
Abstract
Purpose
Plasmacytoid dendritic cells (pDCs) are commonly associated with myeloid malignancies. The association between lymphoblastic leukemia and pDCs has been little explored.
Case presentation
Here, we report a novel case of early T-cell precursor lymphoblastic leukemia (ETP-ALL) accompanied by prominent proliferation of blastic pDCs mimicking BPDCN. The diagnosis was established based on a comprehensive analysis of morphology, immunophenotype and clinical implications. We also present a literature review and discussion on the differential expression of reactive and neoplastic pDCs, the functional role of pDCs in lymphoblastic leukemia, and the etiological association of normal pDCs and BPDCN.
Conclusions
The current case demonstrates for the first time that prominent pDC proliferation can be associated with lymphoid neoplasms and can exhibit blastic morphology and immunophenotype. The underlying mechanism of the coexistence of these two blastic populations remains unknown. Further genetic profiling may be required to denote the progressive development of tumor stem cells to the lymphoid, myeloid or dendritic cell lineage. Moreover, the prognostic value of pDCs in hematological neoplasms needs further investigation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Early T-cell precursor lymphoblastic leukemia (ETP-ALL) is a subtype of T-lymphoblastic leukemia (T-ALL) derived from a subset of cells that have immigrated to the thymus from the bone marrow (BM). The blasts of ETP-ALL share a similar morphology to other types of ALL, and the definition is mainly based on expression of CD7 but lack of CD8 and CD1a and are simultaneously positive for one or more of the myeloid/stem cell markers, including CD34, CD117, HLA-DR, CD13, CD33 and CD11b (Steven et al. 2017). This immunophenotype usually permits delineation of ETP-ALL from other T-ALL subtypes and from mixed phenotype acute leukemia (MPAL) T/myeloid. The prognosis of ETP-ALL remains controversial in adult patients (Bond et al. 2017; Coustan-Smith et al. 2009; Jain et al. 2016; Ma et al. 2012). Differential diagnosis and the use of response-based risk stratification and therapy intensification are vital for treatment and prognosis prediction (Bond et al. 2017; Jain et al. 2016).
Dendritic cells (DCs) mainly contain two subsets: plasmacytoid dendritic cells (pDCs) and conventional DCs (cDCs) or myeloid DCs (mCDs). Functional and nonmalignant pDCs are typically lineage (Lin)-HLA-DR+CD56-CD123+CD11c- cells that play critical roles in immune responses by secreting interferon (IFN) type I (Grouard et al. 1997). PDCs may have antitumor effects, for example, in neuroblastoma (Cordeau et al. 2016), or protumor effects, such as in breast cancer mouse models (Sawant et al. 2012), depending on the malignancy context. Normal pDCs account for a low percentage (less than 1%) of total nucleated cells in both BM and peripheral blood (Derolf et al. 2014; Lucas et al. 2019; Xiao et al. 2019).
The two types of hematological neoplasms related to pDCs are blastic pDC neoplasms (BPDCN) and mature pDC proliferations (MPDCP), both of which are commonly associated with myeloid malignancies, especially chronic myelomonocytic leukemia (CMML) (Facchetti et al. 2016; Vermi et al. 2004). Expansion of pDCs in acute myeloid leukemia (AML) patients (pDC-AML) was observed in approximately 5% of AML cases and is characterized by a high frequency of RUNX1 mutations (Xiao et al. 2021; Zalmai et al. 2021). Conversely, cases of BPDCN associated with lymphoid neoplasms are rare and never show common genetic events, suggesting co-occurrence instead of a common clonal origin (Harvell et al. 2000; Khan et al. 2019; Milley et al. 2016). Here, we report an exceptional case of ETP-ALL accompanied by prominent blastic pDC proliferation that mimics BPDCN. We also reviewed previously reported articles to learn as much as possible about pDCs, BPDCN and lymphoid neoplasms.
Case report
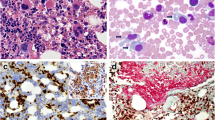
A 54-year-old female patient presented to the emergency department with a short history of inguinal lymph node enlargement, sleep disorders and diarrhea. A complete blood test showed a white blood cell (WBC) count of 2.0 × 109/L (normal, 3.5–9.5 × 109/L), neutrophil count of 0.36 × 109/L (normal, 1.8–6.3 × 109/L), hemoglobin concentration of 123 g/L (normal, 115–150 g/L), and platelet count of 49 × 109/L (normal, 100–300 × 109/L). Peripheral blood smear revealed 24% blast cells. The BM aspirate smear showed 52.5% blasts, with medium to large cell sizes, circular or irregular shapes, oval nuclei, fine chromatin, 1–2 prominent nucleoli, and moderate amphophilic and agranular cytoplasm. Notably, 10% of the blast cells displayed strip or hand-mirror shapes with obvious tailing (Fig. 1a). Multiparametric flow cytometry (FCM) revealed two abnormal cell populations. The first population (approximately 45%) was positive for CD5 (dim), CD7, cytoplasmic CD3 (cCD3), CD99, CD34, CD117, CD13 (partial), and CD33 and negative for CD1a, CD2, CD3, CD4 and CD8, consistent with ETP-ALL. The second population (approximately 15%) at the low-side scatter blast-gate with strong CD45 expression was positive for HLA-DR, CD4, CD56, and CD123 and negative for CD13, CD14, CD64, cytoplasmic MPO (cMPO), CD2, CD8 and cCD3 (Fig. 1e), consistent with the blastic pDC phenotype. BM biopsy also revealed two types of cell populations (Fig. 1b). One showed a nodular pattern with a diffuse, monomorphous infiltration of medium-sized blast cells that expressed CD303 (Fig. 1c), CD4, CD123, CD56 and HLA-DR, compatible with blastic pDCs. Such cells were negative for TCL1 and BCL2 staining. Another population with focal infiltration was positive for CD3 (Fig. 1d), CD99, CD34 (dispersed), CD117 (dispersed), CD5 and CD7, suggestive of ETP-ALL. A G-banded chromosome analysis showed a normal female karyotype. Sanger sequencing and next-generation sequencing (NGS) revealed the DNMT3a R882 mutation but not other BPDCN-associated mutations (Menezes et al. 2014; Neumann et al. 2013). Overall, the findings were consistent with the diagnosis of ETP-ALL with blastic pDC proliferation.

Identification of early T-cell precursor lymphoblastic leukemia (ETP-ALL) accompanied by prominent blastic plasmacytoid dendritic cell (pDC) proliferation. a Bone marrow (BM) aspirate smear showed marked blast cells, with medium to large cell size, circular or irregular shape, oval nucleus, fine chromatin, 1–2 prominent nucleoli, moderate amphophilic and agranular cytoplasm; some displayed strip or hand-mirror shape with obvious tailing. Magnification: ×1000. b–d BM biopsy showed a nodular pattern with a diffuse, monomorphous infiltration of medium-sized blast cells, which were positive for CD303, and focal distribution of cells positive for CD3. Magnification: ×400. Inserts display a magnification of 200. e Flow cytometry revealed two populations of blast cells in the BM, one of which (gated in red) was positive for CD5 (dim), CD7, cCD3, CD99, CD34, CD117, CD13 (partial), and CD33 and negative for CD1a, CD2, CD3, CD4 and CD8. The other population (gated in blue) was positive for CD45, HLA-DR, CD4, CD56, and CD123 and negative for CD13, CD14, CD64, cMPO, CD2, CD8 and cCD3. Lymphocytes were gated in green
Induction therapy containing cyclophosphamide, vincristine and prednisone was given, followed by consolidation chemotherapy. The patient achieved complete remission (CR). T-lymphoblasts or pDCs in the BM were not detected by subsequent FCM. The patient later underwent hematopoietic stem cell transplantation (HSCT). During follow-up, over 3 years, the patient remained in continuous CR.
Methods
Patient samples were withdrawn for routine diagnostic tests. Cytology stainings, immunophenotyping on a FACS Canto II flow cytometer, karyotyping and gene mutation analysis were performed in our department. NGS analysis on Illumina platform was performed in Kindstar (Kindstar Global, Chengdu, China). Histomorphology and immunohistologic stainings were performed in the Department of Pathology, West China Hospital of Sichuan University.
Discussion
We searched the literature and identified no previous articles reporting lymphoblastic leukemia accompanied by prominent pDC proliferation or BPDCN. There were only sporadic cases describing the co-occurrence of BPDCN and another separate lymphoid neoplasm, including small lymphocytic lymphoma and Waldenstrom macroglobulinemia (Harvell et al. 2000; Khan et al. 2019; Milley et al. 2016). Thus, this is the first case showing concomitant ETP-ALL and prominent blastic pDC proliferation.
Normally, circulating pDCs are limited, ranging from 0.01 to 0.5%, and the percentage decreases with age (Jing et al. 2009). They are rare in the BM and nearly absent in the nonlymphoid tissues, where they can be recruited and accumulated in ongoing immune reactions and inflammatory or neoplastic diseases (Facchetti et al. 2016; Sozzani et al. 2010). The percentage of pDCs can increase in a wide range of hematological malignancies, such as classical Hodgkin’s lymphoma, CMML and AML (Facchetti et al. 2016; Song et al. 2012; Vermi et al. 2004). In one study, the median pDC value in the healthy controls was 0.28% of the total BM cell count (range 0.14–0.53%), while in the T-ALL group, three patients (43%) had detectable pDC (mean 0.11% of total BM cell count) (Laane et al. 2007), which was low. The same study also showed an even lower level of pDCs in pediatric B-ALL than T-ALL (Laane et al. 2007). The infiltrated pDCs found in BM and lymph nodes in patients with myeloid neoplasms are described as “tumor-forming pDCs” (TF-PDCs) (Dargent et al. 2011; Wang et al. 2018, 2017). In 62 cases of TF-PDC-positive AML patients, the pDCs are composed of an average of 2.8% of the mononuclear cells in the BM (Zhu et al. 2020). Therefore, our case demonstrated that the expansion of an extraordinarily high percentage of pDCs can occur in lymphoblastic leukemia.
The distinction of normal pDCs and neoplastic DCs in BPDCN is primarily based on the evaluation of the morphological features, immunophenotype and proliferation index (Jegalian et al. 2009; Renosi et al. 2021). Neoplastic DCs generally have blastic cytomorphology that may resemble myeloid blasts or lymphoblasts. They commonly display morphological features, including a monotonous appearance of medium-sized tumor cells or large atypical monocytoid cells resembling lymphoblasts or myeloblasts, irregular nuclei, finely dispersed chromatin, a conspicuous nucleolus and agranular cytoplasm. BPDCN cells may also have cytoplasmic protrusions or pseudopods (hand-mirror cells) or microvacuoles in a string-of-pearls pattern (Fig. 2). A consensus guideline for the immunophenotype of BPDCN has not been established. According to previous reports, 87% of BPDCN cases showed CD4+CD56+CD123+ by immunohistochemistry (Facchetti et al. 2016), and 46% of patients simultaneously expressed CD4, CD56, CD123, TCL1, and CD303 (Julia et al. 2014). In a cohort of patients with BM involvement, cells were positive for CD4, CD123, TCL1, and HLA-DR and negative for CD3, CD8, CD13, CD19, CD34, and myeloperoxidase (MPO) (Alayed et al. 2013). Other antigens expressed by subsets of BPDCN cases included the following: CD56 (81%), CD33 (70%), CD7 (69%), TdT (33%), CD2 (31%), CD117 (22%), and CD5 (15%) (Alayed et al. 2013). It was raised that a confident diagnosis of neoplastic DCs can be made when four of the five antigens (CD4, CD56, CD123, TCL1, and CD303) are expressed (Julia et al. 2014; Trimoreau et al. 2003), and they may express TdT, while Ki67 is usually > 30% (Jegalian et al. 2009). In comparison, normal pDCs are mainly CD123+CD4+ TCL1 + CD56-TdT-(Ki67 < 5%). Although rare CD56- BPDCN cases have been reported, CD56 positivity is a key feature of malignant pDCs compared to normal pDCs. Meanwhile, BPDCN may express other antigens that may be negative in normal pDCs, such as BCL2 (Cota et al. 2010). The immunophenotypic characterization of BPDCN is detailed in Table 1.

Morphology of BPDCN in peripheral blood and bone marrow smear. a and b Peripheral blood involvement shows atypical blastic cells displaying a monotonous appearance of large atypical monocytoid cells with abundant cytoplasm and some microvacuoles; resembling lymphoblasts or myeloblasts; Magnification: ×1000; c and d bone marrow involvement oshows blasts with irregular nuclei, finely dispersed chromatin, conspicuous nucleolus, and abundant and agranular cytoplasm with dendritic-like protrusions and intracytoplasmic microvacuoles in a string-of-pearls pattern; Magnification: ×400
The genetic landscape and clinical presentations also assist in differentiating neoplastic DCs from normal pDCs. Simple and complex recurrent cytogenetic abnormalities have been reported in BPDCN, of which 5q-, 12p- and 12p/ETV6 are the most frequent aberrations (Fu et al. 2013; Tang et al. 2016, 2018). Recurrent mutations associated with BPDCN include point mutations on TET2, ASXL1, NPM1, NRAS and Ikaros (IKZF) (Menezes et al. 2014). The transcriptional signature of BPDCN is also clearly different from the normal pDC signature in that cell proliferation and division were upregulated in BPDCN, whereas regulation of cell shape and signal transduction was downregulated (Renosi et al. 2021). Clinically, skin involvement occurs in almost all BPDCN cases (Petrella and Facchetti 2010), and leukemia as the first presenting symptom without any cutaneous involvement is rare. Moreover, BPDCN is known to be highly malignant, leading to death within a few months. Radiation therapy or chemotherapy regimens did not significantly affect the course of BPDCN, although BM transplantation and targeted therapy against CD123 may improve survival (Dalle et al. 2010; Frankel et al. 2014).
In the current case, none of the following features support the diagnosis of BPDCN: TCL/BCL2 negativity, less aggressive clinical course, no cutaneous involvement, chemosensitivity and good prognosis. Therefore, our case is underscored by the proliferation of reactive pDCs rather than neoplastic pDCs. This finding teaches us that the diagnosis of BPDCN may not solely depend on immunophenotypic markers. Whether CD56 + pDCs are equal to malignant pDCs is worth investigating. A diagnostic consensus and standardized treatment guidelines have also yet to be established.
Etiologically, how stem cells could simultaneously develop into both blastic lymphoid and pDC lineages in this case is not clear. There has been evidence that pDCs may be derived from either myeloid or lymphoid progenitors (Ishikawa et al. 2007; Karsunky et al. 2005; Yang et al. 2005); however, pDCs seem to be closer to the myeloid lineage, and their accumulation frequently occurs in patients with myeloid neoplasms (Facchetti et al. 2016; Vermi et al. 2004). In fact, approximately 10–20% of cases of BPDCN are associated with or develop into other myeloid neoplasms, most commonly CMML, myelodysplastic syndrome and AML (Facchetti et al. 2016; Feuillard et al. 2002; Herling et al. 2003; Khoury et al. 2002; Petrella et al. 2005; Reichard et al. 2005; Vitte et al. 2012). Even MPDCP is invariably associated with a myeloid disorder (Dargent et al. 2011; Petrella and Facchetti 2010; Vermi et al. 2004; Vitte et al. 2012). In parallel, the origin of neoplastic DCs is also controversial. The cellular oncogenic origin of BPDCN is heterogeneous. Previously, pDCs were considered to be the normal counterpart of BPDCNs (Sapienza et al. 2014); however, a subsequent study indicated that BPDCN is closer to CD56 + DCs, a novel mDC subset mixed with some pDC features, by global gene expression profiling(Yu et al. 2015). A recent study on the transcriptomic signature indicated that BPDCN is closer to B-cell acute lymphoblastic leukemia (ALL) in most cases, with enrichment in pDC and B-cell signatures (Renosi et al. 2021). When cCD3 is positive, the differential diagnosis of T-ALL and BPDCN is challenging because both rely heavily on the immunophenotype (Renosi et al. 2021). Notably, the blasts of ETP-ALL are not yet irreversibly committed to the T-cell lineage and retain the potential for myeloid/dendritic cell differentiation (Steven et al. 2017). The common expression of myeloid markers in ETP-ALL may support the notion of lymphocyte-primed multipotent progenitors.
Studies on the role of pDCs in the context of acute lymphoblastic leukemia (ALL) are limited (Table 2). In two separate studies on ALL cases, patients with T-ALL had normal or high pDC levels, while patients with B-ALL had low BM DC levels and impaired pDC function at diagnosis (Laane et al. 2007; Mami et al. 2004). Consistently, pDCs from B-ALL patients display significant changes in phenotype and suppressed function, including depressed pDC proportions, limited antigen-presenting function and pDC function, which are gradually restored following therapy and remission (Zhou et al. 2019). These works suggest that DC development is probably affected in B-ALL but not in T-ALL, which may partially explain the impaired immune system and poor anti-leukemia immune responses in B-ALL. Furthermore, DC proportions could respond to patient conditions and may be useful for monitoring disease progression. Interestingly, DCs within tumor microenvironments may have aberrant phenotypes and have been implicated in the suppression of antitumor adaptive immune responses (Gabrilovich 2004). A later study provided the first evidence that endogenous tumor-associated DCs in the tumor microenvironment can contribute to T-ALL growth through an IGF1R-dependent mechanism (Triplett et al. 2016). However, they never determined which specific DC subsets directly support T-ALL growth. PDC is one subset of DCs that are responsible for the initiation and regulation of immune responses and can promote the polarization of naive T cells (Rissoan et al. 1999). Further investigation of DC phenotypes and functions in ALL may help identify potential therapeutic targets for these specific DC subsets and their tumor-promoting signals. Whether pDC proliferation in ETP-ALL is of prognostic value requires longitudinal analyses on more cases.
Conclusion
To the best of our knowledge, this is the first reported case of ETP-ALL accompanied by prominent CD56 + blastic pDC proliferation mimicking BPDCN. The current case demonstrates that significant pDC proliferation can be associated with lymphoid neoplasms and can exhibit blastic morphology and immunophenotype. The underlying mechanism of the coexistence of these two blast populations remains unknown and may require further genetic profiling of the sorted cells to denote the progressive development of tumor stem cells to the lymphoid, myeloid or dendritic cell lineage. Whether pDC proliferation has an impact on ETP-ALL prognosis remains to be explored. Considering that the incidence of BPDCN is low, more retrospective studies are needed to obtain clinical, molecular, diagnostic, and therapeutic aspects of the disease.
References
Alayed K, Patel KP, Konoplev S, Singh RR, Routbort MJ, Reddy N et al (2013) TET2 mutations, myelodysplastic features, and a distinct immunoprofile characterize blastic plasmacytoid dendritic cell neoplasm in the bone marrow. Am J Hematol 88(12):1055–1061. https://doi.org/10.1002/ajh.23567
Bond J, Graux C, Lhermitte L, Lara D, Cluzeau T, Leguay T et al (2017) Early response-based therapy stratification improves survival in adult early thymic precursor acute lymphoblastic leukemia: a group for research on adult acute lymphoblastic leukemia study. J Clin Oncol off J Am Soc Clin Oncol 35(23):2683–2691. https://doi.org/10.1200/JCO.2016.71.8585
Cordeau M, Belounis A, Lelaidier M, Cordeiro P, Sartelet H, Herblot S et al (2016) Efficient killing of high risk neuroblastoma using natural killer cells activated by plasmacytoid dendritic cells. PLoS One 11(10):e0164401. https://doi.org/10.1371/journal.pone.0164401
Cota C, Vale E, Viana I, Requena L, Ferrara G, Anemona L et al (2010) Cutaneous manifestations of blastic plasmacytoid dendritic cell neoplasm-morphologic and phenotypic variability in a series of 33 patients. Am J Surg Pathol 34(1):75–87. https://doi.org/10.1097/PAS.0b013e3181c5e26b
Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D et al (2009) Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 10(2):147–156. https://doi.org/10.1016/S1470-2045(08)70314-0
Dalle S, Beylot-Barry M, Bagot M, Lipsker D, Machet L, Joly P et al (2010) Blastic plasmacytoid dendritic cell neoplasm: is transplantation the treatment of choice? Br J Dermatol 162(1):74–79. https://doi.org/10.1111/j.1365-2133.2009.09373.x
Dargent JL, Delannoy A, Pieron P, Husson B, Debecker C, Petrella T (2011) Cutaneous accumulation of plasmacytoid dendritic cells associated with acute myeloid leukemia: a rare condition distinct from blastic plasmacytoid dendritic cell neoplasm. J Cutan Pathol 38(11):893–898. https://doi.org/10.1111/j.1600-0560.2011.01777.x
Derolf AR, Laane E, Bjorklund E, Saft L, Bjorkholm M, Porwit A (2014) Dendritic cells in bone marrow at diagnosis and after chemotherapy in adult patients with acute myeloid leukaemia. Scand J Immunol 80(6):424–431. https://doi.org/10.1111/sji.12223
Diaz-Rodriguez Y, Cordeiro P, Belounis A, Herblot S, Duval M (2017) In vitro differentiated plasmacytoid dendritic cells as a tool to induce anti-leukemia activity of natural killer cells. Cancer Immunol Immunother 66(10):1307–1320. https://doi.org/10.1007/s00262-017-2022-y
Facchetti F, Cigognetti M, Fisogni S, Rossi G, Lonardi S, Vermi W (2016) Neoplasms derived from plasmacytoid dendritic cells. Mod Pathol off J U S Can Acad Pathol Inc 29(2):98–111. https://doi.org/10.1038/modpathol.2015.145
Feuillard J, Jacob MC, Valensi F, Maynadie M, Gressin R, Chaperot L et al (2002) Clinical and biologic features of CD4(+)CD56(+) malignancies. Blood 99(5):1556–1563. https://doi.org/10.1182/blood.v99.5.1556
Frankel AE, Woo JH, Ahn C, Pemmaraju N, Medeiros BC, Carraway HE et al (2014) Activity of SL-401, a targeted therapy directed to interleukin-3 receptor, in blastic plasmacytoid dendritic cell neoplasm patients. Blood 124(3):385–392. https://doi.org/10.1182/blood-2014-04-566737
Fu Y, Fesler M, Mahmud G, Bernreuter K, Jia D, Batanian JR (2013) Narrowing down the common deleted region of 5q to 6.0 Mb in blastic plasmacytoid dendritic cell neoplasms. Cancer Genet 206(7–8):293–298. https://doi.org/10.1016/j.cancergen.2013.07.002
Gabrilovich D (2004) Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol 4(12):941–952. https://doi.org/10.1038/nri1498
Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ (1997) The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med 185(6):1101–1111. https://doi.org/10.1084/jem.185.6.1101
Harvell JD, Fulton R, Jones CD, Terris DJ, Warnke RA (2000) Composite dendritic cell neoplasm (NOS) and small lymphocytic lymphoma. Appl Immunohistochem Mol Morphol 8(4):322–328
Herling M, Teitell MA, Shen RR, Medeiros LJ, Jones D (2003) TCL1 expression in plasmacytoid dendritic cells (DC2s) and the related CD4+ CD56+ blastic tumors of skin. Blood 101(12):5007–5009. https://doi.org/10.1182/blood-2002-10-3297
Ishikawa F, Niiro H, Iino T, Yoshida S, Saito N, Onohara S et al (2007) The developmental program of human dendritic cells is operated independently of conventional myeloid and lymphoid pathways. Blood 110(10):3591–3660. https://doi.org/10.1182/blood-2007-02-071613
Jain N, Lamb AV, O’Brien S, Ravandi F, Konopleva M, Jabbour E et al (2016) Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood 127(15):1863–1869. https://doi.org/10.1182/blood-2015-08-661702
Jegalian AG, Facchetti F, Jaffe ES (2009) Plasmacytoid dendritic cells: physiologic roles and pathologic states. Adv Anat Pathol 16(6):392–404. https://doi.org/10.1097/PAP.0b013e3181bb6bc2
Jing Y, Shaheen E, Drake RR, Chen N, Gravenstein S, Deng Y (2009) Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum Immunol 70(10):777–784. https://doi.org/10.1016/j.humimm.2009.07.005
Julia F, Dalle S, Duru G, Balme B, Vergier B, Ortonne N et al (2014) Blastic plasmacytoid dendritic cell neoplasms: clinico-immunohistochemical correlations in a series of 91 patients. Am J Surg Pathol 38(5):673–680. https://doi.org/10.1097/PAS.0000000000000156
Karsunky H, Merad M, Mende I, Manz MG, Engleman EG, Weissman IL (2005) Developmental origin of interferon-alpha-producing dendritic cells from hematopoietic precursors. Exp Hematol 33(2):173–181. https://doi.org/10.1016/j.exphem.2004.10.010
Khan AM, Munir A, Raval M, Mehdi S (2019) Blastic plasmacytoid dendritic cell neoplasm in the background of myeloproliferative disorder and chronic lymphocytic leukaemia. BMJ Case Rep. https://doi.org/10.1136/bcr-2019-230332
Khoury JD, Medeiros LJ, Manning JT, Sulak LE, Bueso-Ramos C, Jones D (2002) CD56(+) TdT(+) blastic natural killer cell tumor of the skin: a primitive systemic malignancy related to myelomonocytic leukemia. Cancer 94(9):2401–2408. https://doi.org/10.1002/cncr.10489
Laane E, Bjorklund E, Mazur J, Lonnerholm G, Soderhall S, Porwit A (2007) Dendritic cell regeneration in the bone marrow of children treated for acute lymphoblastic leukaemia. Scand J Immunol 66(5):572–583. https://doi.org/10.1111/j.1365-3083.2007.02007.x
Lucas N, Duchmann M, Rameau P, Noel F, Michea P, Saada V et al (2019) Biology and prognostic impact of clonal plasmacytoid dendritic cells in chronic myelomonocytic leukemia. Leukemia 33(10):2466–2480. https://doi.org/10.1038/s41375-019-0447-3
Ma M, Wang X, Tang J, Xue H, Chen J, Pan C et al (2012) Early T-cell precursor leukemia: a subtype of high risk childhood acute lymphoblastic leukemia. Front Med 6(4):416–420. https://doi.org/10.1007/s11684-012-0224-4
Maecker B, Mougiakakos D, Zimmermann M, Behrens M, Hollander S, Schrauder A et al (2006) Dendritic cell deficiencies in pediatric acute lymphoblastic leukemia patients. Leukemia 20(4):645–649. https://doi.org/10.1038/sj.leu.2404146
Mami NB, Mohty M, Chambost H, Gaugler B, Olive D (2004) Blood dendritic cells in patients with acute lymphoblastic leukaemia. Br J Haematol 126(1):77–80. https://doi.org/10.1111/j.1365-2141.2004.04989.x
Menezes J, Acquadro F, Wiseman M, Gomez-Lopez G, Salgado RN, Talavera-Casanas JG et al (2014) Exome sequencing reveals novel and recurrent mutations with clinical impact in blastic plasmacytoid dendritic cell neoplasm. Leukemia 28(4):823–829. https://doi.org/10.1038/leu.2013.283
Milley S, Thomas L, Balme B, Dalle S (2016) Blastic plasmacytoid dendritic cell neoplasm following Waldenstrom macroglobulinemia. J Eur Acad Dermatol Venereol 30(5):904–905. https://doi.org/10.1111/jdv.13074
Neumann M, Heesch S, Schlee C, Schwartz S, Gokbuget N, Hoelzer D et al (2013) Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood 121(23):4749–4752. https://doi.org/10.1182/blood-2012-11-465138
Petrella T, Facchetti F (2010) Tumoral aspects of plasmacytoid dendritic cells: what do we know in 2009? Autoimmunity 43(3):210–214. https://doi.org/10.3109/08916930903510898
Petrella T, Bagot M, Willemze R, Beylot-Barry M, Vergier B, Delaunay M et al (2005) Blastic NK-cell lymphomas (agranular CD4+CD56+ hematodermic neoplasms): a review. Am J Clin Pathol 123(5):662–675
Reichard KK, Burks EJ, Foucar MK, Wilson CS, Viswanatha DS, Hozier JC et al (2005) CD4(+) CD56(+) lineage-negative malignancies are rare tumors of plasmacytoid dendritic cells. Am J Surg Pathol 29(10):1274–1283. https://doi.org/10.1097/01.pas.0000172194.32918.5c
Renosi F, Roggy A, Giguelay A, Soret L, Viailly PJ, Cheok M et al (2021) Transcriptomic and genomic heterogeneity in blastic plasmacytoid dendritic cell neoplasms: from ontogeny to oncogenesis. Blood Adv 5(5):1540–1551. https://doi.org/10.1182/bloodadvances.2020003359
Rissoan MC, Soumelis V, Kadowaki N, Grouard G, Briere F, de Waal MR et al (1999) Reciprocal control of T helper cell and dendritic cell differentiation. Science 283(5405):1183–1186. https://doi.org/10.1126/science.283.5405.1183
Sapienza MR, Fuligni F, Agostinelli C, Tripodo C, Righi S, Laginestra MA et al (2014) Molecular profiling of blastic plasmacytoid dendritic cell neoplasm reveals a unique pattern and suggests selective sensitivity to NF-kB pathway inhibition. Leukemia 28(8):1606–1616. https://doi.org/10.1038/leu.2014.64
Sawant A, Hensel JA, Chanda D, Harris BA, Siegal GP, Maheshwari A et al (2012) Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. J Immunol 189(9):4258–4265. https://doi.org/10.4049/jimmunol.1101855
Song HL, Huang WY, Chen YP, Chang KC (2012) Tumorous proliferations of plasmacytoid dendritic cells and Langerhans cells associated with acute myeloid leukaemia. Histopathology 61(5):974–983. https://doi.org/10.1111/j.1365-2559.2012.04282.x
Sozzani S, Vermi W, Del Prete A, Facchetti F (2010) Trafficking properties of plasmacytoid dendritic cells in health and disease. Trends Immunol 31(7):270–277. https://doi.org/10.1016/j.it.2010.05.004
Steven H, Swerdlow EC, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Arber DA, Hasserjian RP, Le Beau MM, Orazi A, Siebert R (2017) WHO Classification of tumours of haematopoietic and lymphoid tissues (Revised, 4th edn. International Agency for Research on Cancer, Lyon, France
Tang Z, Tang G, Wang SA, Lu X, Young KH, Bueso-Ramos CE et al (2016) Simultaneous deletion of 3’ETV6 and 5’EWSR1 genes in blastic plasmacytoid dendritic cell neoplasm: case report and literature review. Mol Cytogenet 9:23. https://doi.org/10.1186/s13039-016-0232-1
Tang Z, Li Y, Wang W, Yin CC, Tang G, Aung PP et al (2018) Genomic aberrations involving 12p/ETV6 are highly prevalent in blastic plasmacytoid dendritic cell neoplasms and might represent early clonal events. Leuk Res 73:86–94. https://doi.org/10.1016/j.leukres.2018.09.006
Trimoreau F, Donnard M, Turlure P, Gachard N, Bordessoule D, Feuillard J (2003) The CD4+ CD56+ CD116- CD123+ CD45RA+ CD45RO- profile is specific of DC2 malignancies. Haematologica 88(3):ELT10
Triplett TA, Cardenas KT, Lancaster JN, Hu Z, Selden HJ, Jasso GJ et al (2016) Endogenous dendritic cells from the tumor microenvironment support T-ALL growth via IGF1R activation. Proc Natl Acad Sci U S A 113(8):E1016-1025. https://doi.org/10.1073/pnas.1520245113
Vermi W, Facchetti F, Rosati S, Vergoni F, Rossi E, Festa S et al (2004) Nodal and extranodal tumor-forming accumulation of plasmacytoid monocytes/interferon-producing cells associated with myeloid disorders. Am J Surg Pathol 28(5):585–595. https://doi.org/10.1097/00000478-200405000-00004
Vitte F, Fabiani B, Benet C, Dalac S, Balme B, Delattre C et al (2012) Specific skin lesions in chronic myelomonocytic leukemia: a spectrum of myelomonocytic and dendritic cell proliferations: a study of 42 cases. Am J Surg Pathol 36(9):1302–1316. https://doi.org/10.1097/PAS.0b013e31825dd4de
Wang M, Chen YJ, Wang LR, Wang YZ, Lu J (2018) Plasmacytoid dendritic cells proliferation coexisted with acute myeloid leukemia. Chin Med J 131(15):1866–1867. https://doi.org/10.4103/0366-6999.237404
Wang P, Feng Y, Deng X, Liu S, Qiang X, Gou Y et al (2017) Tumor-forming plasmacytoid dendritic cells in acute myelocytic leukemia: a report of three cases and literature review. Int J Clin Exp Pathol 10(7):7285–7291
Xiao W, Goldberg AD, Famulare CA, Devlin SM, Nguyen NT, Sim S et al (2019) Loss of plasmacytoid dendritic cell differentiation is highly predictive for post-induction measurable residual disease and inferior outcomes in acute myeloid leukemia. Haematologica 104(7):1378–1387. https://doi.org/10.3324/haematol.2018.203018
Xiao W, Chan A, Waarts MR, Mishra T, Liu Y, Cai SF et al (2021) Plasmacytoid dendritic cell expansion defines a distinct subset of RUNX1-mutated acute myeloid leukemia. Blood 137(10):1377–1391. https://doi.org/10.1182/blood.2020007897
Yang GX, Lian ZX, Kikuchi K, Moritoki Y, Ansari AA, Liu YJ et al (2005) Plasmacytoid dendritic cells of different origins have distinct characteristics and function: studies of lymphoid progenitors versus myeloid progenitors. J Immunol 175(11):7281–7287. https://doi.org/10.4049/jimmunol.175.11.7281
Yu H, Zhang P, Yin X, Yin Z, Shi Q, Cui Y et al (2015) Human BDCA2+CD123+CD56+ dendritic cells (DCs) related to blastic plasmacytoid dendritic cell neoplasm represent a unique myeloid DC subset. Protein Cell 6(4):297–306. https://doi.org/10.1007/s13238-015-0140-x
Zalmai L, Viailly PJ, Biichle S, Cheok M, Soret L, Angelot-Delettre F et al (2021) Plasmacytoid dendritic cells proliferation associated with acute myeloid leukemia: phenotype profile and mutation landscape. Haematologica 106(12):3056–3066. https://doi.org/10.3324/haematol.2020.253740
Zhou Z, Lin W, Li X, Huang Y, Ren J, Gao Y et al (2019) Aberrant phenotype and function of dendritic cells in adult B lineage acute lymphoblastic leukemia. Immunol Invest 48(8):781–793. https://doi.org/10.1080/08820139.2019.1610428
Zhu L, Wang P, Zhang W, Li Q, Xiong J, Li J et al (2020) Plasmacytoid dendritic cell infiltration in acute myeloid leukemia. Cancer Manag Res 12:11411–11419. https://doi.org/10.2147/CMAR.S260825
Funding
This work was supported by grants from the National Natural Science Foundation of China (81902129) and Science and Technology Agency of Sichuan Province (2019YFS0313 and 2019YFS0321).
Author information
Authors and Affiliations
Contributions
HL, NJ, and QZ: designed the research; HL, YL and JY: performed the research; SZ, HZ and DX: contributed vital new reagents or analytical tools; all authors analyzed the data and wrote the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethics approval
The patient provided consent for publication of the case. The current study has been approved by the Institutional Review Board of West China Hospital, Sichuan University and has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liao, H., Yu, J., Liu, Y. et al. Early T-cell precursor lymphoblastic leukemia accompanied by prominent blastic plasmacytoid dendritic cell proliferation mimicking blastic plasmacytoid dendritic cell neoplasm: an exceptional case report and literature review. J Cancer Res Clin Oncol 148, 2911–2919 (2022). https://doi.org/10.1007/s00432-022-04238-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-022-04238-0