
Abstract
Prolactinoma is a rare pituitary adenoma secreting prolactin. Studies on diagnostics, treatment, and prognosis in pediatric prolactinoma patients are rare. We analyzed clinical presentation, response to treatment, and prognosis of 27 pediatric prolactinoma patients (10 m/17 f. based on patients’ records. Tumors included 6 microadenomas (tumor volume: median 0.2 cm3, range 0.01–0.4 cm3; serum prolactin at diagnosis: median 101 ng/ml, range 33–177 ng/ml), 15 macroadenomas (volume: median 3.3 cm3, range 0.4–25.8 cm3; prolactin: median 890 ng/ml, range 87–8624), and 3 giant adenomas (volume: median 44.5 cm3, range 38.6–93.5 cm3; prolactin: median 4720 ng/ml, range 317–10,400); data for 3 patients were not available. Dopamine agonist treatment (n = 22) was safe and effective, leading to reductions in tumor size (p < 0.01) and prolactin levels (p < 0.01). Threat to vision was the indication for decompressing surgery in three of seven operated patients. No patient was irradiated. Long-term functional capacity was not impaired when compared with other sellar masses (n = 235).
Conclusion: In pediatric prolactinoma, diagnosis is based on hyperprolactinemia and imaging. Dopamine agonist treatment is effective and safe. Overall survival and functional capacity as a measure of quality of survival were not impaired, indicating an optimistic prognosis. Surgery should be considered only in emergency situations of threatened visual function, not presenting a fast response to dopamine agonist treatment. Severe side effects of medication and lack of efficacy should be considered as contraindications.
What is Known: • In pediatric prolactinoma—a very rare pediatric neuroendocrinological disease—gender-related differences in terms of clinical presentation at initial diagnosis are known. • Due to the rareness of the disease, reports on long-term outcome and prognosis after childhood-onset prolactinoma based on prospective follow-up are not published. |
What is New: • Dopamine agonist treatment is efficient and safe for tumor volume reduction in pediatric prolactinoma and surgical interventions are recommended only for decompression of the optic chiasm in case of threat to vision. In case of inefficient response to medication, side effects or parental refuse, alternative therapeutic options should be considered. • Quality of life in terms of survival and functional capacity was not impaired in pediatric prolactinoma patients when compared with 235 long-term survivors of different sellar masses. |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pediatric prolactinoma (PP), a benign pituitary adenoma secreting prolactin, typically occurs at the age when puberty begins [1, 4, 7, 11, 12, 18, 19, 22, 28, 33]. Although there are rare functional effects, the most common complaint at diagnosis is headache, and a large tumor mass can cause visual impairment. Furthermore, galactorrhea and delayed pubertal development are observed. With an incidence of < 0.1/1.000.000 population, they are rare yet represent 50% of all pediatric pituitary adenomas and 2% of all pediatric intracranial tumors [4, 7, 11, 19, 22, 28, 33]. Girls are affected more frequently than boys, but prolactinoma in boys tends to be larger and more aggressive—occurring at an earlier age, achieving a larger mass, and having higher prolactin serum levels [1, 11]. Hence, microadenomas (tumors < 10 mm in diameter) are typical in females and macroadenomas (10–40 mm in diameter) are typical in males [12].
Reports on PP are rare. Only general algorithms for diagnosis and treatment exist, which recommend measuring for elevated prolactin levels, performing magnetic resonance imaging (MRI), and treatment with dopamine agonists. Dose and duration of drug therapy remain inconsistently defined, with the most successful therapies varying according to prolactin levels and tumor growth rate [12]. Overall survival rates and functional capacity as a measure of quality of survival have not been reported for PP so far.
In this study, clinical symptoms, laboratory parameters, treatment, and outcome of 27 PP patients were retrospectively analyzed. The aim of this study was to gain more insight and knowledge of this disease and its prognosis in the specific group of pediatric patients as a basis for age- and gender-adapted treatment recommendations.
Patients and methods
Patients
German patients with pediatric neuro oncological diseases are registered since 1990 with a high degree of completeness in the German Childhood Cancer Registry (DKKR). All patients with sellar masses registered in the DKKR were recruited and prospectively analyzed in the German international trials HIT-Endo and KRANIOPHARYNGEOM 2000/2007 (Clinical trial no. KRANIOPHARYNGEOM 2000—NCT00258453; KRANIOPHARYNGEOM 2007—NCT01272622). Twenty-seven German PP patients recruited since 1990 were retrospectively analyzed for history before diagnosis, treatment, and initial presenting manifestations as well as the influence of these factors on prognosis [25]. Diagnoses of prolactinoma were confirmed either by hyperprolactinemia plus typical radiographic findings or by reference-confirmed histological diagnosis in rare cases. Tumor sizes were calculated using maximal tumor diameters (A, B, C) in three dimensions (A × B × C / 2) based on results of computed tomography (n = 4) or MRI (n = 27). Microadenomas are defined by a maximal diameter < 10 mm, macroadenomas measure 10–40 mm, while giant pituitary adenomas measure > 40 mm in maximum diameter [14, 21].
Functional capacity assessment
The German daily life ability scale, Fertigkeitenskala-Münster-Heidelberg (FMH), was used for self-assessment of functional capacity [34]. The FMH measures the capability for routine actions. The average time required was 4.5 min in first-time users [26]. Fourteen of 27 PP patients (52%) answered the FMH questionnaire. FMH-scores in PP patients were compared with FMH-scores of patients with different sellar masses recruited in the German Craniopharyngioma Registry (174 craniopharyngioma, 27 hypothalamic glioma, 14 optic/chiasm glioma, 11 germinoma, 9 cyst of Rathke pouch).
Statistical analyses
Statistical analyses were performed using SPSS19.0 (SPSS, INC., Chicago, IL). The Mann–Whitney U test was used for comparison of two independent groups for a continuous variable. The chi-square test was used for comparison of different groups for categorical variables. Correlation between two variables was analyzed using the Spearman correlation coefficient. Overall survival (OS) rates were estimated by the Kaplan–Meier method. Groups were compared using the log-rank test. p values of ≤ 0.05 were chosen as being statistically significant. Inferential statistics are intended to be exploratory, not confirmatory, and were interpreted accordingly.
Results
Twenty-seven PP patients (17 female), recruited between 1990 and 2015 in HIT-Endo and KRANIOPHARYNGEOM 2000/2007, were analyzed. Median age at diagnosis was 15.6 years, ranging from 11.4 to 17.7 years. Median follow-up time was 27 months (range 0.5–117 months). In terms of age at diagnosis, we observed a younger age (p = 0.012) of boys (median age 14.0 years) compared to girls (median age 16.5 years). Duration of history before initial diagnosis was recorded in 19 patients. The median duration of history was 12 months (range 2–48 months). The median prolactin serum concentration at the time of diagnosis was 494.9 ng/ml (range 87.6–10,400.0 ng/ml). In 24 patients, measurement of tumor volume was performed at the time of diagnosis, showing a median volume of 2345.1 mm3 (range 9.4–93,549.2 mm3). Tumor size was categorized by maximum diameter (Ø) in microadenoma with Ø < 10 mm (n = 6; 168 mm3 (range 9–449 mm3)), macroadenoma with Ø = 10–40 mm (n = 15; 3.312 mm3 (range 385–25.800 mm3)), and giant adenoma with Ø > 40 mm (n = 3; 44.548 mm3 (range 38.640–93.549 mm3)) (Table 1).
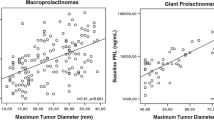
The diagnosis of prolactinoma was confirmed by hyperprolactinemia in combination with clinical information and imaging in 25 PP patients. In four PP patients (15%), additional histological verification was performed. Prolactin serum levels and tumor size at the time of diagnosis were positively correlated (r 0.825; p = 0.01). As reported in other studies, PP occurred in our 27 PP patients more often in females (17 patients; 63%) than in males (10 patients; 37%). Male patients presented with higher serum prolactin concentrations and larger tumor size at the time of primary diagnosis when compared with female patients (p = 0.024; p = 0.046) (Table 1). Fifteen patients (55%) presented with headache, which was the most frequent symptom at diagnosis.
Seven of the 27 analyzed PP patients (26%) received resection surgery. In four patients (15%), incomplete resection was achieved; in three cases (11%), surgery resulted in complete resection as confirmed by normalization of prolactin and by postoperative MRI. The decision on surgical treatment appears to be related to tumor size, meaning surgery was performed more frequently in patients with macroadenoma and giant adenoma (Table 2). All seven patients, who were treated by surgery, received immediate dopamine agonist medication at the time of prolactinoma diagnosis. In three cases with severe threat to vision, the immediate medication did not result in visual improvement and tumor response as measured by tumor volume and prolactin serum concentration, so that after short-term treatment with dopamine agonists (for 2-, 3-, and 3-week duration) upfront surgery was performed. After resection, visual function stabilized in all cases. In the other four of seven patients, who underwent surgery, dopamine agonist medication was performed for 3, 6, 9, and 10 months without significant response in two patients after 3 and 6 months treatment. In two patients, the residual tumor was surgically removed after 9 and 10 months of efficient dopamine agonist medication. After resection, tumor progressions or relapses were observed in four cases (57%). Two patients needed subsequent surgery after tumor progression.
Irradiation was not performed in our cohort. Treatment with a dopamine agonist was documented in 22 PP (81%) and started at a median interval of 11 days after diagnosis (range 1–42 days). In 18 of these cases (82%), cabergoline was used, with a median initial dose of 0.5 mg (range 0.25–3.0 mg) per week. Bromocriptine was applied in four (18%) and quinagolide in two (11%) of all dopamine agonist treatment cases. The dosage was adjusted throughout treatment according to changes in individual patient’s tumor size and prolactin serum levels.
Side effects were documented in 25% of all cases, which included hypotension (14%) and headache (11%) as the most frequently noted side effects. Obesity in one patient and psychotic symptoms in another were detected in two patients under bromocriptine medication. Thirty-seven percent of all PP patients did not experience any side effects. A change of medication was performed for four patients (14%) due to inefficacy of treatment, i.e., lack of tumor shrinkage and persistent elevated prolactin serum concentrations (n = 2) after 6 months median duration of dopamine agonist therapy (range 4–8 months) or side effects manifesting during early course of medication (n = 2; hypotension, psychotic symptoms). In one patient with a macroadenoma and two patients with giant adenoma, medication was changed from cabergoline to quinagolide, whereas the medication for one patient with giant adenoma was changed from bromocriptine to cabergoline. In the majority of patients (67%), the type of medication was not changed during therapy.
During treatment with dopamine agonists, prolactin levels decreased over time. Baseline levels of serum prolactin at the time of diagnosis (n = 22; median 494 ng/ml) rapidly decreased (p = 0.002) to 175.9 ng/ml (n = 14) about 1 to 2 weeks after initiation of treatment. During follow-up with an interval of 1 to 2 months after diagnosis, the median serum prolactin concentration was 80 ng/ml (n = 17; p = 0.000), decreasing further on to a median concentration of 13.2 ng/ml (n = 11; p = 0.003) after more than 1 year of treatment (Fig. 1a). The median tumor size at the time of diagnosis was 2345.1 mm3 and, similar to prolactin levels, decreased over time in response to dopamine agonist treatment. Two to six months after diagnosis, median tumor size was 780 mm3 (n = 15; p = 0.001), decreasing further to a median size of 282.3 mm3 (n = 14; p = 0.001) after 1 year of treatment (Fig. 1b).

Monitoring of serum prolactin concentrations (a) and tumor size (b) at the time of diagnosis and during follow-up treatment with dopamine agonist or after surgery in patients with pediatric prolactinoma recruited in the sellar mass trials HIT-Endo and KRANIOPHARYNGEOM 2000/2007. The horizontal line in the middle of the boxes depicts the median. The edges of the boxes mark the 25th and 75th percentiles. Whiskers indicate the range of values that fall within 1.5 box-lengths
No deaths were recorded during follow-up. Overall survival (OS) rates were not impaired in PP (3 years, OS 1.0). Functional capacity as measured by FMH ability scale was not impaired in 14 of 27 PP patients who answered the FMH questionnaire, as compared with other sellar mass patients (174 craniopharyngioma, 27 hypothalamic glioma, 14 optic/chiasma glioma, 11 germinoma, 9 cyst of Rathke pouch) (Fig. 2).

Functional capacity as measured by German ability scale Fertigkeitenskala-Münster-Heidelberg (FMH) in pediatric patients recruited with prolactinoma, craniopharyngioma, hypothalamic glioma, optic/chiasma glioma, germinoma, and cyst of Rathke pouch in the sellar mass trials HIT-Endo and KRANIOPHARYNGEOM 2000/2007
Discussion
We observed a higher rate of PP in girls (as in adults: ratio ≈ 2:1), and earlier onset, larger tumor volume, and higher prolactin serum levels in boys. Accordingly, our results support earlier studies showing these gender-related differences in PP [1, 10, 12, 19]. We speculate that this could be a result of different physiology in hormonal balance in boys and girls. At the time of diagnosis, our PP patients were all in puberty, supporting the speculation that pubertal modifications in hormonal homeostasis might promote tumor growth in PP. Rare cases of younger children have been reported [7].
Median duration of history before diagnosis was 12 months with a wide range from 2 to 48 months. The long period between disease onset and PP patients seeking medical support corresponds with the benign character and low proliferation index in prolactinoma. In our pediatric cohort, no hydrocephalus was observed at the time of initial diagnosis, which distinguishes PP from other sellar tumors, such as craniopharyngioma [23, 24]. Visual impairment at the time of diagnosis was an important predictor for the necessity of tumor mass-reduction resection. If visual impairment was present, surgery was performed in 50% of cases, compared to 12% of cases with non-impaired vision at the time of diagnosis. In most cases, a combination of clinical symptoms, hyperprolactinemia, and typical MRI findings confirmed the diagnosis. This diagnostic approach is appropriate and clinicians should measure initial prolactin serum levels in all cases of sellar masses, as this can prevent unnecessary invasive interventions. In some PP, histological investigation was necessary to confirm the diagnosis, and in rare cases surgery was performed despite diagnostic confirmation in order to rapidly release compression on the optic chiasm.
Since 20 of the 27 analyzed PP patients did not need any surgical intervention, we emphasize the use of efficient dopamine agonist medication for clinical improvement, normalization of prolactin levels, and tumor shrinkage [6, 8, 27, 30]. This is also suggested by Catli et al. [3], who point out that medical treatment should be the first-line treatment option in both microadenoma and macroadenoma. We support this approach, as we observed in our cohort that tumor mass in most PP patients could be reduced by dopamine agonist treatment within months. Furthermore, prolactin levels decreased rapidly in a matter of weeks. In addition, with dopamine agonist medication, a preservation of the pituitary and visual function is made possible in many cases [1, 7, 22].
Dopamine agonist medication was well tolerated and drug-induced side effects occurred only in rare cases. As medication is frequently needed for a long time period [9, 16, 17] side effects should be taken seriously. PP patients under prolonged high-dose cabergoline medication should be monitored by echocardiography for assessment of potential cardiac side effects (valvular abnormalities) [13, 15, 20, 29, 31]. In our cohort, we report only one case in which medication could be successfully discontinued without reoccurrence of symptoms. In our PP patients, throughout therapy the dosage of medication was adjusted according to reductions in prolactin levels and tumor sizes.
Earlier studies showed that cabergoline is more efficient than bromocriptine, while causing less and milder side effects [32]. This difference was not detectable in our study. However, the low number of patients in our study reduces a predictive value in this context [2, 32]. A novel finding in our cohort was that, during therapy with bromocriptine, psychotic symptoms were observed in a child. This side effect is seldom seen in puerperal use and the connection to the child’s bromocriptine therapy cannot be verified.
Our results support earlier suggestions that surgery should be performed only if an urgent decompression of the optic chiasm is required for conservation of threatened visual function [3]. After surgery, progression or relapse of the tumor occurs in most cases [16]. Radiotherapy was not used in our PP patients. In adult prolactinoma patients, irradiation is used for treatment of medically and surgically refractory tumors. However, side effects of irradiation such as neurological sequelae, hypopituitarism, and increased risk for second malignancies make radiotherapy less appropriate for treatment of children [5]. Overall survival rates and functional capacity as a measure of quality of survival were not impaired in PP patients, indicating that patients with this rare disease have an optimistic prognosis.
Severe deterioration of visual function was not observed after surgery and during dopamine agonist medication. However, comparison of visual outcome after surgery vs. dopamine agonist therapy is difficult due to the fact that resection was mainly performed because of imminent threat to vision, which was absent or at least less severe in most patients treated by dopamine agonist medication alone.
Retrospective analysis and the variability of documentation in the medical records of our patients are limitations of our study. We analyzed a small cohort of patients. However, as prolactinomas occur very rarely in childhood, PP studies are lacking and the number of patients is limited due to rareness of disease. Therefore, a clinical study such as this that includes outcome data can bring a unique, updated perspective especially with regard to prognosis of PP.
We conclude that PP should be considered as a disease with specific requirements for treatment. Dopamine agonist medication as a first choice of treatment is efficient in PP. In contrast to adult prolactinoma, irradiation is not recommended in PP due to risks of long-term sequelae such as second malignancies. We would favor to initiate pharmaceutical treatment immediately at the time of initial prolactinoma diagnosis especially in case of severe visual impairment. If there is no short-term response in terms of visual improvement and prolactinoma serum concentrations, early surgical treatment should be considered in case of further visual deterioration and/or lab/imaging signs of progression. Risk-adapted guidelines for treatment of PP patients with these requirements in mind need to be developed. Overall survival rates and functional capacity as a measure of quality of survival indicate that PP has an optimistic prognosis.
Abbreviations
- MRI:
-
Magnetic resonance imaging
- CT:
-
Computed tomography
- FMH:
-
Fertigkeitenskala-Münster-Heidelberg
- OS:
-
Overall survival
References
Acharya SV, Gopal RA, Bandgar TR, Joshi SR, Menon PS, Shah NS (2009) Clinical profile and long term follow up of children and adolescents with prolactinomas. Pituitary 12:186–189
Atmaca M, Korkmaz S, Ustundag B, Ozkan Y (2015) Increased serum prolactin in borderline personality disorder. Int J Psychiatry Med 49:169–175
Catli G, Abaci A, Altincik A, Demir K, Can S, Buyukgebiz A, Bober E (2012) Hyperprolactinemia in children: clinical features and long-term results. J Pediatr Endocrinol Metab 25:1123–1128
Ciccarelli A, Daly AF, Beckers A (2005) The epidemiology of prolactinomas. Pituitary 8:3–6
Cohen-Inbar O, Xu Z, Schlesinger D, Vance ML, Sheehan JP (2015) Gamma knife radiosurgery for medically and surgically refractory prolactinomas: long-term results. Pituitary 18:820–830
Colao A, Di Sarno A, Landi ML, Scavuzzo F, Cappabianca P, Pivonello R, Volpe R, Di Salle F, Cirillo S, Annunziato L, Lombardi G (2000) Macroprolactinoma shrinkage during cabergoline treatment is greater in naive patients than in patients pretreated with other dopamine agonists: a prospective study in 110 patients. J Clin Endocrinol Metab 85:2247–2252
Colao A, Loche S, Cappa M, Di Sarno A, Landi ML, Sarnacchiaro F, Facciolli G, Lombardi G (1998) Prolactinomas in children and adolescents. Clinical presentation and long-term follow-up. J Clin Endocrinol Metab 83:2777–2780
Colao A, Vitale G, Cappabianca P, Briganti F, Ciccarelli A, De Rosa M, Zarrilli S, Lombardi G (2004) Outcome of cabergoline treatment in men with prolactinoma: effects of a 24-month treatment on prolactin levels, tumor mass, recovery of pituitary function, and semen analysis. J Clin Endocrinol Metab 89:1704–1711
Dekkers OM, Lagro J, Burman P, Jorgensen JO, Romijn JA, Pereira AM (2010) Recurrence of hyperprolactinemia after withdrawal of dopamine agonists: systematic review and meta-analysis. J Clin Endocrinol Metab 95:43–51
Eren E, Yapici S, Cakir ED, Ceylan LA, Saglam H, Tarim O (2011) Clinical course of hyperprolactinemia in children and adolescents: a review of 21 cases. J Clin Res Pediatr Endocrinol 3:65–69
Fideleff HL, Boquete HR, Suarez MG, Azaretzky M (2009) Prolactinoma in children and adolescents. Horm Res 72:197–205
Guaraldi F, Storr HL, Ghizzoni L, Ghigo E, Savage MO (2014) Paediatric pituitary adenomas: a decade of change. Horm Res Paediatr 81:145–155
Herring N, Szmigielski C, Becher H, Karavitaki N, Wass JA (2009) Valvular heart disease and the use of cabergoline for the treatment of prolactinoma. Clin Endocrinol 70:104–108
Jane JA Jr, Laws ER Jr (2000) Surgical treatment of pituitary adenomas. In: De Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, Koch C, McLachlan R, New M, Rebar R, Singer F, Vinik A, Weickert MO (eds) Endotext. MDText.com, Inc., South Dartmouth
Kars M, Delgado V, Holman ER, Feelders RA, Smit JW, Romijn JA, Bax JJ, Pereira AM (2008) Aortic valve calcification and mild tricuspid regurgitation but no clinical heart disease after 8 years of dopamine agonist therapy for prolactinoma. J Clin Endocrinol Metab 93:3348–3356
Kharlip J, Salvatori R, Yenokyan G, Wand GS (2009) Recurrence of hyperprolactinemia after withdrawal of long-term cabergoline therapy. J Clin Endocrinol Metab 94:2428–2436
Klibanski A (2009) Dopamine agonist therapy in prolactinomas: when can treatment be discontinued? J Clin Endocrinol Metab 94:2247–2249
Kreutzer J, Buslei R, Wallaschofski H, Hofmann B, Nimsky C, Fahlbusch R, Buchfelder M (2008) Operative treatment of prolactinomas: indications and results in a current consecutive series of 212 patients. Eur J Endocrinol 158:11–18
Kunwar S, Wilson CB (1999) Pediatric pituitary adenomas. J Clin Endocrinol Metab 84:4385–4389
Lancellotti P, Livadariu E, Markov M, Daly AF, Burlacu MC, Betea D, Pierard L, Beckers A (2008) Cabergoline and the risk of valvular lesions in endocrine disease. Eur J Endocrinol 159:1–5
Maiter D, Delgrange E (2014) Therapy of endocrine disease: the challenges in managing giant prolactinomas. Eur J Endocrinol 170:R213–R227
Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, Wass JA (2011) Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 96:273–288
Muller HL (2013) Childhood craniopharyngioma. Pituitary 16:56–67
Muller HL (2014) Craniopharyngioma. Endocr Rev 35(3):513–543
Muller HL, Bueb K, Bartels U, Roth C, Harz K, Graf N, Korinthenberg R, Bettendorf M, Kuhl J, Gutjahr P, Sorensen N, Calaminus G (2001) Obesity after childhood craniopharyngioma—German multicenter study on pre-operative risk factors and quality of life. Klin Padiatr 213:244–249
Muller HL, Gebhardt U, Faldum A, Emser A, Etavard-Gorris N, Kolb R, Sorensen N (2005) Functional capacity and body mass index in patients with sellar masses—cross-sectional study on 403 patients diagnosed during childhood and adolescence. Childs Nerv Syst 21:539–545
Ono M, Miki N, Kawamata T, Makino R, Amano K, Seki T, Kubo O, Hori T, Takano K (2008) Prospective study of high-dose cabergoline treatment of prolactinomas in 150 patients. J Clin Endocrinol Metab 93:4721–4727
Sauder SE, Frager M, Case GD, Kelch RP, Marshall JC (1984) Abnormal patterns of pulsatile luteinizing hormone secretion in women with hyperprolactinemia and amenorrhea: responses to bromocriptine. J Clin Endocrinol Metab 59:941–948
Vallette S, Serri K, Rivera J, Santagata P, Delorme S, Garfield N, Kahtani N, Beauregard H, Aris-Jilwan N, Houde G, Serri O (2009) Long-term cabergoline therapy is not associated with valvular heart disease in patients with prolactinomas. Pituitary 12:153–157
Verhelst J, Abs R, Maiter D, van den Bruel A, Vandeweghe M, Velkeniers B, Mockel J, Lamberigts G, Petrossians P, Coremans P, Mahler C, Stevenaert A, Verlooy J, Raftopoulos C, Beckers A (1999) Cabergoline in the treatment of hyperprolactinemia: a study in 455 patients. J Clin Endocrinol Metab 84:2518–2522
Wakil A, Rigby AS, Clark AL, Kallvikbacka-Bennett A, Atkin SL (2008) Low dose cabergoline for hyperprolactinaemia is not associated with clinically significant valvular heart disease. Eur J Endocrinol 159:R11–R14
Wang AT, Mullan RJ, Lane MA, Hazem A, Prasad C, Gathaiya NW, Fernandez-Balsells MM, Bagatto A, Coto-Yglesias F, Carey J, Elraiyah TA, Erwin PJ, Gandhi GY, Montori VM, Murad MH (2012) Treatment of hyperprolactinemia: a systematic review and meta-analysis. Syst Rev 1:33
Winters SJ, Troen P (1984) Altered pulsatile secretion of luteinizing hormone in hypogonadal men with hyperprolactinaemia. Clin Endocrinol 21:257–263
Wolff JE, Daumling E, Dirksen A, Dabrock A, Hartmann M, Jurgens H (1996) Munster Heidelberg abilities scale—a measuring instrument for global comparison of illness sequelae. Klin Padiatr 208:294–298
Acknowledgements
We are grateful for the help of Margarita Neff-Heinrich, Göttingen, Germany, in proofreading and editing the manuscript.
Funding
This study was funded by the German Childhood Cancer Foundation, Bonn, Germany (grant DKS 2014.13). The authors have no financial relationship with the organization that sponsored the research.
Author information
Authors and Affiliations
Contributions
Hoffmann A: Dr. Hoffmann has written and reviewed the manuscript and supervised data acquisition and analyses. She is the study assistant of the German Registry.
Adelmann S: Mrs. Adelmann analyzed retrospective data, and participated in writing and reviewing the manuscript.
Lohle K: Mrs. Lohle performed the statistical analyses, created the figures and tables, and participated in writing and reviewing the manuscript.
Claviez A: Dr. Claviez initiated the study, contributed patients, and participated in analyzing data and writing and reviewing the manuscript.
Müller HL: Dr. Müller is chairman of the German Registry. He supervised data collection, analyses, and participated in writing and review of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Hermann L. Müller and coauthors declare that they have no conflict of interest.
This manuscript was composed in the absence of any commercial or financial relationships that could be perceived as a potential conflict of interest.
Ethical approval
All procedures performed in KRANIOPHARYNGEOM 2000—NCT00258453 and KRANIOPHARYNGEOM 2007—NCT01272622 involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the studies in KRANIOPHARYNGEOM 2000—NCT00258453 and KRANIOPHARYNGEOM 2007—NCT01272622.
Trial registration: KRANIOPHARYNGEOM 2000/2007 (NCT00258453; NCT01272622)
Additional information
Communicated by Peter de Winter
Rights and permissions
About this article

Cite this article
Hoffmann, A., Adelmann, S., Lohle, K. et al. Pediatric prolactinoma: initial presentation, treatment, and long-term prognosis. Eur J Pediatr 177, 125–132 (2018). https://doi.org/10.1007/s00431-017-3042-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-017-3042-5