
Abstract
Purpose
Hepatocellular carcinoma (HCC) is a complicated disease with low survival rate due to frequent recurrence and the lack of efficient therapies. For advanced HCC, sorafenib, as the only approved first-line drug for HCC, improves the survival to some extent, but depressingly with severe adverse effects and emerging resistance conditions, which cause a poor prognosis. Ferroptosis is a new recognized way of non-apoptosis-regulated cell death, characterized by the iron-dependent accumulation of lipid hydroperoxides, showing a tremendous promising in the therapy of cancer, especially in HCC. To provide ideas for the diagnosis and treatment of HCC, we summarized the role of ferroptosis in HCC.
Methods
The relevant literature from PubMed is reviewed in this article.
Results
Interestingly enough, investigators have found sorafenib can induce ferroptosis in HCC. Moreover, recent researches reported increasing pathways and mechanisms related to ferroptosis in HCC such as TP53 and Rb, and strategies to improve sorafenib resistance by targeting ferroptosis. In addition, other drugs were reported to induce ferroptosis in HCC such as erastin and showed good efficacy in vivo and in vitro.
Conclusion
In this review, we summarize pathways and mechanisms of ferroptosis in HCC and other digestive system neoplasms such as gastric cancer, pancreatic cancer and colorectal cancer and point out the trends of ferroptosis in HCC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Hepatocellular carcinoma (HCC) is the most common type of liver cancer and is the second leading cause of cancer death worldwide (Torre et al. 2015). It is estimated that 782, 500 new liver cancer cases and 745, 500 deaths occurred worldwide during 2012 (Torre et al. 2015). Only few people are diagnosed at early stage, most people are diagnosed at middle-late stage, when they lose the opportunity of surgical therapy. For those patients, molecular targeted agents such as sorafenib (Llovet et al. 2008) and regorafenib (Bruix et al. 2017), which is used after sorafenib failure, have been implicated. Sorafenib, a multikinase inhibitor, is the first drug to be used for the systematic treatment of advanced HCC and can prolong the survival of HCC patients (Hsu et al. 2009). In two randomized phase III clinical trials of advanced HCC patients (Cheng et al. 2009; Llovet et al. 2008), sorafenib treatment improved the time to progression and extended overall survival only by 2.8 and 2.3 months compared to the placebo group, suggesting that drug resistance makes sorafenib an unsatisfactory effect. Thus, how to improve sorafenib resistance and seeking more effective new drugs have been an emerging event for advanced HCC patients and postoperative adjuvant chemotherapy patients. Recently, sorafenib was found to induce a new type of regulated cell death (RCD)-ferroptosis (Louandre et al. 2013), which is distinct from apoptosis, necroptosis and autophagy (Dixon et al. 2012). Ferroptosis is a new identified cell death and has been found in many physiological and pathological diseases such as neurodegenerative diseases (Do Van et al. 2016; Hambright et al. 2017), ischemia reperfusion injury (Tonnus and Linkermann 2016) and a series of cancers (Alvarez et al. 2017; Hao et al. 2017; Kinowaki et al. 2018; Louandre et al. 2013; Woo et al. 2018). Researchers have found ferroptosis plays a vital part in HCC (Sun et al. 2016a) and some new drugs (Ou et al. 2017) can induce ferroptosis and more importantly, some regulators that can regulate ferroptosis in HCC, such as Rb (Louandre et al. 2015), p53 (Jennis et al. 2016), and nuclear factor erythroid 2-related factor 2 (NRF2) (Sun et al. 2016b). What role on earth does ferroptosis play in HCC and what clues have researchers found and the future directions about ferroptosis in HCC can be found in this article.
Ferroptosis and its regulation in cancer
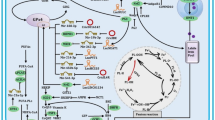
Ferroptosis was recently identified as a new form of RCD by Brent R. Stockwell’s laboratory in 2012 (Dixon et al. 2012). Ferroptosis differs from apoptosis and other major forms of RCD in many aspects. Morphologically, it is characterized by smaller mitochondria than normal and increased membrane density (Dixon et al. 2012). Mechanismly, it is an iron-dependent with accumulation of lipid peroxidation cell death and is regulated by a distinct set of genes, such as RPL8 (ribosomal protein L8), IREB2 (iron-responsive element-binding protein 2), ATP5G3 (ATP synthase F0 complex subunit C3), CS (citrate synthase), TTC35 (tetratricopeptide repeat domain 35) and ACSF2 (acyl-CoA synthetase family member 2) (Dixon et al. 2012). From the perspective of death inhibitor, ferroptosis is blocked by ferrostatin-1, not ZVAD-FMK (a potent apoptosis inhibitor) and necrosulfonamide (a potent necroptosis inhibitor). Erastin, a typical inducer of ferroptosis, inhibits SLC7A11 (Dixon et al. 2012), which is a member of cystine/glutamate antiporter, thus to inhibit synthesis of glutathione (GSH), then causing the accumulation of reactive oxygen species (ROS) and following ferroptosis. Glutathione peroxidase 4 (GPX4) is an essential negative regulator in ferroptosis in many cancers (Yang et al. 2014). In more detail, GPX4 is a selenoprotein that has a key selenocysteine residue within its catalytic site and ebselen, a GPX4 mimetic, is able to partially protect the HCC cells from ferroptosis (Ou et al. 2017). In short, ferroptosis is a new way of cell death and will bring new targets in the therapies and prognosis of various diseases (Fig. 1).

The mechanism of ferroptosis. Ferroptosis is characterized by production of ROS from accumulated iron and lipid peroxidation. This illustration shows the process of ferroptosis and summarizes the molecule pathways that regulate iron and lipid perioxidation, respectively. PUFA polyunsaturated fatty acids, PHKG2 phosphorylase kinase G2, NCOA4 nuclear receptor coactivator 4, BSO buthionine sulfoximine
As mentioned above, ferroptosis was found in several types of tumor cells such as diffuse large B-cell lymphoma (DLBCL) (Kinowaki et al. 2018), renal cell carcinoma(RCC) (Woo et al. 2018), breast cancer (Ma et al. 2016), pancreatic cancer(PC) (Eling et al. 2015) and lung cancer (Alvarez et al. 2017), in particular HCC. As we know, induction of cell death is an emerging approach for cancer therapy. Since the first demonstration in 2012, a series of strategies have been developed to induce ferroptosis of cancer cells, including the use of nanomaterials (Ou et al. 2017), clinical drugs (such as sorafenib (Louandre et al. 2013), sulfasalazine (SSZ) (Dixon et al. 2012) and artesunate (Eling et al. 2015), experimental compounds (Dixon et al. 2012) and deprivation of cystine (Hayano et al. 2016). Ferroptosis is emerging as a potential weapon against tumor growth and opens new avenue for cancer therapies.
Potential roles of ferroptosis in HCC
The fact that sorafenib can induce ferroptosis adds a piece to the puzzle of sorafenib anti-tumor mechanisms in HCC. Moreover, targeting ferroptosis can improve sorafenib resistance from a new perspective. More than sorafenib, researchers also have found other drugs which can induce ferroptosis and new targets which can regulate ferroptosis in HCC. In short, ferroptosis brings a new angle for the therapy and prognosis for HCC.
Ferroptosis and sorafenib in HCC
The multikinase inhibitor sorafenib is the only first-line drug for patients with advanced HCC (Llovet et al. 2008). As we know, sorafenib exerts its anticancer effects by induction of apoptosis and inhibition of proliferation as well as inhibition of angiogenesis (Liu et al. 2006) mainly by its kinase inhibitory effect. However, sorafenib is a weak apoptosis inducer as a single agent (Galmiche et al. 2014). Delightedly, investigators have found sorafenib can induce a novel way of RCD–ferroptosis. A recent study reported that the depletion of the intracellular iron stores achieved using the iron chelator deferoxamine (DFX) protected HCC cells from the cytotoxic effects of sorafenib and DFX prevented sorafenib from inducing oxidative stress (Louandre et al. 2013), consisting with the iron and lipid peroxidation dependences of ferroptosis. Likewise, Louandre et al. (Louandre et al. 2013) suggested that both pharmacological ferroptosis inhibitors (ferrostatin-1) or genetic procedures (RNA interference against IREB2), readily inhibited the cytotoxic effects of sorafenib in HCC cells. Beyond that, Lachaier et al. (Lachaier et al. 2014) reported that in comparison with other kinases inhibitors, sorafenib is the only drug that displayed ferroptotic efficacy, suggesting that the induction of ferroptosis is a specific property of sorafenib and unrelated to the RAF kinases inhibitory effect of sorafenib. By the way, researchers also found sorafenib-induced ferroptosis not only in HCC, but also in pancreatic adenocarcinoma, colon carcinoma, and kidney tumors (Lachaier et al. 2014), also providing a new perspective in the therapy of these tumors. In terms of mechanism, another study showed sorafenib can also inhibit SLC7A11 to induce ferroptosis (Dixon et al. 2014), such as erastin. Collectively, sorafenib-induced ferroptosis is an effective mechanism for the induction of cell death in HCC independent its kinases inhibitory effect. However, more studies are still needed to illuminate the precise mechanisms in this process.
Rb inhibits sorafenib-induced ferroptosis in HCC
We already know that retinoblastoma (Rb) protein regulates cell proliferation and plays a vital role in G1/S checkpoint via its ability to regulate the activity of the transcription factors of the E2F family (Knudsen and Knudsen 2008). The loss of function of the Rb protein is an important event during liver carcinogenesis (Mayhew et al. 2007), but it is unclear whether the Rb status modulates the response of HCC cells to sorafenib. Recently, Louandre et al. found that HCC cells with reduced levels of Rb exhibited a two- to threefold increase in cell death induction upon exposure to sorafenib compared with controls (Louandre et al. 2015). They also found that upon exposure to sorafenib, the Rb-negative status of HCC cells promoted the occurrence of ferroptosis with high expression of mitochondria ROS (Louandre et al. 2015). Furthermore, the Rb status of individual HCC patients is associated with the prognosis of these patients when they receive sorafenib. The findings illuminate the role of Rb in the response of HCC cells to sorafenib and in the regulation of ferroptosis.
NRF2 protects HCC from sorafenib-induced ferroptosis
We already know the NRF2 is a key regulator of the antioxidant response (Ma 2013) and NRF2 overexpression inhibits apoptosis and contributes to chemoresistance in several cancers (Wang et al. 2008). However, it is still unclear whether NRF2 activation is involved in the regulation of other forms of RCD, such as ferroptosis. A new study indicated NRF2 plays a central role in protecting HCC cells against sorafenib-induced ferroptosis (Sun et al. 2016b). Upon exposure to erastin and sorafenib, p62 prevented NRF2 degradation and enhanced subsequent NRF2 nuclear accumulation through inactivation of Kelch-like ECH-associated protein 1(Keap1). Additionally, nuclear NRF2 interacted with the transcriptional coactivator small v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog (Maf) proteins such as MafG (Suzuki et al. 2013) and then activated transcription of quinone oxidoreductase 1 (NQO1), heme oxygenase-1 (HO1), and ferritin heavy chain 1 (FTH1), which are antioxidants, then causing the resistance to ferroptosis. FTH1 and ferritin light chain make up ferritin heteropolymers, where ferrous iron stores (Harrison and Arosio 1996). Knockdown of p62, NQO1, HO1, and FTH1 in HCC cells promoted ferroptosis in response to erastin or sorafenib and genetic or pharmacologic inhibition of NRF2 expression/activity in HCC cells rendered HCC more sensitive to erastin and sorafenib in vitro and in vivo. Furthermore, NRF2 inhibitors–alkaloid trigonelline (Arlt et al. 2013) had the potency to be used in combination therapy for HCC by overcoming chemoresistance with the induction of ferroptosis (Sun et al. 2016b). Above all, activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in HCC cells and the research results provide an approach for combination therapy to improve sorafenib resistance.
MT-1G inhibits ferroptosis thus facilitating sorafenib resistance in HCC
Acquired resistance to sorafenib has been found in HCC patients, which results in a poor prognosis (Liu et al. 2017), thus elucidating mechanisms underlying sorafenib resistance has great significance for improving the efficacy of sorafenib. A recent study suggested that metallothionein-1G (MT-1G) is a critical negative regulator of ferroptosis and has been a promising therapeutic target of sorafenib resistance in human HCC cells (Sun et al. 2016a). Studies reported the expression of MT-1G is remarkably induced by sorafenib, but not other clinically relevant kinase inhibitors (e.g., erlotinib, gefitinib, tivantinib, and vemurafenib, etc.). Importantly, genetic and pharmacological inhibition of MT-1G enhanced the anticancer activity of sorafenib in vitro and in tumor xenograft models. The molecular mechanisms underlying the action of MT-1G in sorafenib resistance involved the inhibition of ferroptosis. In detail, knockdown of MT-1G increased GSH depletion and lipid peroxidation without altering iron levels, which contribute to sorafenib-induced ferroptosis. Investigators also found the upstream regulator in this process and they showed that activation of NRF2, but not p53 and hypoxia-inducible factor 1-alpha (HIF1α), was essential for induction of MT-1G expression via the cystathionase pathway following sorafenib treatment. Moreover, the study found high induction of MT1 in the serum was indicative of poor prognosis in HCC patients treated with sorafenib with low overall survival, which has a significant clinical meaning for predicting prognosis. In conclusion, MT-1G is a novel molecular mechanism of sorafenib resistance and also a new negative regulator of ferroptosis in HCC.
Haloperidol promotes sorafenib-induced ferroptosis in HCC
Sigma 1 receptor (S1R) is a protein modulator which is related to oxidative stress metabolism. Lately, Bai et al. discovered that haloperidol, as a antagonist, promoted both erastin and sorafenib-induced ferroptosis, even with both drugs at relatively lower doses, indicating that haloperidol may benefit HCC patients treated with sorafenib by reducing the dosage or potentiating the effectiveness of this drug (Bai et al. 2017). In terms of mechanism, haloperidol increased the cellular levels of Fe2+ and lipid peroxidation. Otherwise, haloperidol influenced many ferrotosis-related targets such as NRF2, HO-1 and GPX4. In conclusion, this finding provides a novel strategy for the combination of drugs for HCC therapy.
CISD1 negatively regulates ferroptosis in HCC
As we know, the occurrence of ferroptosis is accompanied by the morphologic change of mitochondria (Dixon et al. 2012), so we can assume that mitochondria is vital in the ferroptosis, but the key regulator of ferroptosis in mitochondria remains unknown. Currently, Yuan et al. have found that CDGSH iron sulfur domain 1 (CISD1, also termed mitoNEET), an iron-containing outer mitochondrial membrane iron sulfur protein, negatively regulates ferroptosis and is upregulated by erastin in an iron-dependent manner in human HCC cells (Yuan et al. 2016). They found inhibition of CISD1 contributed to erastin-induced ferroptosis and in contrast, stabilization of the iron sulfur cluster of CISD1 by pioglitazone inhibited mitochondrial iron uptake, lipid peroxidation, and subsequent ferroptosis in HCC. To sum up, CISD1 protects against mitochondrial injury in ferroptosis in HCC.
Polymorphism of TP53 genes (S47 variant) inhibits ferroptosis in HCC
TP53 gene is a well-known tumor suppressor gene and regulates apoptosis (Shi et al. 2014), necrosis (Khan and Xiang 2017), and autophagy (Mrakovcic and Frohlich 2018), but whether it regulates ferroptosis is unclear. As we know, phosphorylation of Ser46 is important to maintain the tumor suppression function of p53 (Taira et al. 2007). A recent study showed that mutant p53 with Pro47 losing its function to phosphorylate Ser46, which is referred to as Ser 47 (S47), inhibits ferroptosis in HCC (Jennis et al. 2016). Compared with wide-type p53 HCC cells, SLC7A11 was increased and PTGS2 (a ferroptosis biomarker in vivo) was decreased in the S47 cells after exposure to cisplatin indicating a resistance to ferroptosis. They showed that this variant showed a defect in ferroptosis induction and conferred increased cancer risk in a mouse model (Jennis et al. 2016). Notably, this mutation just existed in Africans and African-Americans, so the genetic typing has a significance to assess the risk of cancer in these people. In brief, S47 variant shows a defect in ferroptosis induction in HCC.
Regulation of lipid metabolism in ferroptosis in HCC
ACSL4 contributes to ferroptosis in HCC
Although lipid peroxidation plays a central role in triggering ferroptosis, the essential regulator of lipid metabolism in ferroptosis remains poorly defined. Now, Yuan et al. have identified acyl-CoA synthetase long-chain family member 4 (ACSL4) is required for ferroptosis in HCC (Jennis et al. 2016). Compared with ferroptosis-sensitive cells, the expression of ACSL4 was remarkably downregulated in ferroptosis-resistant cells, so expression of ACSL4 may be a useful biomarker for monitoring ferroptosis. Moreover, knockdown of ACSL4 inhibited erastin-induced ferroptosis in HepG2 cells. Mechanically, ACSL4-mediated production of 5-hydroxyeicosatetraenoic acid (5-HETE) contributed to ferroptosis and pharmacological inhibition of 5-HETE production by zileuton limited ACSL4 overexpression-induced ferroptosis. The production of 5-HETE has been observed in the induction of ferroptosis previously (Friedmann Angeli et al. 2014). Happily, compared with normal tissue, ACSL4 is overexpressed in several cancers such as liver, kidney, colorectal, and head and neck cancer (Chen et al. 2016), so induction of ferroptosis may be an anticancer approach with fewer side effects for these cancers. Collectively, ACSL4 plays a key role in promoting erastin-induced ferroptosis through 5-HETE-mediated lipotoxicity. ACSL4 is not only a sensitive monitor of ferroptosis, but also an important contributor of ferroptosis.
LDL–DHA nanoparticles can induce ferroptosis in HCC
Researchers have found some drugs can induce ferroptosis in cancers, and they improve the therapy of cancer significantly. Recently, Ou et al. have found low-density lipoprotein–docosahexaenoic acid (LDL–DHA) (Firestone 1994) can induce ferroptosis to kill HCC cells (Ou et al. 2017). LDL–DHA were cytotoxic to both rat hepatoma and human HCC cell lines with pronounced lipid peroxidation, depletion of GSH and inactivation of the GPX4 prioring to cell death. In keeping with the mechanisms of ferroptosis, GPX4 was also found to be a central regulator of LDL–DHA nanoparticle-induced tumor cell killing. On one hand, DHA degraded GPX4 directly. On the other hand, LDL–DHA nanoparticles decreased intracellular GSH by reducing redox couples GSH/GSSG and NADPH/NADP+ and removing GSH-aldehyde adducts (Hayes and McLellan 1999), then GPX4 cannot exert its enzymatic activity because of the depletion of substrate. In conclusion, LDL–DHA nanoparticles induce cell death in HCC cells through the ferroptosis pathway, which represents a novel molecular mechanism of anticancer activity for LDL–DHA nanoparticles. We believe more drugs will be discovered to induce ferroptosis for HCC patients.
Ferroptosis in other digestive system neoplasms
Digestive system tumors mainly include esophagus cancer, gastric cancer (GC), colorectal cancer (CRC), liver cancer and PC. These tumors have high morbidity and mortality rates (Torre et al. 2015) but lack effective drugs, so seeking new targets for them is very important. Recently, increasing studies have indicated that ferroptosis is strongly implicated in GC (Hao et al. 2017), CRC (Guo et al. 2018; Hong et al. 2017; Xie et al. 2017), and PC (Eling et al. 2015; Kasukabe et al. 2016; Shintoku et al. 2017; Wang et al. 2016; Xie et al. 2016; Yamaguchi et al. 2018; Zhu et al. 2017). For example, artesunate induces ferroptosis in RAS-activated human pancreatic ductal adenocarcinoma (PDAC); HSPA5 (Heatshock 70-kDa protein 5) negatively regulates ferroptosis and inhibition of HSPA5-GPX4 pathway enhances gemcitabine sensitivity. We list a table to generalize progress of ferroptosis in GC, CRC and PC (Table 1). Up to now, the function of ferroptosis in esophageal cancer has not been studied. We believe the study of ferroptosis in digestive system neoplasms will provide new insights into the therapies for them.
Conclusion and perspective
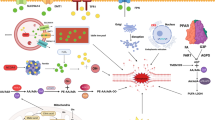
Taken together, we can see that ferroptosis has a very vital role in cancers, especially in HCC. Sorafenib can induce ferroptosis, as a new mechanism to mediate its cytotoxicity. Rb, NRF2, and MT-IG inhibit sorafenib-induced ferroptosis, so inhibition of the three regulators can improve sorafenib resistance, which provide promising strategies for HCC therapy. On the contrary, haloperidol promotes sorafenib-induced ferroptosis and provides a strategy for combination therapy with sorafenib in HCC. In addition, CISD1 is also a negative regulator in HCC and gives us a better understanding about iron metabolism of ferroptosis. In addition to iron metabolism, lipid metabolism is also critical for ferroptosis in HCC, for example, LDL–DHA can induce ferroptosis and ACSL4 is a monitor and contributor to ferroptosis (Fig. 2). Some progress has been made about ferroptosis in HCC; however, detailed signal transduction pathways and key transcriptional regulators of ferroptosis in HCC remain unknown. For example, p53 can enhance ferroptosis by inhibiting SLC7A11 in breast cancer, but TP53 inhibits erastin-induced ferroptosis via blocking DPP4 activity in a transcription-independent manner in CRC. What role does p53 play in human HCC is still needed to be clarified. And more work is needed to investigate, in addition to drug resistance, what other roles ferroptosis play in other phenotypes such as metastasis, energy metabolism and autophagy in HCC. We believe the understandings of ferroptosis in HCC can bring us a hope to improve the therapy and prognosis of HCC.

The role of ferroptosis in HCC. Erastin, sorafenib and LDL–DHA can induce ferroptosis in HCC and the schematic diagram represents regulators and pathways of ferroptosis in HCC
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- RCD:
-
Regulated cell death
- NRF2:
-
Nuclear factor erythroid 2-related factor 2
- RPL8:
-
Ribosomal protein L8
- IREB2:
-
Iron-responsive element-binding protein 2
- ATP5G3:
-
ATP synthase F0 complex subunit C3
- CS:
-
Citrate synthase
- TTC35:
-
Tetratricopeptide repeat domain 35
- ACSF2:
-
Acyl-CoA synthetase family member 2
- GSH:
-
Glutathione
- ROS:
-
Reactive oxygen species
- GPX4:
-
Glutathione peroxidase 4
- DLBCL:
-
Diffuse large B-cell lymphoma
- RCC:
-
Renal cell carcinoma
- PC:
-
Pancreatic cancer
- SSZ:
-
Sulfasalazine
- DFX:
-
Deferoxamine
- Rb:
-
Retinoblastoma
- Keap1:
-
Kelch-like ECH-associated protein 1
- Maf:
-
V-maf avian musculoaponeurotic fibrosarcoma oncogene homolog
- NQO1:
-
Quinone oxidoreductase 1
- HO1:
-
Heme oxygenase-1
- FTH1:
-
Ferritin heavy chain 1
- MT:
-
Metallothionein
- HIF1α:
-
Hypoxia-inducible factor 1-alpha
- S1R:
-
Sigma 1 receptor
- CISD1:
-
CDGSH iron sulfur domain 1
- S47:
-
Ser 47
- ACSL4:
-
Acyl-CoA synthetase long-chain family member 4
- 5-HETE:
-
5-Hydroxyeicosatetraenoic acid
- LDL–DHA:
-
Low-density lipoprotein docosahexaenoic acid
- GC:
-
Gastric cancer
- CRC:
-
Colorectal cancer
- CDO1:
-
Cysteine dioxygenase 1
- PDAC:
-
Pancreatic ductal adenocarcinoma
- CN-A:
-
Cotylenin A
- PEITC:
-
Phenethyl isothiocyanate
- PL:
-
Piperlongumine
- HSPA5:
-
Heatshock 70-kDa protein 5
- LOX:
-
Lipoxygenases
- ALOX:
-
Arachidonate lipoxygenase
- NCOA4:
-
Nuclear receptor coactivator 4
- CHOP:
-
C/EBP-homologous protein
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- DPP4:
-
Dipeptidyl-peptidase-4
References
Alvarez SW et al (2017) NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551:639–643. https://doi.org/10.1038/nature24637
Arlt A et al (2013) Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 32:4825–4835. https://doi.org/10.1038/onc.2012.493
Bai T, Wang S, Zhao Y, Zhu R, Wang W, Sun Y (2017) Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem Biophys Res Commun 491:919–925. https://doi.org/10.1016/j.bbrc.2017.07.136
Bruix J et al (2017) Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (London England) 389:56–66. https://doi.org/10.1016/s0140-6736(16)32453-9
Chen WC, Wang CY, Hung YH, Weng TY, Yen MC, Lai MD (2016) Systematic analysis of gene expression alterations and clinical outcomes for long-chain acyl-coenzyme a synthetase family in cancer. PloS One 11:e0155660. https://doi.org/10.1371/journal.pone.0155660
Cheng AL et al (2009) Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 10:25–34. https://doi.org/10.1016/s1470-2045(08)70285-7
Dixon SJ et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Dixon SJ et al (2014) Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress ferroptosis. Elife 3:e02523. https://doi.org/10.7554/eLife.02523
Do Van B et al (2016) Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis 94:169–178. https://doi.org/10.1016/j.nbd.2016.05.011
Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR (2015) Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2:517–532. https://doi.org/10.18632/oncoscience.160
Firestone RA (1994) Low-density lipoprotein as a vehicle for targeting antitumor compounds to cancer cells. Bioconj Chem 5:105–113
Friedmann Angeli JP et al (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16:1180–1191. https://doi.org/10.1038/ncb3064
Galmiche A, Chauffert B, Barbare JC (2014) New biological perspectives for the improvement of the efficacy of sorafenib in hepatocellular carcinoma. Cancer Lett 346:159–162. https://doi.org/10.1016/j.canlet.2013.12.028
Guo J et al (2018) Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat 50:445–460. https://doi.org/10.4143/crt.2016.572
Hambright WS, Fonseca RS, Chen L, Na R, Ran Q (2017) Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol 12:8–17 https://doi.org/10.1016/j.redox.2017.01.021
Hao S et al (2017) Cysteine dioxygenase 1 mediates erastin-induced ferroptosis in human gastric cancer cells. Neoplasia (New York NY) 19:1022–1032. https://doi.org/10.1016/j.neo.2017.10.005
Harrison PM, Arosio P (1996) The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1275:161–203
Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR (2016) Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Diff 23:270–278. https://doi.org/10.1038/cdd.2015.93
Hayes JD, McLellan LI (1999) Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Rad Res 31:273–300
Hong SH et al (2017) Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent. PUMA Exp Oncotarget 8:115164–115178. https://doi.org/10.18632/oncotarget.23046
Hou W et al (2016) Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12:1425–1428. https://doi.org/10.1080/15548627.2016.1187366
Hsu C, Shen YC, Cheng AL (2009) Sorafenib for the treatment of hepatocellular carcinoma across geographic regions. Expert Rev Clin Pharmacol 2:129–136. https://doi.org/10.1586/17512433.2.2.129
Jennis M et al (2016) An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 30:918–930 https://doi.org/10.1101/gad.275891.115
Kasukabe T, Honma Y, Okabe-Kado J, Higuchi Y, Kato N, Kumakura S (2016) Combined treatment with cotylenin A and phenethyl isothiocyanate induces strong antitumor activity mainly through the induction of ferroptotic cell death in human pancreatic cancer cells. Oncol Rep 36:968–976. https://doi.org/10.3892/or.2016.4867
Khan MR, Xiang S (2017) The p53-inducible long noncoding RNA TRINGS protects cancer cells from necrosis under glucose starvation. EMBO J 36:3483–3500. https://doi.org/10.15252/embj.201696239
Kinowaki Y et al (2018) Glutathione peroxidase 4 overexpression inhibits ROS-induced cell death in diffuse large B-cell lymphoma Laboratory investigation. J Tech Methods Pathol. https://doi.org/10.1038/s41374-017-0008-1
Knudsen ES, Knudsen KE (2008) Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 8:714–724. https://doi.org/10.1038/nrc2401
Lachaier E et al (2014) Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res 34:6417–6422
Liu L et al (2006) Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 66:11851–11858. https://doi.org/10.1158/0008-5472.can-06-1377
Liu J, Liu Y, Meng L, Liu K, Ji B (2017) Targeting the PD-L1/DNMT1 axis in acquired resistance to sorafenib in human hepatocellular carcinoma. Oncol Rep 38:899–907. https://doi.org/10.3892/or.2017.5722
Llovet JM et al (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359:378–390. https://doi.org/10.1056/NEJMoa0708857
Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B, Galmiche A (2013) Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer 133:1732–1742. https://doi.org/10.1002/ijc.28159
Louandre C et al (2015) The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett 356:971–977. https://doi.org/10.1016/j.canlet.2014.11.014
Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426. https://doi.org/10.1146/annurev-pharmtox-011112-140320
Ma S, Henson ES, Chen Y, Gibson SB (2016) Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis 7:e2307. https://doi.org/10.1038/cddis.2016.208
Mayhew CN et al (2007) RB loss abrogates cell cycle control and genome integrity to promote liver tumorigenesis. Gastroenterology 133:976–984. https://doi.org/10.1053/j.gastro.2007.06.025
Mrakovcic M, Frohlich LF (2018) p53-mediated molecular control of autophagy in tumor cells. Biomolecules. https://doi.org/10.3390/biom8020014
Ou W, Mulik RS, Anwar A, McDonald JG, He X, Corbin IR (2017) Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free Rad Biol Med 112:597–607. https://doi.org/10.1016/j.freeradbiomed.2017.09.002
Shi X et al (2014) Nutlin-3 downregulates p53 phosphorylation on serine392 and induces apoptosis in hepatocellular carcinoma cells. BMB Rep 47:221–226
Shintoku R et al (2017) Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci 108:2187–2194 https://doi.org/10.1111/cas.13380
Sun X, Niu X, Chen R, He W, Chen D, Kang R, Tang D (2016a) Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology (Baltimore, Md) 64:488–500. https://doi.org/10.1002/hep.28574
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D (2016b) Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology (Baltimore, Md) 63:173–184. https://doi.org/10.1002/hep.28251
Suzuki T, Motohashi H, Yamamoto M (2013) Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci 34:340–346. https://doi.org/10.1016/j.tips.2013.04.005
Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K (2007) DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to. DNA Damage Mol Cell 25:725–738. https://doi.org/10.1016/j.molcel.2007.02.007
Tonnus W, Linkermann A (2016) “Death is my Heir"—ferroptosis connects cancer pharmacogenomics and ischemia-reperfusion injury. Cell Chem Biol 23:202–203. https://doi.org/10.1016/j.chembiol.2016.02.005
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA Cancer J Clin 65:87–108. https://doi.org/10.3322/caac.21262
Wang XJ et al (2008) Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs the dark side of Nrf2. Carcinogenesis 29:1235–1243. https://doi.org/10.1093/carcin/bgn095
Wang D, Peng Y, Xie Y, Zhou B, Sun X, Kang R, Tang D (2016) Antiferroptotic activity of non-oxidative dopamine. Biochem Biophys Res Commun 480:602–607. https://doi.org/10.1016/j.bbrc.2016.10.099
Woo SM et al (2018) Corosolic acid induces non-apoptotic cell death through generation of lipid reactive oxygen species production in human renal carcinoma caki. Cells 19 https://doi.org/10.3390/ijms19051309
Xie Y et al (2016) Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem Biophys Res Commun 473:775–780. https://doi.org/10.1016/j.bbrc.2016.03.052
Xie Y et al (2017) The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4. Activity Cell Rep 20:1692–1704. https://doi.org/10.1016/j.celrep.2017.07.055
Yamaguchi Y, Kasukabe T, Kumakura S (2018) Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol 52:1011–1022. https://doi.org/10.3892/ijo.2018.4259
Yang WS et al (2014) Regulation of ferroptotic cancer cell death by GPX. 4 Cell 156:317–331. https://doi.org/10.1016/j.cell.2013.12.010
Yuan H, Li X, Zhang X, Kang R, Tang D (2016) CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun 478:838–844. https://doi.org/10.1016/j.bbrc.2016.08.034
Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R, Tang D (2017) HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res 77:2064–2077. https://doi.org/10.1158/0008-5472.can-16-1979
Acknowledgements
We are grateful to the staff at the GI oncology of cancer hospital of HMU. And we thank Kainan Kang for his critical reading of the manuscript.
Funding
This study was funded by the National Natural Scientific Foundation of China (No. 81302060, No.81472322 and No. 81672930), the national youth talent support program for TS Zheng, the Natural Science Foundation of Heilongjiang Province (LC201437/H1617), China Postdoctoral Science Foundation (No. 2015T80369 and No. 2014M560271), and Heilongjiang Postdoctoral Science Foundation (No. LBH-Z14142 and No. LBH-TZ1615), the Fok Ying Tung Education Foundation (No. 151037), the Academician Yu Weihan Outstanding youth foundation of Harbin Medical University for TS Zheng. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Nie, J., Lin, B., Zhou, M. et al. Role of ferroptosis in hepatocellular carcinoma. J Cancer Res Clin Oncol 144, 2329–2337 (2018). https://doi.org/10.1007/s00432-018-2740-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-018-2740-3