCR was carried out, prior to (Buchanan et al. 2014) or during this study (Online Resource 1), for a subset of 13 CNVs predicted, using the four CNV calling algorithms, to overlap the known Lynch Syndrome genes MLH1, MSH2, MSH6 and PMS2, or 7 other selected genes (Online Resource 1A). During this study, we predicted a total of nine deletions [including four in MMR genes previously identified using MLPA testing (Buchanan et al. 2014)], and four duplications (including one duplication in MSH6). MLPA for MMR genes was carried out with SALSA kits P003, P003-B2, P008, P072-B2, and P248-A2 (MRC-Holland, Amsterdam, the Netherlands), and qPCR validation studies for all genes tested were performed using the Roche Light Cycler 480 (LC480) (Hoffmann-La Roche Ltd, Basel, Switzerland) and Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen, California, United States of America). Primers (Online Resource 3) were designed to target coding regions overlapping CNVs using previously documented parameters (D’Haene et al. 2010). Genomic regions overlapping ZNF80 (D’Haene et al. 2010) and ALB (Meijerink et al. 2001) were targeted as internal references for normalisation. Each sample was run with four independent replicates and at least three non-CNV carrying controls assayed alongside. Normalised copy number values calculated using the Lightcycler 480 Gene Scanning software. qPCR analysis validated seven deletions and three duplications, including all four MMR gene CNVs previously identified using MLPA. Only CNVs predicted by PennCNV were consistently validated by qPCR (Online Resource 1 A, B, C).
Identification of genes, CpG islands and small nuclear RNAs overlapping CNVs and defining rare CNVs
To avoid examining multiple isoforms from genes, we annotated 39,544 UCSC RefSeq (NCBI36/Hg18) transcripts using the SOURCE database (Diehn et al. 2003) and defined the genomic intervals for a total of 18,791 unique genes. Thus, each gene interval encompassed the start and end of all associated RefSeq transcripts (Online Resource 4). CNVs and gene regions that were estimated to overlap by at least one base pair were identified in a genome-wide scan using Intersect and Join tools from the Galaxy web server (Blankenberg et al. 2010; Giardine et al. 2005; Goecks et al. 2010).
Since CNV calls do not typically conform to discrete genomic regions in different individuals, we used the genome coordinates of 18,791 RefSeq gene (NCBI36/Hg18) boundaries to define a CNV region (Online Resource 4). Each of these regions, therefore, represented a cluster of one or more CNVs overlapping a well-characterised gene in the human genome and was used to measure the frequency of CNVs in our study cohort. Rare gene-overlapping CNVs were defined as those with frequency <1 % of the total cohort. To identify rare CNVs overlapping CpG islands and small RNA genomic regions, a similar approach was carried out using coordinates of CpG islands and sno/miRNA [UCSC Genome Browser (NCBI36/hg18); http://genome.ucsc.edu/cgi-bin/hgGateway] instead of gene regions. The CpG island track used by UCSC Genome Browser defined 20,338 unique coordinates across the genome using the following criteria: GC content of ≥50 %, length >200 bp, ratio of observed to expected CpG dinucleotides >0.6. The data track for sno/miRNA from UCSC Genome Browser (NCBI36/hg18) defined 1,120 regions across the genome.
Statistical analysis of CNV load
The statistical package R Project version R 2.14.2 (http://www.r-project.org/) was used to perform statistical analyses. T tests with the Satterthwaite adjustment for unequal variances were conducted to establish the level of significance associated with the difference in CNV carrier frequencies between the cases and controls. Mid-P exact test was used as a measure of association between CNV load and disease status. P values were considered significant if P value <0.05.
Results
CNV frequency in endometrial cancer cases and controls
After stringent quality control measures were applied, genotype data from Illumina 610 K SNP arrays suitable for genome-wide CNV scans were available for 1,209 cases and 528 female controls (Online Resource 2). Using the PennCNV algorithm, a total of 30,663 and 13,399 CNV calls were generated for cases and controls, respectively (Table 1), ranging in size from 0.5 kb to 998 kb. The average number of CNVs observed per individual did not differ significantly between cases and controls (25.36 vs 25.38, P = 0.97). Likewise, no difference was observed when considering deletions and gains separately (Table 1).
The average number of CNVs predicted to overlap genes was marginally greater in cases versus controls (8.56 vs 8.17, P = 0.04), with this effect attributable to deletions (case average = 5.07 vs control average = 4.70, P = 0.01) rather than DNA gains (case average = 3.48 vs control average = 3.48, P = 0.98) (Table 1). There was no statistically significant difference between cases and controls for deletions or gains of CpG islands. Increased frequency in cases versus controls for CNVs overlapping sno/miRNA regions (0.10 vs 0.04, P = 6 × 10−5) was attributable to a 2.53-fold increase of deletions overlapping sno/miRNA regions. The average number of sno/miRNAs disrupted by deletions was 4.3-fold higher in cases compared to controls (P = 0.001) (Table 1). Additionally, 34 miRNA regions predicted to be deleted in at least one case sample were not found in controls, with the most frequently affected regions encompassing hsa-mir-661 (n = 14 cases) and hsa-mir-203 (n = 11 cases) (Online Resource 5). For copy number variants overlapping sno/miRNAs but not genes, there was no significant difference between cases and controls when assessing the number of deletions (P = 0.09) or duplications (P = 0.61), however, there was limited power due to small CNV numbers (Online Resource 6). The average size of CNVs did not differ between cases versus controls (Online Resource 7).
Rare CNVs in endometrial cancer cases and controls
To examine the prevalence of rare CNVs overlapping genes in our study cohort, CNVs overlapping RefSeq gene regions in 18 or more individuals (≥1 % frequency in the study cohort) were excluded, all remaining rare CNVs either fully or partially overlapped at least one RefSeq gene region. The number of rare deletions per individual that overlapped at least one gene was 1.73-fold greater in cases compared to controls (1.63 vs 0.94; P = 8 × 10−10), but no statistically significant difference was seen for copy number gains between the two groups (P = 0.69) (Table 2). Conversely, the average number of RefSeq genes predicted to be disrupted by a rare genomic deletion was 2.26-fold higher in cases compared to controls (2.76 vs 1.22; P = 4 × 10−10). Thus, on average, rare deletions in endometrial cancer cases are disrupting one additional gene compared to controls.
A similar approach was used to identify rare CNVs (<1 % frequency) overlapping CpG islands. The average number of rare deletions predicted to fully or partially overlap at least one CpG island was 1.96-fold greater in cases than controls (0.78 vs 0.40; P = 1 × 10−7), and the number of gains overlapping CpG islands was 1.16-fold greater in cases than controls (0.72 vs 0.62; P = 0.05) (Table 2). Likewise, the average number of CpG islands predicted to be disrupted by a rare genomic deletion was 3.38-fold higher in cases compared to controls (3.01 vs 0.89; P = 2 × 10−7), and the average number of CpG islands impacted by rare genomic copy number gains was 1.41-fold higher in cases compared to controls (2.08 vs 1.47; P = 3 × 10−3). Results of this analysis suggest that rare CNVs in endometrial cancer cases are disrupting two additional CpG islands compared to controls. Interestingly, 64 % of these CpG islands are located within genes also disrupted by rare CNVs (data not shown). To extricate the contribution of CpG islands (falling within the coordinates of a Refseq gene) from the observed results, the loading effect of CNVs overlapping only apparently intergenic CpG islands was assessed. There were more rare CNVs disrupting intergenic CpG islands in cases compared to controls (deletions P = 0.001; gains P = 0.31) (Online Resource 8). Further, intergenic CpG islands were disrupted significantly more often in cases than controls by both rare deletions (0.88 vs 0.55; P = 0.002) and rare DNA gains (1.09 vs 0.77; P = 0.008).
Rare deletions (<1 % frequency) overlapping sno/miRNAs occurred at a frequency 7.69-fold higher in cases than controls (0.07 vs 0.01; P = 3 × 10−9). This equated to a 13.90-fold increase in the number of sno/miRNAs disrupted by rare CNVs (0.16 vs 0.01; P = 6 × 10−4), and there were 191 sno/miRNAs disrupted in cases while only six sno/miRNAs were affected in controls (Table 2). That is, over 30 sno/miRNAs were disrupted by rare deletions in cases for every single disruption event in controls.
DNA repair genes and known cancer susceptibility genes disrupted by CNVs
Given the role of many known high-risk cancer susceptibility genes in DNA repair, we extended our analysis to identify rare CNVs overlapping genes acting in the DNA repair pathway. One hundred and seventy-six DNA repair genes used in this analysis were sourced through an updated version of an online inventory (http://sciencepark.mdanderson.org/labs/wood/dna_repair_genes.html; updated 4th March 2013), and included the MMR genes MLH1, MSH2, MSH6, and PMS2. There were more cases than controls carrying rare deletions overlapping DNA repair genes (19 vs 1, P = 0.007, OR = 8.40, 95 % CI 1.54–177.0) (Table 3), and this finding remained nominally significant even after removing four samples with deletions overlapping MLH1, MSH2, and MSH6 (15 cases versus 1 control, P = 0.03, OR = 6.64, 95 % CI 1.18–141.60) (Online Resource 9). A total of 24 DNA repair genes (including the MMR genes MLH1, MSH2, and MSH6) overlapped rare CNVs across 39 cases (out of 1,209; 3.2 %) compared to eight genes across ten controls (out of 528; 1.9 %). There was no evidence for an increase in rare CNV gains overlapping DNA repair genes in cases versus controls. There was evidence for an increase in CNV deletions between cases with (n = 15) or without (n = 1,190) CNV disruption of DNA repair genes (Online Resource 10), and this difference was observed overall (P = 0.01), for CNVs overlapping all genomic features assessed (P = 0.01 for genes, P = 0.005 for CpG islands, P = 0.05 for sno/miRNAs). Mean CNV size of CNVs overlapping genes and sno/miRNAs was greater in cases without DNA repair gene disruption (P = 0.04), however, no size difference was observed for all CNVs and CNVs overlapping CpG islands (Online Resource 11).
Discussion
Relatively little is known about inherited factors that influence endometrial cancer risk, and to our knowledge this is the first genome-wide study to explore the role of germline CNV load in endometrial cancer. We used the PennCNV algorithm for assessing CNVs detected on the Illumina platform, and compared frequencies of CNVs stratified by functional annotation and frequency in a well-characterised set of endometrial cancer cases and controls. More deletions than gains were observed in both cases and controls in our study, consistent with the fact that probe intensity differences for most DNA gains are typically smaller than those for deletions; hemizygous deletions will reduce the probe intensity by half (2:1 ratio) compared to a 1.5-fold increase in probe intensity (3:2 ratio) for duplications.
For large-scale studies where experimental validation of every predicted CNV is impractical, such as this load analysis, accuracy of CNV calling algorithms is of great importance. PennCNV was designed for use with Illumina array data (Wang et al. 2007), and has been reported in several studies to have a high true positive call rate for CNVs called using a five probe minimum (Dellinger et al. 2010; Marenne et al. 2011). Specificity of 98 % was observed for PennCNV in a controlled study validating CNVs by MLPA analysis in paired samples (Marenne et al. 2011). In support of this, we have found PennCNV to demonstrate high concordance with MLPA-identified and/or qPCR-verified gene deletions in our study. Another important consideration when characterising CNVs, in particular rare CNVs, is the control group selected for comparison. A number of published reports define rare CNVs as those that do not occur in the Database of Genomic Variants (DGV). However, DGV is a compilation of validated and unvalidated CNVs found in studies of individuals from various populations of different ethnicities, and using a variety of array and sequencing platforms (Iafrate et al. 2004). Importantly, there is no medical information available for most of the samples in DGV which precludes filtering for non-diseased individuals to serve as appropriate controls, and the database includes variants overlapping known cancer susceptibility genes (e.g. MSH2 and MSH6) (Iafrate et al. 2004). Therefore, DGV is not an appropriate control dataset to define rare CNVs of clinical importance.
Rare CNV load has been reported to be relevant for pancreatic (Al-Sukhni et al. 2012), breast (Krepischi et al. 2012a) and ovarian (Yoshihara et al. 2011) cancers, in studies that have not considered regulatory regions or defined rare CNVs using a large, well-annotated control dataset. In this study, we provide evidence for a role of germline structural alterations in endometrial cancer risk. We found that overall CNV load of deletions or DNA gains did not differ significantly between cases and controls, but cases presented with an excess of rare germline copy number deletions disrupting genes, CpG islands and sno/miRNAs.
The observation that effects were most obvious when considering rare deletions of functionally important gene regions is supported in the literature. In particular, Krepischi et al. (2012a) postulate that overtly deleterious germline CNVs are removed from the population and those remaining are a result of less stringent selection or alternatively they indicate inefficient DNA repair and apoptosis in relevant individuals. This is corroborated by our finding of an elevated frequency of germline loss of a DNA repair gene in cases versus controls and demonstrates that genes in DNA repair pathways other than the four mismatch repair genes are also worthy of further investigation in future endometrial cancer genetic studies. It was also supported by our observation that cases with CNV disruption of DNA repair genes were themselves more likely to demonstrate increased CNV deletion load. These results are also consistent with whole genome and exome sequencing studies of other cancers, many of which provide evidence for a role for DNA repair gene variants in cancer predisposition (Thompson et al. 2012).
The number of CpG islands disrupted by rare deletions was approximately threefold higher in cases versus controls. In some instances, the CpG gain/loss occurs in tandem with gain/loss of nearby gene(s). However, CpG island loss/gain does not always directly impact the exonic coding sequence, and there are several other ways by which CpG island disruption could contribute to gene dysregulation and tumourigenesis. First, disruption may directly alter transcription factor binding sites and thus gene regulation. For example, deletion of a CpG island in the promoter for AMCAR results in gene upregulation and promotes colon carcinogenesis, whereas simultaneous deletion of another two promoter-located CpG islands was associated with the opposite effect (Zhang et al. 2009). Furthermore, length of CpG islands has been found to correlate with gene expression levels (Elango and Yi 2011). Second, deletions or gains of CpG islands could contribute to endometrial cancer development via abnormal methylation, given that CNVs overlapping CpG islands are reported to directly influence methylation patterns (Robinson et al. 2010) and that abnormal methylation patterns are frequently observed in endometrial tumours (Banno et al. 2006; Ghabreau et al. 2004).
This study found a 2.5-fold increase in deletions overlapping sno/miRNA regions in cases versus controls. Deletions of sno/miRNAs are anticipated to disrupt regulation of multiple genes simultaneously and thus have widespread downstream effects. Both upregulation and downregulation of miRNAs have been implicated in gynaecological cancer development (Torres et al. 2011), but the germline loss of miRNAs is yet to be characterised in endometrial cancer. These findings are supported by literature detailing increased complexity in cellular processes and miRNA regulation due to normal cycling of endometrial tissue (Kuokkanen et al. 2010), and that miRNA dysregulation is well documented in endometrial cancer development (Torres et al. 2011).
The miRNA, hsa-miR-661, deleted in 14 cases and no controls, is a relatively well-characterised regulatory molecule. Transfection studies of the MCF-7 wildtype TP53 breast cancer cell line showed that miR-661 siRNA-mediated inhibition leads to decreased expression of MDM2 and MDM4, both negative regulators of TP53, in a tumour-suppressive manner. In addition, increased expression of hsa-miR-661 was reported to be associated with better outcome for breast cancer patients (Hoffman et al. 2014). hsa-miR-661 expression in MDA-MB-231 breast cancer cells was reported to decrease cell motility, invasiveness, and anchorage, and decrease tumour formation in nude mice (Reddy et al. 2009). In our case cohort, germline loss of hsa-miR-661 is a recurring event, supporting a potential role of miR-661 as a tumour suppressor in endometrial cancer predisposition.
miRNA hsa-miR-203 was deleted in 11 cases and no controls and is known to be intricately involved in regulating endometrial cell cycling. It is downregulated in the late proliferative endometrium and upregulated in the mid-term secretion phase (Kuokkanen et al. 2010). Literature to date describes a decrease of hsa-miR-203 expression in most malignancies, including hormone-related tumours (Viticchie et al. 2011; Wang et al. 2012). There are only two reports that examine the expression of hsa-miR-203 in normal and malignant endometrium. Huang et al. (2014) found that hsa-miR-203 was significantly hypermethylated in tumour samples, correlating with MLH1 methylation status, microsatellite instability, and decreased expression of pro-oncogene SOX4 in Ishikawa cells (Huang et al. 2014). Conversely, a smaller study by Chung et al. (2009) reported increased expression of hsa-miR-203 in endometrial adenocarcinomas compared to normal endometrium samples (Chung et al. 2009). The germline loss of the miR-203 in multiple cases but not controls in our study supports a role of hsa-miR-203 as a tumour suppressor in endometrial cancer predisposition, consistent with most previous studies on hormonal cancers.
Apart from CpG island analysis of rare CNVs, there was no evidence that DNA gains were involved in endometrial cancer predisposition. While this suggests the duplications in general are not as disruptive to gene function as deletions, it may also reflect that SNP arrays are not able to define the genomic location of duplicated material. For example, duplicated genomic material may lie in tandem, within itself, or be inserted at another part of the genome in either a benign or disruptive manner. Thus, the functional relevance of DNA gains would be better assessed using alternative technologies, possibly by determining if they are associated with altered expression of the gene(s) implicated, or directly assessing their effects on gene transcription. We also acknowledge that detection of CNVs in our study was limited by the resolution of the microarray used for genotyping. Future analyses performed at higher resolution with CNV specific arrays, or next-generation sequencing as this becomes more affordable, would improve the potential for detecting causal variants along the genome, both deletions and duplications.
Our results implicate rare germline deletions of functional and regulatory regions as possible mechanisms conferring risk in endometrial cancer. As such, this study provides a baseline for future validation studies that consider the functional relevance of predicted CNVs in assessing the role of CNV load in cancer predisposition. This study has also identified specific regulatory elements as candidates for further investigation in endometrial cancer predisposition.
References
Al-Sukhni W, Joe S, Lionel AC, Zwingerman N, Zogopoulos G, Marshall CR, Borgida A, Holter S, Gropper A, Moore S, Bondy M, Klein AP, Petersen GM, Rabe KG, Schwartz AG, Syngal S, Scherer SW, Gallinger S (2012) Identification of germline genomic copy number variation in familial pancreatic cancer. Hum Genet 131:1481–1494. doi:10.1007/s00439-012-1183-1
Banno K, Yanokura M, Susumu N, Kawaguchi M, Hirao N, Hirasawa A, Tsukazaki K, Aoki D (2006) Relationship of the aberrant DNA hypermethylation of cancer-related genes with carcinogenesis of endometrial cancer. Oncol Rep 16:1189–1196
Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J (2010) Current protocols in molecular biology. In: Ausubel FM et al (eds) Galaxy: a web-based genome analysis tool for experimentalists, Chap 19: Unit 19, vol 10., pp 1–21. doi:10.1002/0471142727.mb1910s89
Buchanan DD, Tan YY, Walsh MD, Clendenning M, Metcalf AM, Ferguson K, Arnold ST, Thompson BA, Lose FA, Parsons MT, Walters RJ, Pearson SA, Cummings M, Oehler MK, Blomfield PB, Quinn MA, Kirk JA, Stewart CJ, Obermair A, Young JP, Webb PM, Spurdle AB (2014) Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J Clin Oncol 32:90–100. doi:10.1200/jco.2013.51.2129
Chung TK, Cheung TH, Huen NY, Wong KW, Lo KW, Yim SF, Siu NS, Wong YM, Tsang PT, Pang MW, Yu MY, To KF, Mok SC, Wang VW, Li C, Cheung AY, Doran G, Birrer MJ, Smith DI, Wong YF (2009) Dysregulated microRNAs and their predicted targets associated with endometrioid endometrial adenocarcinoma in Hong Kong women. Int J Cancer 124:1358–1365. doi:10.1002/ijc.24071
Clement PB, Young RH (2002) Endometrioid carcinoma of the uterine corpus: a review of its pathology with emphasis on recent advances and problematic aspects. Adv Anat Pathol 9:145–184
Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, Ragoussis J (2007) QuantiSNP: an objective Bayes Hidden-Markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res 35:2013–2025. doi:10.1093/nar/gkm076
Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, Vukcevic D, Barnes C, Conrad DF, Giannoulatou E, Holmes C, Marchini JL, Stirrups K, Tobin MD, Wain LV, Yau C, Aerts J, Ahmad T, Andrews TD, Arbury H, Attwood A, Auton A, Ball SG, Balmforth AJ, Barrett JC, Barroso I, Barton A, Bennett AJ, Bhaskar S, Blaszczyk K, Bowes J, Brand OJ, Braund PS, Bredin F, Breen G, Brown MJ, Bruce IN, Bull J, Burren OS, Burton J, Byrnes J, Caesar S, Clee CM, Coffey AJ, Connell JM, Cooper JD, Dominiczak AF, Downes K, Drummond HE, Dudakia D, Dunham A, Ebbs B, Eccles D, Edkins S, Edwards C, Elliot A, Emery P, Evans DM, Evans G, Eyre S, Farmer A, Ferrier IN, Feuk L, Fitzgerald T, Flynn E, Forbes A, Forty L, Franklyn JA, Freathy RM, Gibbs P, Gilbert P, Gokumen O, Gordon-Smith K, Gray E, Green E, Groves CJ, Grozeva D, Gwilliam R, Hall A, Hammond N, Hardy M, Harrison P, Hassanali N, Hebaishi H, Hines S, Hinks A, Hitman GA, Hocking L, Howard E, Howard P, Howson JM, Hughes D, Hunt S, Isaacs JD, Jain M, Jewell DP, Johnson T, Jolley JD, Jones IR, Jones LA et al (2010) Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464:713–720. doi:10.1038/nature08979
Dellinger AE, Saw SM, Goh LK, Seielstad M, Young TL, Li YJ (2010) Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res 38:e105. doi:10.1093/nar/gkq040
D’Haene B, Vandesompele J, Hellemans J (2010) Accurate and objective copy number profiling using real-time quantitative PCR. Methods 50:262–270. doi:10.1016/j.ymeth.2009.12.007
Diehn M, Sherlock G, Binkley G, Jin H, Matese JC, Hernandez-Boussard T, Rees CA, Cherry JM, Botstein D, Brown PO, Alizadeh AA (2003) SOURCE: a unified genomic resource of functional annotations, ontologies, and gene expression data. Nucleic Acids Res 31:219–223
Dowdy SC, Borah BJ, Bakkum-Gamez JN, Kumar S, Weaver AL, McGree ME, Haas LR, Cliby WA, Podratz KC (2012) Factors predictive of postoperative morbidity and cost in patients with endometrial cancer. Obstet Gynecol 120:1419–1427. doi:10.1097/AOG.0b013e3182737538
Elango N, Yi SV (2011) Functional relevance of CpG island length for regulation of gene expression. Genetics 187:1077–1083. doi:10.1534/genetics.110.126094
Evans T, Sany O, Pearmain P, Ganesan R, Blann A, Sundar S (2011) Differential trends in the rising incidence of endometrial cancer by type: data from a UK population-based registry from 1994 to 2006. Br J Cancer 104:1505–1510. doi:10.1038/bjc.2011.68
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer (Journal international du cancer) 127:2893–2917. doi:10.1002/ijc.25516
Ghabreau L, Roux JP, Niveleau A, Fontaniere B, Mahe C, Mokni M, Frappart L (2004) Correlation between the DNA global methylation status and progesterone receptor expression in normal endometrium, endometrioid adenocarcinoma and precursors. Virchows Arch 445:129–134. doi:10.1007/s00428-004-1059-4
Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A (2005) Galaxy: a platform for interactive large-scale genome analysis. Genome Res 15:1451–1455. doi:10.1101/gr.4086505
Goecks J, Nekrutenko A, Taylor J (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11:R86. doi:10.1186/gb-2010-11-8-r86
Hoffman Y, Bublik DR, Pilpel Y, Oren M (2014) miR-661 downregulates both Mdm2 and Mdm4 to activate p53. Cell Death Differ 21:302–309. doi:10.1038/cdd.2013.146
Huang YW, Kuo CT, Chen JH, Goodfellow PJ, Huang TH, Rader JS, Uyar DS (2014) Hypermethylation of miR-203 in endometrial carcinomas. Gynecol Oncol. doi:10.1016/j.ygyno.2014.02.009
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C (2004) Detection of large-scale variation in the human genome. Nat Genet 36:949–951. doi:10.1038/ng1416
Krepischi AC, Achatz MI, Santos EM, Costa SS, Lisboa BC, Brentani H, Santos TM, Goncalves A, Nobrega AF, Pearson PL, Vianna-Morgante AM, Carraro DM, Brentani RR, Rosenberg C (2012a) Germline DNA copy number variation in familial and early-onset breast cancer. Breast Cancer Res 14:R24. doi:10.1186/bcr3109
Krepischi AC, Pearson PL, Rosenberg C (2012b) Germline copy number variations and cancer predisposition. Future Oncol 8:441–450. doi:10.2217/fon.12.34
Kuiper RP, Ligtenberg MJ, Hoogerbrugge N, Geurts van Kessel A (2010) Germline copy number variation and cancer risk. Curr Opin Genet Dev 20:282–289. doi:10.1016/j.gde.2010.03.005
Kuokkanen S, Chen B, Ojalvo L, Benard L, Santoro N, Pollard JW (2010) Genomic profiling of microRNAs and messenger RNAs reveals hormonal regulation in microRNA expression in human endometrium. Biol Reprod 82:791–801. doi:10.1095/biolreprod.109.081059
Long J, Zheng W, Xiang YB, Lose F, Thompson D, Tomlinson I, Yu H, Wentzensen N, Lambrechts D, Dork T, Dubrowinskaja N, Goodman MT, Salvesen HB, Fasching PA, Scott RJ, Delahanty R, Zheng Y, O’Mara T, Healey CS, Hodgson S, Risch H, Yang HP, Amant F, Turmanov N, Schwake A, Lurie G, Trovik J, Beckmann MW, Ashton K, Ji BT, Bao PP, Howarth K, Lu L, Lissowska J, Coenegrachts L, Kaidarova D, Durst M, Thompson PJ, Krakstad C, Ekici AB, Otton G, Shi J, Zhang B, Gorman M, Brinton L, Coosemans A, Matsuno RK, Halle MK, Hein A, Proietto A, Cai H, Lu W, Dunning A, Easton D, Gao YT, Cai Q, Spurdle AB, Shu XO (2012) Genome-wide association study identifies a possible susceptibility locus for endometrial cancer. Cancer Epidemiol Biomark Prev 21:980–987. doi:10.1158/1055-9965.EPI-11-1160
Long J, Delahanty RJ, Li G, Gao YT, Lu W, Cai Q, Xiang YB, Li C, Ji BT, Zheng Y, Ali S, Shu XO, Zheng W (2013) A common deletion in the APOBEC3 genes and breast cancer risk. J Natl Cancer Inst 105:573–579. doi:10.1093/jnci/djt018
Marcinkowska M, Szymanski M, Krzyzosiak WJ, Kozlowski P (2011) Copy number variation of microRNA genes in the human genome. BMC Genomics 12:183. doi:10.1186/1471-2164-12-183
Marenne G, Rodriguez-Santiago B, Closas MG, Perez-Jurado L, Rothman N, Rico D, Pita G, Pisano DG, Kogevinas M, Silverman DT, Valencia A, Real FX, Chanock SJ, Genin E, Malats N (2011) Assessment of copy number variation using the Illumina Infinium 1 M SNP-array: a comparison of methodological approaches in the Spanish Bladder Cancer/EPICURO study. Hum Mutat 32:240–248. doi:10.1002/humu.21398
McEvoy M, Smith W, D’Este C, Duke J, Peel R, Schofield P, Scott R, Byles J, Henry D, Ewald B, Hancock S, Smith D, Attia J (2010) Cohort profile: the Hunter Community Study. Int J Epidemiol 39:1452–1463. doi:10.1093/ije/dyp343
Meijerink J, Mandigers C, van de Locht L, Tonnissen E, Goodsaid F, Raemaekers J (2001) A novel method to compensate for different amplification efficiencies between patient DNA samples in quantitative real-time PCR. J Mol Diagn 3:55–61. doi:10.1016/s1525-1578(10)60652-6
Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, Sherborne A, Chubb D, Carvajal-Carmona LG, Ma Y, Kaur K, Dobbins S, Barclay E, Gorman M, Martin L, Kovac MB, Humphray S, Lucassen A, Holmes CC, Bentley D, Donnelly P, Taylor J, Petridis C, Roylance R, Sawyer EJ, Kerr DJ, Clark S, Grimes J, Kearsey SE, Thomas HJ, McVean G, Houlston RS, Tomlinson I (2013) Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45:136–144. doi:10.1038/ng.2503
Pylkas K, Vuorela M, Otsukka M, Kallioniemi A, Jukkola-Vuorinen A, Winqvist R (2012) Rare copy number variants observed in hereditary breast cancer cases disrupt genes in estrogen signaling and TP53 tumor suppression network. PLoS Genet 8:e1002734. doi:10.1371/journal.pgen.1002734
Reddy SD, Pakala SB, Ohshiro K, Rayala SK, Kumar R (2009) MicroRNA-661, a c/EBPalpha target, inhibits metastatic tumor antigen 1 and regulates its functions. Cancer Res 69:5639–5642. doi:10.1158/0008-5472.can-09-0898
Robinson MD, Stirzaker C, Statham AL, Coolen MW, Song JZ, Nair SS, Strbenac D, Speed TP, Clark SJ (2010) Evaluation of affinity-based genome-wide DNA methylation data: effects of CpG density, amplification bias, and copy number variation. Genome Res 20:1719–1729. doi:10.1101/gr.110601.110
Setiawan VW, Doherty JA, Shu XO, Akbari MR, Chen C, De Vivo I, Demichele A, Garcia-Closas M, Goodman MT, Haiman CA, Hankinson SE, Henderson BE, Horn-Ross PL, Lacey JV Jr, Le Marchand L, Levine DA, Liang X, Lissowska J, Lurie G, McGrath M, Narod SA, Rebbeck TR, Ursin G, Weiss NS, Xiang YB, Yang HP, Zheng W, Olson SH (2009) Two estrogen-related variants in CYP19A1 and endometrial cancer risk: a pooled analysis in the Epidemiology of Endometrial Cancer Consortium. Cancer Epidemiol Biomarkers Prev 18:242–247. doi:10.1158/1055-9965.EPI-08-0689
Shlien A, Tabori U, Marshall CR, Pienkowska M, Feuk L, Novokmet A, Nanda S, Druker H, Scherer SW, Malkin D (2008) Excessive genomic DNA copy number variation in the Li-Fraumeni cancer predisposition syndrome. Proc Natl Acad Sci USA 105:11264–11269. doi:10.1073/pnas.0802970105
Spurdle AB, Thompson DJ, Ahmed S, Ferguson K, Healey CS, O’Mara T, Walker LC, Montgomery SB, Dermitzakis ET, Fahey P, Montgomery GW, Webb PM, Fasching PA, Beckmann MW, Ekici AB, Hein A, Lambrechts D, Coenegrachts L, Vergote I, Amant F, Salvesen HB, Trovik J, Njolstad TS, Helland H, Scott RJ, Ashton K, Proietto T, Otton G, Tomlinson I, Gorman M, Howarth K, Hodgson S, Garcia-Closas M, Wentzensen N, Yang H, Chanock S, Hall P, Czene K, Liu J, Li J, Shu XO, Zheng W, Long J, Xiang YB, Shah M, Morrison J, Michailidou K, Pharoah PD, Dunning AM, Easton DF (2011) Genome-wide association study identifies a common variant associated with risk of endometrial cancer. Nat Genet 43:451–454. doi:10.1038/ng.812
Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, Tyler-Smith C, Carter N, Scherer SW, Tavare S, Deloukas P, Hurles ME, Dermitzakis ET (2007) Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315:848–853. doi:10.1126/science.1136678
Talseth-Palmer BA, Holliday EG, Evans TJ, McEvoy M, Attia J, Grice DM, Masson AL, Meldrum C, Spigelman A, Scott RJ (2013) Continuing difficulties in interpreting CNV data: lessons from a genome-wide CNV association study of Australian HNPCC/lynch syndrome patients. BMC Med Genomics 6:10. doi:10.1186/1755-8794-6-10
Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DY, Tothill RW, Thorne H, Barnes DR, Li J, Ellul J, Philip GK, Antill YC, James PA, Trainer AH, Mitchell G, Campbell IG (2012) Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet 8:e1002894. doi:10.1371/journal.pgen.1002894
Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, Bapat B, Bernstein I, Capella G, den Dunnen JT, du Sart D, Fabre A, Farrell MP, Farrington SM, Frayling IM, Frebourg T, Goldgar DE, Heinen CD, Holinski-Feder E, Kohonen-Corish M, Robinson KL, Leung SY, Martins A, Moller P, Morak M, Nystrom M, Peltomaki P, Pineda M, Qi M, Ramesar R, Rasmussen LJ, Royer-Pokora B, Scott RJ, Sijmons R, Tavtigian SV, Tops CM, Weber T, Wijnen J, Woods MO, Macrae F, Genuardi M (2014) Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 46:107–115. doi:10.1038/ng.2854
Torres A, Torres K, Maciejewski R, Harvey WH (2011) MicroRNAs and their role in gynecological tumors. Med Res Rev 31:895–923. doi:10.1002/med.20205
Viticchie G, Lena AM, Latina A, Formosa A, Gregersen LH, Lund AH, Bernardini S, Mauriello A, Miano R, Spagnoli LG, Knight RA, Candi E, Melino G (2011) MiR-203 controls proliferation, migration and invasive potential of prostate cancer cell lines. Cell Cycle 10:1121–1131
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M (2007) PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17:1665–1674. doi:10.1101/gr.6861907
Wang C, Zheng X, Shen C, Shi Y (2012) MicroRNA-203 suppresses cell proliferation and migration by targeting BIRC5 and LASP1 in human triple-negative breast cancer cells. J Exp Clin Cancer Res 31:58. doi:10.1186/1756-9966-31-58
Yang R, Chen B, Pfutze K, Buch S, Steinke V, Holinski-Feder E, Stocker S, von Schonfels W, Becker T, Schackert HK, Royer-Pokora B, Kloor M, Schmiegel WH, Buttner R, Engel C, Lascorz Puertolas J, Forsti A, Kunkel N, Bugert P, Schreiber S, Krawczak M, Schafmayer C, Propping P, Hampe J, Hemminki K, Burwinkel B (2014) Genome-wide analysis associates familial colorectal cancer with increases in copy number variations and a rare structural variation at 12p12.3. Carcinogenesis 35:315–323. doi:10.1093/carcin/bgt344
Yoshihara K, Tajima A, Adachi S, Quan J, Sekine M, Kase H, Yahata T, Inoue I, Tanaka K (2011) Germline copy number variations in BRCA1-associated ovarian cancer patients. Genes Chromosomes Cancer 50:167–177. doi:10.1002/gcc.20841
Zhang X, Leav I, Revelo MP, Deka R, Medvedovic M, Jiang Z, Ho SM (2009) Deletion hotspots in AMACR promoter CpG island are cis-regulatory elements controlling the gene expression in the colon. PLoS Genet 5:e1000334. doi:10.1371/journal.pgen.1000334
Acknowledgments
LW is funded by the HRC Sir Charles Hercus Health Research Fellowship. ABS is funded by a National Health and Medical Research Council (NHMRC) Senior Research Fellowship. EGH was funded by an NHMRC Postdoctoral Fellowship. This work was supported by the NHMRC (NHMRC; ID 552402), the Wellcome Trust and by Cancer Research UK grants C1287/A10118, C490/A1021, C8197/A10865 and C8197/A10123.
Conflict of interest
There are no conflicts of interest to be disclosed by any author of this manuscript.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
A. B. Spurdle and L. C. Walker contributed equally (for last authorship position).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Moir-Meyer, G.L., Pearson, J.F., Lose, F. et al. Rare germline copy number deletions of likely functional importance are implicated in endometrial cancer predisposition. Hum Genet 134, 269–278 (2015). https://doi.org/10.1007/s00439-014-1507-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-014-1507-4