
Abstract
Main conclusion
Co-expression networks based on transcriptomes of Populus trichocarpa major tissues and specific cell types suggest redundant control of cell wall component biosynthetic genes by transcription factors in wood formation.
We analyzed the transcriptomes of five tissues (xylem, phloem, shoot, leaf, and root) and two wood forming cell types (fiber and vessel) of Populus trichocarpa to assemble gene co-expression subnetworks associated with wood formation. We identified 165 transcription factors (TFs) that showed xylem-, fiber-, and vessel-specific expression. Of these 165 TFs, 101 co-expressed (correlation coefficient, r > 0.7) with the 45 secondary cell wall cellulose, hemicellulose, and lignin biosynthetic genes. Each cell wall component gene co-expressed on average with 34 TFs, suggesting redundant control of the cell wall component gene expression. Co-expression analysis showed that the 101 TFs and the 45 cell wall component genes each has two distinct groups (groups 1 and 2), based on their co-expression patterns. The group 1 TFs (44 members) are predominantly xylem and fiber specific, and are all highly positively co-expressed with the group 1 cell wall component genes (30 members), suggesting their roles as major wood formation regulators. Group 1 TFs include a lateral organ boundary domain gene (LBD) that has the highest number of positively correlated cell wall component genes (36) and TFs (47). The group 2 TFs have 57 members, including 14 vessel-specific TFs, and are generally less correlated with the cell wall component genes. An exception is a vessel-specific basic helix-loop-helix (bHLH) gene that negatively correlates with 20 cell wall component genes, and may function as a key transcriptional suppressor. The co-expression networks revealed here suggest a well-structured transcriptional homeostasis for cell wall component biosynthesis during wood formation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wood is one of the most abundant biological materials on the Earth (Carlowicz 2012). It has played an essential role during the history of human civilization as fuel and raw material for buildings and tools (Perlin 2005). Wood is still used directly for heating and cooking by a large fraction of the world’s population (Rehfuess et al. 2006). Although fossil fuels have dominated our energy-based industrial economy in recent centuries, burning nonrenewable fossil fuels has led to significant CO2 accumulation in the atmosphere and contributed to global warming (Vitousek et al. 1997). Wood can be a renewable feedstock for pulp/paper and biofuels and a key ecosystem carbon sink; therefore, wood production plays important industrial and environmental roles in addition to its use as a raw material (Sarkanen 1976; Chiang 2002; Ragauskas et al. 2006).
Wood in angiosperm tree species (hardwoods), such as poplars, is derived from the vascular cambium, which divides and differentiates into secondary xylem (Esau 1965; Larson 1994; Evert 2006). Differentiating secondary xylem cells mature into long thick-walled fibers, thin walled vessel elements, and isodiametric ray cells with highly active biosynthesis and deposition of the major cell wall components, cellulose, hemicelluloses, and lignin (Sarkanen 1976; Evert 2006). The secondary cell walls of the xylem cell types are the major constituents in wood. The density and strength of wood depend on considerable part on the volume occupied by fibers and the thickness of the fiber secondary cell walls. Delignified fiber cells are the major source of material for the production of pulp and paper (Sarkanen 1976; Chiang 2002; Ragauskas et al. 2006).
Wood formation is controlled by hierarchical transcription factor networks that regulate specific pathway genes in cell wall component biosynthesis (Li et al. 2012; Lin et al. 2013; Lu et al. 2013; Zhong and Ye 2014). An understanding of the networks of regulatory genes in wood formation is important for improving feedstock sustainability and productivity. However, genetic network analysis by transgenics in woody plants is challenging due to the lack of efficient gene transformation and the absence of mutant collections for tree species (Merkle and Dean 2000; Song et al. 2006; Liu et al. 2014a, b).
Gene expression profiling aids in the identification and evaluation of gene function. Transcript profiling reveals hierarchical structure of gene regulation via the construction of gene expression correlation networks (D’haeseleer et al. 2000; Stuart et al. 2003). Brady et al. (2007) constructed a high-resolution root spatiotemporal gene expression map for Arabidopsis root development, based on microarray data of all root cell types isolated by cell sorting. Taylor-Teeples et al. (2015) identified 50 transcription factor (TF) genes for xylem cell specification, and combined them with genes for cell wall component biosynthesis to construct a regulatory network for secondary cell wall biosynthesis in roots of Arabidopsis.
Microarray data have been used to build co-expression networks for poplars (Yang et al. 2011; Cai et al. 2014; Netotea et al. 2014). Comparative analysis of Arabidopsis and poplars has identified many functional gene homologs for cell wall component biosynthesis. Subnetworks or modules of genes associated with cell wall biosynthesis, including TF genes, were defined based on GO ontology (Yang et al. 2011; Cai et al. 2014). Netotea et al. (2014) inferred co-expression networks by comparison of poplar with Arabidopsis and rice to identify orthologs with conserved regulation among these species. Street et al. (2011) identified transcriptional modules in leaves of poplar. However, the study of transcriptional regulatory networks for stem differentiating xylem (SDX) (i.e., wood formation tissue) is still limited.
To carry out a systematic study of stem wood formation in P. trichocarpa, we first constructed RNA-seq libraries for five major tissues: SDX and four nonwoody tissues (shoot, phloem, leaves, and primary roots). We also studied the transcriptomes of specific cell types in SDX isolated by laser capture microdissection (LCM) (Chen et al. 2014a, b) and constructed RNA-seq libraries of the fiber and vessel cell types (Wang et al. 2015). These resources provide an opportunity to investigate gene correlation networks for wood formation at the cell-type level. In this report, we analyzed the transcriptomes of tissues and wood forming cell types in P. trichocarpa. We used these transcriptomic data to construct a co-expression network and subnetworks of cell wall component genes and associated TF genes. This study not only provides a comprehensive view of a wood formation associated regulatory network, but also provides new insights into fiber cell and vessel element differentiation and development.
Materials and methods
Plant material and RNA-seq library construction
Populus trichocarpa Nisqually-1 plants were grown in a greenhouse according to Li et al. (2011). Soil composition was 1/2 Miracle-Gro Soil (Scotts Miracle-Gro products, Maysville, OH, USA) and 1/2 Metro-Mix 200 (Sun Gro, Bellevue, WA, USA). The greenhouse was set to a 16-h light/8-h dark cycle with supplemental light of ~300 µE m−2 s−1, and the temperature was held between 17 and 26 °C (Li et al. 2011).
Tissue samples were collected from 1-year-old trees. Shoot tip samples were obtained from the first to third internode, leaf samples were fully expanded young leaves, phloem was scraped from the inner part of the bark, and xylem samples were stem secondary differentiating xylem collected by scraping the surface of debarked stems as we did previously (Shi et al. 2010). Root samples were primary roots collected from young seedlings rooted in water. Total RNA was isolated using the Qiagen RNeasy Plant RNA isolation kit (Qiagen, Valencia, CA) as described in Shi et al. (2010, 2013). Samples containing fiber cells, vessel cells, or a mixture of three different cell types (fibers, vessels, and rays) were collected from 6-month-old greenhouse grown P. trichocarpa by LCM using an LMD7000 microscope system (Leica, Wetzlar, Germany) (Chen et al. 2014a, b; Wang et al. 2015). The quality of the RNA was estimated using an Agilent 2100 Bioanalyzer and RNA 6000 Pico Assay chips (Agilent, Santa Clara, CA). One microgram total RNA from collected samples was used for the RNA-seq library construction following the Illumina TruSeq RNA Sample Prep v2 LS protocol (Illumina, San Diego, CA).
Gene annotation
Monolignol genes were annotated as we did previously (Shi et al. 2010). Two xylem-specific caffeoyl shikimate esterase (CSE) genes were also added as monolignol genes (Vanholme et al. 2013; Wang et al. 2015). Cellulose and hemicellulose biosynthetic genes were annotated based on the update of the nomenclature for the cellulose synthase genes in poplars (Kumar et al. 2009) and on the previously report of Suzuki et al. (2006). TF annotation is based on PlantTFDB (http://planttfdb.cbi.pku.edu.cn/index.php?sp=Pth), a plant transcription factor database.
RNA-seq data processing and analysis
Multiple RNA-seq libraries with unique tags were pooled at equal concentrations for multiplex sequencing. Libraries were assayed for quality and quantity (NCSU Genomic Sciences Laboratory, GSL) and sequenced at the GSL (NCSU) or by the Beijing Genomics Institute (BGI) on an Illumina HiSeq 2000. The resulting sequences (100 nt) were analyzed following our previous pipeline (Li et al. 2013; Lin et al. 2013; Lu et al. 2013; Chen et al. 2014a, b). The RNA-seq sequences of each library were mapped to the P. trichocarpa genome v3.0 (http://www.phytozome.org) using TOPHAT (Trapnell et al. 2009). The frequency of raw counts was determined by BEDtools (Quinlan and Hall 2010) for all annotated genes. Genes with at least two counts-per-million (cpm) in at least three samples were retained and normalized using the trimmed mean of M value (TMM) (Robinson and Oshlack 2010). Differential expressed genes (DEGs) were identified based on the transcript fold change between different samples and an appropriate false discovery rate (FDR) using the edgeR package (Chen et al. 2014a, b). G:Profiler Web server (http://biit.cs.ut.ee/gprofiler; Reimand et al. 2011) was used for the GO analysis. The GO functional enrichment is based on v2.0 annotation for P. trichocarpa genes. The statistical significance of functional enrichment was calculated for selected genes using known P. trichocarpa genes as background (Lin et al. 2013).
Network construction and visualization
A matrix of all correlated gene expression data was constructed by calculating pairwise Pearson correlation coefficients (PCC) based on log2 transformed normalized expression data across all samples. The co-expression measurement was calculated using the basic Pearson correlation inference option of cyni Toolbox (Guitart-Pla et al. 2015), a plugin app in the Cytoscape platform (version 3.2.1; Shannon et al. 2003). The threshold for new edges was set to an absolute value of 0.7. The associated gene network in the form of a matrix was visualized in Cytoscape following yFiles Organic layout, which uses a spring-embedded algorithm to show the clustered structure for a network in graph form (Cline et al. 2007). The absolute value of the PCC between nodes was used as the weight parameter in layout analysis if required.
Cluster analysis
The Markov cluster algorithm (MCL) is a fast and scalable unsupervised cluster algorithm for network construction based on simulation of stochastic flow (Van Dongen and Abreu-Goodger 2012). MCL analysis was conducted to dissect the network into different correlated subnetworks using clusterMaker, a Cytoscape plugin that integrated the MCL option under default parameters. Hierarchical cluster analyses were also conducted using “hclust”, an R script for grouping co-expressed genes based on the correlation coefficients.
Results
Transcriptomes from different tissues and xylem cell types
To study gene regulation in wood formation, we collected RNA from different tissues and cell types of wood forming tissue of P. trichocarpa. The shoot tissue (S) samples included shoot tips from the first to the third internode, fully expanded young leaves (L), stem differentiating phloem (P), and stem differentiating xylem (SDX, also abbreviated as xylem, or X) from 1-year-old greenhouse grown plants. Primary roots (R) were collected from cuttings grown hydroponically. Cell-type samples included stem differentiating fiber cells (F), vessel cells (V), and a combination of fiber, vessel, and ray cells, which we named as “three-cell-types” (T). All the cell-type samples were collected from the stem differentiating xylem (within 16 cell layers from the cambium) using LCM as reported previously (Chen et al. 2014a, b; Wang et al. 2015).
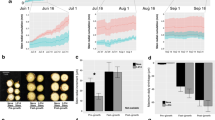
Total RNAs from these tissues and cell-type samples (three biological replicates) were extracted. While the RNAs from different tissues were used directly for RNA-seq, the RNAs of the cell-type samples were amplified to ensure that the amounts of RNA were sufficient for sequencing. Twenty-four RNA-seq libraries for these samples were constructed and sequenced, yielding 6.3–20 million reads per library (Fig. 1a) (GEO accession number: GSE81077) with 80.2–92.1% of the sequences mapping to the P. trichocarpa genome (v3.0) (Supplementary Table S1). The depth and coverage of these RNA-seq libraries are typical (Liu et al. 2014a, b) and consistent with our previous results (Lin et al. 2013; Lu et al. 2013; Shi et al. 2013).

RNA-seq analyses of S, L, P, R, X, V, F, and T libraries of shoot tip, leaf, phloem, root, xylem, vessel cell, fiber cell, and three-cell-type samples. a Number of raw sequence reads (blue) and mapped reads (orange) to the P. trichocarpa genome v3.0 for each RNA-seq library. b Scatter plots of replicated pairs of libraries for the same tissue or cell type. The numbers are Pearson correlation coefficients (r). c Multidimensional scaling plot for the relationship of each library based on distance calculated as the leading log fold change for biological variation between libraries (Chen et al. 2014a, b). Dimension 1 separates tissue or cell type, and dimension 2 roughly corresponds to individual libraries
Biological replicates of the tissue and cell-type mapped reads were compared by pairwise correlation (Fig. 1b). The Pearson coefficients (r) of biological replicates for the tissue samples of xylem, shoot, phloem, leaf, and root range from 0.97 to 0.98 (±<0.008) (Fig. 1b). The coefficients between replicates of the same cell types are 0.85 ± 0.018, 0.73 ± 0.026, and 0.94 ± 0.007 for fibers, vessels, and mixtures of the three-cell-type libraries, respectively (Fig. 1b). The variation within the cell-type samples is greater than that of tissue samples, most likely due to amplification of the RNAs before library preparation.
Relationships of all library sequences to each other were evaluated using a multidimensional plot (Fig. 1c) generated using the plot2MDS function in the Limma R package (Chen et al. 2014a, b). This plot illustrates the biological variation between RNA samples (libraries). The replicates of the same tissue or cell-type group together and libraries from different tissues or cell types are separated. Therefore, differences in gene expression between different tissues are greater than those of replicates of the same tissue, suggesting that the differential expressed genes (DEGs) between tissues can be clearly resolved (McCarthy et al. 2012) (Fig. 1c).
Identification of xylem-specific gene transcripts associated with cell wall component biosynthesis in wood formation
To identify genes with specific expression in wood formation, we first screened our transcriptomes for genes showing tissue specificity. We defined DEGs where the relative abundance of transcript is at least twofold greater than that in any other tissues, and the false discovery rate (FDR) is below 0.05 (Zheng and Moriyama 2013). There were 2206, 1208, 834, 3200, and 4086 genes specifically expressed in xylem (X), phloem (P), shoot tip (S), leaf (L), and root (R), respectively (Table 1). Expression of 26,953 genes was not tissue specific; their transcripts are similar in abundance among multiple tissues. 2848 previously annotated gene transcripts were not detected in any of our RNA-seq libraries.
Xylem tissue is specialized for wood formation; therefore, many genes associated with wood formation should be more active in xylem than in other tissues. We can identify candidate genes for cell wall component biosynthesis or other genes associated with wood formation, as we did to identify the monolignol pathway genes, which represent lignin production in P. trichocarpa (Shi et al. 2010). All core monolignol pathway genes identified by Shi et al. (2010) were also identified here.
One recently identified gene encoding caffeoyl shikimate esterase (CSE) is expressed in Arabidopsis at high levels (Vanholme et al. 2013). Two CSE homologs were detected in P. trichocarpa as xylem specific but show relatively low transcript levels (Fig. 2a). Two PAL family members, PAL1 and PAL3, are abundant but are not xylem specific (Fig. 2a; Table 1). We added PAL1 and PAL3 to the 20 xylem specific (Table 1) monolignol gene list (for a total of 22), because these genes are likely to contribute to total PAL activity and monolignol biosynthesis in wood forming tissue (Shi et al. 2013) (Fig. 2a). All other monolignol genes are highly specific in xylem (Shi et al. 2010). Analysis of candidate genes for the biosynthesis of cellulose and hemicelluloses in wood formation identified 5 cellulose synthase (CesA) genes and 18 genes for hemicellulose biosynthesis that are xylem-specific DEGs (Fig. 2b). Our gene list (45 genes) contains 22, 5, and 18 genes for the biosynthesis of monolignols, cellulose, and hemicelluloses, the three major components in secondary cell wall biosynthesis (Supplementary Table S2).

Relative transcript abundance of xylem-specific genes for cell wall component biosynthesis in SDX (X). a Xylem-specific gene expression for monolignol biosynthesis. b Xylem-specific gene expression for biosynthesis of cellulose and hemicelluloses. The scale of transcript abundance on the y-axis of the figures is the normalized counts-per-million (cpm). Height of each bar represents the average value of transcript abundance in three libraries of the same tissue
Identification of fiber and vessel cell-type specific genes
Cell wall component gene families often contain multiple members. To determine the specific functions of the family members or the level of redundancy in the gene family, it is important to know if they are expressed in the same cell types during wood formation. We have used LCM to isolate specific cells from SDX followed by the RNA-seq analysis to determine whether expression of all cell wall component genes occurs in the same cell types. Expression of all 45 genes on our cell wall component gene list was detected in both fiber and vessel cell types, but some at substantially different levels (Supplementary Table S2).
We identified fiber cell-specific DEGs using a fiber over vessel (F/V) abundance ratio greater than 2 and an FDR <0.4. A high FDR was adopted as recommended by Zheng and Moriyama (2013), because the cell-type samples had high variation between replicates (Fig. 1b). 565 fiber-specific DEGs, including 17 cell wall component genes (Fig. 3a) and 32 TF genes, were identified (Fig. 3b). Similarly, we identified 930 vessel-specific DEGs using a vessel over fiber cell (V/F) abundance ratio greater than 2 and FDR <0.4 (Table 2). These 930 DEGs did not include any cell wall component genes, but included 43 TF genes (Table 2; Fig. 3c; Supplementary Table S2).

RNA-seq-based transcript abundances for cell-type specific cell wall component biosynthetic genes and TFs. a Fiber cell-specific cell wall component biosynthetic genes. b Fiber cell-specific TFs. c Vessel cell-specific TFs. Y-axis values are normalized transcript abundances as counts-per-million (cpm). Expression in fiber cells is represented by green color and vessel expression is represented by red color. Gene names or gene family names are listed on the x-axis as abbreviations
Gene ontology (GO) functional enrichment analysis for cell-type-specific genes
GO analysis was used to explore the functional significance of fiber or vessel cell-type specific DEGs with g:Profiler (http://biit.cs.ut.ee/gprofiler). 27 GO terms for fiber cell-specific DEGs were enriched in seven major GO hierarchical classes, where the cell wall-related class is the largest. Vessel cell-specific DEGs have 84 GO terms in ten major GO hierarchical classes, the two largest classes were biological processes (BP), cellular components (CC), and molecular function (MF). Cell wall-related functions, especially hemicellulose associated GO terms, are more significantly enriched in fibers compared to vessels. GO terms for glycoside hydrolases are enriched in vessels and have a putative function to degrade cellulose. Glucuronosyltransferase and PAL activities are enriched in fiber cells (Supplementary Table S3).
Co-expression network of TFs and cell wall component biosynthetic genes
We next investigated the relationships of the TFs and cell wall component genes for wood formation. There are a total of 165 TFs representing all xylem-specific TFs (109, Table 1) and all cell-type-specific TFs (75, Table 2), including 19 TFs, that are both xylem specific and cell-type specific. We used the 165 TFs along with the 45 cell wall component genes to build a co-expression network for inferring the function of these TFs in wood cell wall component biosynthesis.
The co-expression network was built using a basic correlation function in the Cyni Toolbox (http://www.proteomics.fr/Sysbio/CyniProject) of the Cytoscape platform (Shannon et al. 2003). Pairwise Pearson correlations were calculated based on log transformed transcript abundance data for the selected genes. A threshold of 0.7 for absolute value of positive or negative correlation coefficients (r) was used as a cutoff to remove low correlations. The final co-expression network included 208 genes (nodes) and 3562 correlations (edges) which include 3366 positive correlations and 196 negative correlations (Fig. 4a).

Co-expression networks of xylem- or cell-type-specific enriched TFs and gene of cell wall component biosynthesis. A node represents a gene in a network, and the node shape of round, hexagon, rectangle, and triangle represents genes for monolignols, cellulose, hemicelluloses, and TFs, respectively. Sizes of nodes are relative to level of gene expression in xylem tissue. Nodes with the filling color of orange, dark green, light green, yellow, brown, and grey represent tissue expression specificity of xylem, leaf, shoot, root, and nontissue specific, respectively. Dark green outlined nodes are fiber-specific genes and red are vessel-specific genes. An edge indicates significant co-expression between two connected genes. Edge color in blue illustrates positive correlations and red edges are negative correlations. Thick edges are high absolute value correlation coefficients. a Globel view of all selected genes. b Subnetwork with most cell wall component biosynthetic genes and associated TFs. c Co-expressed genes of a LBD homolog (Potri.013G156200), a xylem-specific TF and a fiber cell-specific TF (see Supplementary Table S4). d Co-expressed genes of a bHLH homolog (Potri.002G143300), a vessel cell-specific gene and a leaf-specific gene (see Supplementary Table S5). e Includes subnetworks that contain fiber and vessel-specific TFs with negative correlations between different cell-type-specific TFs. f Subnetworks mainly contain vessel-specific TFs. g Subnetworks mainly consist of xylem-specific TFs
A global subnetwork, assembled by Cytoscape (Fig. 4a), indicates that most of cell wall component genes group together with xylem-specific TFs or fiber cell-specific TFs. However, these cell wall component genes are not grouped with the majority of the vessel-specific TFs (Fig. 4a). This pattern can be further illustrated by the subnetwork cluster analysis based on the co-expression correlations. For instance, using a MCL cluster algorithm with default parameters, all genes can be clustered into several subnetworks containing relatively highly correlated genes. The major subnetwork contains most of cell wall component genes (41 of total 45), which also clustered with 58 xylem-specific and cell-type-specific TFs (Fig. 4b). Only three monolignol genes (PAL1, PAL3, and HCT6) and one hemicellulose gene (IRX10-L-A1) are not included in this major subnetwork. In this subnetwork, most of the 58 TFs are positively correlated with each other, and with the 41 cell wall component genes. In the same subnetwork, four vessel-specific TFs are negatively correlated with other subnetwork TFs and cell wall component genes (Fig. 4b).
In the major subnetwork (Fig. 4b), a lateral organ boundary domain gene (LBD, Potri.013G156200) has the highest number of positively correlated cell wall component genes—19, 5, and 12 monolignol, cellulose, and hemicellulose biosynthetic genes, respectively (Fig. 4c; Supplementary Table S4). This TF is also positively correlated with 47 xylem- or fiber-specific TFs, and negatively correlated with 5 vessel-specific TFs (Fig. 4c; Supplementary Table S4). A basic helix-loop-helix gene (bHLH, Potri.002G143300), a vessel-specific TF, is negatively correlated with 9, 4, and 7 monolignol, cellulose, and hemicellulose biosynthetic genes, respectively. It is also negatively correlated with 26 xylem- and fiber-specific TFs (Fig. 4d; Supplementary Table S5).
There are three types of minor subnetworks: Type I are those involving correlations between both fiber- and vessel-specific TFs (Fig. 4e). Type II represents correlated vessel-specific TF subnetworks (Fig. 4f). Type III are correlated xylem-specific TF subnetworks (Fig. 4g). TFs in these three types of subnetworks are only correlated with one cell wall component gene, PAL1 (Fig. 4b). Type I subnetworks contain many negative correlations (Fig. 4e), whereas Type II and III subnetworks contain mostly positive correlations (Fig. 4f, g).
Analysis of TFs co-expressed with genes of cell wall component biosynthesis
For more in depth analysis of the relationship between TFs and cell wall component genes, we extracted the TFs that correlated with cell wall component genes from the global subnetwork (Fig. 4a). 101 TFs were extracted and their co-expression correlations with cell wall component genes are presented as hierarchical cluster trees based on the correlation coefficients (Fig. 5; Supplementary Table S6). Of the 101 TFs, 79 are xylem-specific, providing further support for the relevance of the co-expression network with wood formation.

Correlation coefficients between TFs and genes of cell wall component biosynthesis. TFs are listed vertically, and genes associated with cell wall component biosynthesis are listed horizontally. Correlation coefficients are illustrated in blocks. Green blocks represent positive correlations and red for negative correlations, with a scale shown on the left top of the figure. Genes are listed by name, tissue specificity, and cell-type specificity. TFs (T), genes of biosynthesis of lignin in form of monolignols (M), then cellulose (C) and hemicelluloses (H). Five tissue specificities are described for xylem (X), shoot (S), leaf (L), phloem (P), root (R), and nontissue specificity (N), and are shown with a background color of orange (X), light green (S), dark green (L), yellow (P), brown (R), and grey (N), respectively. Cell-type-specific labels for fibers (F) and vessels (V) are in background of dark green and dark red. A hierarchical cluster tree for TFs and cell wall component biosynthetic genes is also presented next to gene information
Although all the 101 TFs are associated with cell wall component genes, only some of them are highly correlated with a specific group of such genes. For example, the hierarchical cluster tree shows two TF gene groups (groups 1 and 2) and two cell wall component gene groups (groups 1 and 2). TF gene group 1 includes 44 TFs, all but one are xylem-specific. Each of these 44 TFs is highly positively co-expressed with all the 30 group 1 cell wall component genes. These 30 cell wall component genes include 14 monolignol genes, all five secondary cell wall CesA genes (Kumar et al. 2009) and 11 hemicellulose genes. The expression of each of these cell wall component genes is highly correlated with the expression of 30–43 TFs in TF gene group 1. Because of the xylem specificity and the large numbers of co-expressed cell wall component genes, these group 1 TF genes are major wood formation regulators. For example, PtrMYB026, PtrMYB090, PtrMYB152, PtrMYB075, PtrMYB128, PtrSND2/3-A1, PtrSND2/3-A2, PtrSND2/3-B1, and PtrSND2/3-B2, which are in TF group 1, have been implicated in their role in wood formation. TF gene group 2 includes 67 TFs and contains all the 14 vessel-specific TFs (Fig. 5). Group 2 cell wall component genes include 8 monolignol genes and 7 hemicellulose genes. No secondary cell wall CesA genes are in this group. Genes in the TF gene group 2 and the cell wall component gene group 2 are less correlated (Fig. 5). Of the 14 vessel-specific TFs, 9 are negatively correlated with some of the group 2 cell wall component genes, suggesting a likely role as transcriptional suppressors for these TFs.
Discussion
Cell wall component biosynthetic genes for wood formation
In this report, we took advantage of the differential transcription in the major tissues and wood forming cell types of P. trichocarpa to investigate genes associated with wood formation. We assumed that genes abundantly and specifically expressed in wood forming tissue would be key genes for regulation of wood formation. We identified 22, 5, and 18 xylem-specific expressed genes that encode enzymes for the biosynthesis of monolignols, cellulose, and hemicelluloses, the three major components of secondary cell walls. Most of genes implicated in the biosynthesis of monolignols have been confirmed for their biochemical functions (Wang et al. 2014). However, the enzyme activity of CSEs in P. trichocarpa is too low to be detected, probably due to their low transcript level (Fig. 2a). All five xylem enriched genes for cellulose synthesis in wood forming tissue were included; they are homologs of the three Arabidopsis secondary cell wall CesAs (AtCesA4, AtCesA7 and AtCesA8) (Kumar et al. 2009; Carpita 2011). Primary cell wall CesAs were not included. Most of the annotated genes for the biosynthesis of hemicelluloses (18 out of 23) are specifically expressed in the wood forming tissue.
Analysis of gene transcripts for cell-type specificity provided additional information for wood cell-type differentiation. Some of the cell wall component biosynthetic genes can be identified as fiber-specific, but vessel element-specific genes did not include any of the cell wall component biosynthetic genes. GO analysis of cell-type specific genes indicated that wood associated cell wall component biosynthesis is more active in fibers compared with vessels during wood formation. This is consistent with the thicker cell walls of fibers compared to vessels in wood.
Regulators of cell wall component biosynthetic genes
We identified 165 TFs that are xylem tissue-specific and xylem cell-type-specific. We regard these TFs as putative regulators in wood formation. To evaluate the relationship between these TF genes and the cell wall component biosynthetic genes, a global co-expression subnetwork based on Pearson correlation coefficient was constructed (Fig. 4a). Because the co-expressed genes show a similar expression pattern, they may be under the control of functionally related transcriptional regulations (Stuart et al. 2003; Weirauch 2011). Most of the gene co-expressions in the global subnetwork are positively correlated. However, some negative co-expression patterns were found between fiber and vessel cell-type-specific TFs, which indicate distinct regulation of the two cell types.
We focused on 101 TFs for their transcriptional regulation during wood formation in P. trichocarpa, because they were co-expressed with the 45 cell wall component genes. The co-expression analysis showed that 101 TFs can be clustered into two groups (groups 1 and 2), and that the 45 cell wall component genes can also be clustered into two groups (groups 1 and 2). Cell wall component gene group 1 contains most of the late monolignol pathway genes, whereas cell wall component gene group 2 includes the early pathway genes, such as all PAL members. These results suggest that the early and late monolignol pathway genes are regulated differently in wood formation.
The TF gene group 1 includes TFs that are likely to regulate the biosynthesis of secondary cell wall components. Group 2 TF genes are less correlated with wood component genes. Fiber cell-specific TFs can be found in both TF gene groups, but vessel-specific TFs are only found in TF gene group 2. TFs with low or no correlation with genes for wood component biosynthesis may be involved in other aspects of regulation associated with wood formation.
Our co-expression analysis reveals the putative regulators of wood formation. Many of the cell wall component genes are highly correlated with the expression of multiple TFs (Fig. 5), suggesting that the cell wall component biosynthesis during wood formation is redundantly controlled by many distinct TFs. Of particular interest are LBD and bHLH that strongly correlated with 87 and 46 genes (Fig. 4c, d), respectively, related to wood formation. The strong correlation between group 1 TF genes and group 1 cell wall component genes suggests many novel regulators of which manipulation may provide new strategies for improving wood properties for biomaterials and biofuels.
Author contribution statement
RS, YCL, QL, HC, and YHS conducted sample collection and RNA-seq libraries construction; RS conducted the bioinformatics analysis; and RS, JPW, RRS, and VLC designed the research and wrote the manuscript.
References
Brady SM, Orlando DA, Lee JY, Wang JY, Koch J, Dinneny JR, Mace D, Ohler U, Benfey PN (2007) A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 318:801–806
Cai B, Li CH, Huang J (2014) Systematic identification of cell wall related genes in Populus based on analysis of functional modules in co-expression network. PLoS One 9:e95176
Carlowicz M (2012) Seeing forests for the trees and the carbon: Mapping the world’s forests in three dimensions. Earth Observatory (NASA). http://earthobservatory.nasa.gov/Features/ForestCarbon
Carpita NC (2011) Update on mechanisms of plant cell wall biosynthesis: how plants make cellulose and other (1 → 4)-β-d-glycans. Plant Physiol 155:171–184
Chen HC, Song J, Wang JP, Lin YC, Ducoste J, Shuford CM, Liu J, Li Q, Shi R, Nepomuceno A, Isik F (2014a) Systems biology of lignin biosynthesis in Populus trichocarpa: heteromeric 4-coumaric acid: coenzyme A ligase protein complex formation, regulation, and numerical modeling. Plant Cell 26:876–893
Chen Y, Lun AT, Smyth GK (2014b) Differential expression analysis of complex RNA-seq experiments using edgeR. In: Datta S, Nettleton D (eds) Statistical analysis of next generation sequencing data. Springer International Publishing, Switzerland, pp 51–74
Chiang VL (2002) From rags to riches. Nat Biotechnol 20:557–558
Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, Hanspers K (2007) Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2:2366–2382
D’haeseleer P, Liang S, Somogyi R (2000) Genetic network inference: from co-expression clustering to reverse engineering. Bioinformatics 16:707–726
Esau K (1965) Vascular differentiation in plants. Holt Rinehart and Winston, New York
Evert RF (2006) Esau’s plant anatomy: meristems, cells, and tissues of the plant body: their structure, function, and development. Wiley, Hoboken
Guitart-Pla O, Kustagi M, Rügheimer F, Califano A, Schwikowski B (2015) The Cyni framework for network inference in Cytoscape. Bioinformatics 31:1499–1501
Kumar M, Thammannagowda S, Bulone V, Chiang V, Han KH, Joshi CP, Mansfield SD, Mellerowicz E, Sundberg B, Teeri T, Ellis BE (2009) An update on the nomenclature for the cellulose synthase genes in Populus. Trends Plant Sci 14:248–254
Larson PR (1994) The vascular cambium: development and structure. Springer, Berlin
Li Q, Lin YC, Sun YH, Song J, Chen H, Zhang XH, Sederoff RR, Chiang VL (2012) Splice variant of the SND1 transcription factor is a dominant negative of SND1 members and their regulation in Populus trichocarpa. Proc Natl Acad Sci USA 109:14699–14704
Li Q, Min D, Wang JP, Peszlen I, Horvath L, Horvath B, Nishimura Y, Jameel H, Chang HM, Chiang VL (2011) Down-regulation of glycosyltransferase 8D genes in Populus trichocarpa caused reduced mechanical strength and xylan content in wood. Tree Physiol 31:226–236
Lin YC, Li W, Sun YH, Kumari S, Wei H, Li Q, Tunlaya-Anukit S, Sederoff RR, Chiang VL (2013) SND1 transcription factor-directed quantitative functional hierarchical genetic regulatory network in wood formation in Populus trichocarpa. Plant Cell 25:4324–4341
Liu L, Filkov V, Groover A (2014a) Modeling transcriptional networks regulating secondary growth and wood formation in forest trees. Physiol Plant 151:156–163
Liu Y, Zhou J, White KP (2014b) RNA-seq differential expression studies: more sequence or more replication? Bioinformatics 30:301–304
Lu S, Li Q, Wei H, Chang MJ, Tunlaya-Anukit S, Kim H, Liu J, Song J, Sun YH, Yuan L, Yeh TF (2013) Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc Natl Acad Sci USA 110:10848–10853
McCarthy DJ, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucl Acids Res 40:4288–4297
Merkle SA, Dean JF (2000) Forest tree biotechnology. Curr Opin Biotechnol 11:298–302
Netotea S, Sundell D, Street NR, Hvidsten TR (2014) ComPlEx: conservation and divergence of co-expression networks in A. thaliana, Populus and O. sativa. BMC Genom 15:1
Perlin J (2005) A forest journey: the story of wood and civilization. The Countryman Press, Woodstock
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–884
Ragauskas AJ, Nagy M, Kim DH, Eckert CA, Hallett JP, Liotta CL (2006) From wood to fuels: integrating biofuels and pulp production. Ind Biotechnol 2:55–65
Rehfuess E, Mehta S, Prüss-Üstün A (2006) Assessing household solid fuel use: multiple implications for the Millennium development goals. Environ Health Perspect 114:373–378
Reimand J, Arak T, Vilo J (2011) g:Profiler–a web server for functional interpretation of gene lists (2011 update). Nucl Acids Res 39:W307–W315
Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11:R25
Sarkanen KV (1976) Renewable resources for the production of fuels and chemicals. Science 191:428
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Shi R, Sun Y, Li Q, Heber S, Sederoff RR, Chiang VL (2010) Towards a systems approach for lignin biosynthesis in Populus trichocarpa: transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol 51:144–163
Shi R, Shuford CM, Wang JP, Sun YH, Yang Z, Chen HC, Tunlaya-Anukit S, Li Q, Liu J, Muddiman DC, Sederoff RR (2013) Regulation of phenylalanine ammonia-lyase (PAL) gene family in wood forming tissue of Populus trichocarpa. Planta 238:487–497
Song J, Lu S, Chen ZZ, Lourenco R, Chiang VL (2006) Genetic transformation of Populus trichocarpa genotype Nisqually-1: a functional genomic tool for woody plants. Plant Cell Physiol 47:1582–1589
Street NR, Jansson S, Hvidsten TR (2011) A systems biology model of the regulatory network in Populus leaves reveals interacting regulators and conserved regulation. BMC Plant Biol 11:13
Stuart JM, Segal E, Koller D, Kim SK (2003) A gene-coexpression network for global discovery of conserved genetic modules. Science 302:249–255
Suzuki S, Li L, Sun YH, Chiang VL (2006) The cellulose synthase gene superfamily and biochemical functions of xylem-specific cellulose synthase-like genes in Populus trichocarpa. Plant Physiol 142:1233–1245
Taylor-Teeples M, Lin L, de Lucas M, Turco G, Toal TW, Gaudinier A, Young NF, Trabucco GM, Veling MT, Lamothe R, Handakumbura PP (2015) An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature 517:571–575
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111
Van Dongen S, Abreu-Goodger C (2012) Using MCL to extract clusters from networks. In: van Helden J, Toussaint A, Thieffry D (eds) Bacterial molecular networks: methods and protocols. Springer, New York, pp p281–p295
Vanholme R, Cesarino I, Rataj K, Xiao Y, Sundin L, Goeminne G, Kim H, Cross J, Morreel K, Araujo P, Welsh L (2013) Caffeoyl shikimate esterase (CSE) is an enzyme in the lignin biosynthetic pathway in Arabidopsis. Science 341:1103–1106
Vitousek PM, Mooney HA, Lubchenco J, Melillo JM (1997) Human domination of earth’s ecosystems. Science 277:494–499
Wang JP, Chuang L, Loziuk PL, Chen H, Lin YC, Shi R, Qu GZ, Muddiman DC, Sederoff RR, Chiang VL (2015) Phosphorylation is an on/off switch for 5-hydroxyconiferaldehyde O-methyltransferase activity in poplar monolignol biosynthesis. Proc Natl Acad Sci USA 112:8481–8486
Wang JP, Naik PP, Chen HC, Shi R, Lin CY, Liu J, Shuford CM, Li Q, Sun YH, Tunlaya-Anukit S, Williams CM, Muddiman DC, Ducoste JJ, Sederoff RR, Chiang VL (2014) Complete proteomic based enzyme reaction and inhibition kinetics reveal how monolignol biosynthetic enzyme families affect metabolic-flux and lignin. Plant Cell 26:894–914
Weirauch MT (2011) Gene coexpression networks for the analysis of DNA microarray data. In: Dehmer M, Emmert-Streib F, Graber A, Salvador A (eds) Applied statistics for network biology: methods in systems biology. Wiley, Hoboken, pp 215–250
Yang X, Ye CY, Bisaria A, Tuskan GA, Kalluri UC (2011) Identification of candidate genes in Arabidopsis and Populus cell wall biosynthesis using text-mining, co-expression network analysis and comparative genomics. Plant Sci 181:675–687
Zheng X, Moriyama EN (2013) Comparative studies of differential gene calling using RNA-Seq data. BMC Bioinform 14:S7
Zhong R, Ye ZH (2014) Complexity of the transcriptional network controlling secondary wall biosynthesis. Plant Sci 229:193–207
Acknowledgements
This work was supported by the US National Science Foundation, Plant Genome Research Program Grant DBI-0922391, and the US Department of Energy Grant DE-SC000691. We acknowledge additional supports from the NC State University Jordan Family Distinguished Professor Endowment, the NC State University Forest Biotechnology Industrial Research Consortium, and the National Natural Science Foundation of China (NSFC).
Author information
Authors and Affiliations
Corresponding authors
Additional information
R. Shi and J. P. Wang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shi, R., Wang, J.P., Lin, YC. et al. Tissue and cell-type co-expression networks of transcription factors and wood component genes in Populus trichocarpa . Planta 245, 927–938 (2017). https://doi.org/10.1007/s00425-016-2640-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-016-2640-1