
Abstract
SLC26A11 (human)/Slc26a11 (mouse), also known as kidney brain anion transporter (KBAT), is a member of the SLC26 anion transporter family and shows abundant mRNA expression in the brain. However, its exact cellular distribution and subcellular localization in the brain and its functional identity and possible physiological roles remain unknown. Expression and immunostaining studies demonstrated that Slc26a11 is abundantly expressed in the cerebellum, with a predominant expression in Purkinje cells. Lower expression levels were detected in hippocampus, olfactory bulb, cerebral cortex, and subcortical structures. Patch clamp studies in HEK293 cells transfected with mouse cDNA demonstrated that Slc26a11 can function as a chloride channel that is active under basal conditions and is not regulated by calcium, forskolin, or co-expression with cystic fibrosis transmembrane regulator. Single and double immunofluorescent labeling studies demonstrated the localization of vacuolar (V) H+-ATPase and Slc26a11 (KBAT) in the plasma membrane in Purkinje cells. Functional studies in HEK293 cells indicated that transfection with Slc26a11 stimulated acid transport via endogenous V H+-ATPase. We conclude that Slc26a11 (KBAT) is prominently distributed in output neurons of various subcortical and cortical structures in the central nervous system, with specific expression in Purkinje cells and that it may operate as a chloride channel regulating acid translocation by H+-ATPase across the plasma membrane and in intracellular compartments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plasma membrane chloride (Cl−) channels are expressed in numerous tissues including neurons. Their functions are diverse and range from transepithelial transport to cell volume regulation and from mediation of neuronal activity inhibition to intra- and extracellular ion homeostasis [3, 4, 7, 13, 15, 28, 30, 43, 47]. Several known molecules with chloride channel activity have been identified in neurons. In addition to GABA-activated and glycine-activated chloride channels [19, 56], several isoforms of the CLC (voltage-gated chloride channel) family are known to be expressed in various parts of the brain where they often modulate neuronal excitability [33]. Further, the cystic fibrosis transmembrane regulator (CFTR), which functions as a chloride channel, is also expressed in the brain [24, 49]. Finally, potassium chloride cotransporter (KCC2) has been identified as a neuron-specific Cl− transporter [34, 45] that is important in excitability control and postsynaptic inhibition mediated by ligand-gated anion channels, like (GABAA) and glycine receptors [39].
In neurons, V H+-ATPases play an important role in modulating the cytoplasmic pH [57] as well as establishing an electrochemical gradient of protons in subcellular structures in coordination with chloride channels [25, 31]. As a result of the latter, V H+-ATPases are shown to be involved in enhancing the accumulation of neurotransmitters in synaptic vesicles [23, 42].
SLC26 isoforms belong to a conserved family of transporters, with ability to transport various anions, including chloride, sulfate, bicarbonate, and oxalate, with variable specificity [8, 11, 12, 17, 18, 29, 35, 38, 40, 41, 46, 51]. Currently, relatively little is known about the functional identity, cellular distribution, and the role of SLC26A11 in physiologic processes. SLC26A11 mRNA has been detected in several tissues, including placenta, brain, kidney, and venules [46], and more recently, SLC26A11 protein has been shown to function as a chloride transporter [52]. To indicate its high expression in kidney and brain and its affinity for chloride transport, SLC26A11 has been designated as kidney brain anion transporter (KBAT) [52]. However, the cellular distribution and subcellular localization of SLC26A11 in the brain remain unknown. Further, the exact functional modes of KBAT have not been determined.
Our studies demonstrate that KBAT expression in the brain is readily detected in various cortical and subcortical brain structures, with a most prominent localization in cerebellar Purkinje cells. We further demonstrate that KBAT can function as a constitutively active chloride channel. KBAT colocalizes with V H+-ATPase in the plasma membrane and intracellular compartments of Purkinje cells, and functional studies in cultured cells demonstrated the stimulation of H+ transport via V H+-ATPase by KBAT. KBAT is a novel chloride channel and may play an important role in the cell physiological properties of output neurons in various parts of the brain.
Experimental procedures
Animal models
C57BL/6 mice at 25–30 g body weight were used for these studies. Animals were allowed free access to water and food. The use of anesthetics (pentobarbital sodium) and method of euthanasia (pentobarbital sodium overdose) were approved according to the institutional guidelines at Erasmus MC in Rotterdam and University of Cincinnati in Cincinnati, OH. Animals were euthanized and their brains removed and processed for the isolation of RNA or protein and immunohistochemistry studies.
PCR of Slc26a11 in the brain
Total RNA prepared from mouse whole brain, cerebral cortex, cerebellum, and kidney was poly(A)+ selected using Oligotex latex beads (Qiagen) and then reverse transcribed at 47 °C using SuperScript II RT (Life Technologies) and oligo(dT) primers. Oligonucleotide primers (5′-TAT CAT GTC TCT CCT GGT GTC C (sense) and 5′-ATC AAT TGC AGG GAA GTA CAG G (antisense)) were designed based on mouse Slc26a11 sequence (GenBank accession number AF345196). The cycling parameters were 94 °C for 1 min, then 94 °C for 30 s, followed by 68 °C for 2 min for 35 cycles. After PCR, the product was gel purified (revealing a single band of 1.2 kb) and used as a probe for Northern blot hybridizations. Sequence analysis of the PCR product verified the identity of the band as KBAT.
RNA isolation and Northern blot hybridization
Total cellular RNA was extracted from whole brain, cerebral cortex, and cerebellum according to established methods, quantitated spectrophotometrically, and stored at −80 °C. Total RNA samples (30 μg/lane) were fractionated on a 1.2 % agarose formaldehyde gel, transferred to Magna NT nylon membranes, cross-linked by UV light, and baked. RNA isolated from kidney was used for comparison. Hybridization was performed according to established methods. The membranes were washed, blotted dry, and exposed to a PhosphorImager screen (Molecular Dynamics, Sunnyvale, CA). Slc26a11-specific 32P-labeled cDNA (see above) was used as a probe for Northern hybridizations. The band densities on Northern hybridization were quantitated by densitometry using ImageQuaNT software (Molecular Dynamics, Sunnyvale, CA).
Cloning of the full-length mouse Slc26a11
Full-length mouse Slc26a11 cDNA was amplified from mouse kidney RNA using the following PCR primers: 5′-CTC TGT GAA AGG TCT GGG TC (sense) and 5′-TCA GGG GCC GGA GGG AGA CTT (antisense). These primers encode nucleotides 80–1904 of a mouse Slc26a11 cDNA (GenBank accession number AF345196) and contain the entire open reading frame. Amplification of the mouse Slc26a11 cDNA by PCR was performed according to the CLONTECH Advantage 2 PCR kit protocol. The product was gel purified and sequence analysis verified its identity as Slc26a11. The PCR product was ligated into mammalian expression pTarget Vector (Promega, cat. # A1410) for expression studies.
Cell culture and transfection
HEK 293 cells were cultured in DMEM (Gibco, Karlsruhe, Germany) supplemented with 10 % FBS at 37 °C, 5 % CO2, and humidified atmosphere. Cells were plated on fibronectin- and collagen-coated coverslips and cotransfected with cDNA encoding either SLC26a11, CFTR, or empty pcDNA3.1 vector (mock) along with P2Y2 receptor and CD8. Transfections were carried out using Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) according to the manufacturer’s protocol. Patch clamp studies on HEK 293 cells were conducted according to published reports [21] as follows.
Patch clamp studies
Patch clamp studies were performed according to established protocols [21]. At 2 or 3 days after transfection, HEK 293 cells were incubated for 1–2 min with Dynabeads CD8 (Invitrogen). Coverslips were mounted on the stage of an inverted microscope (IM35; Carl Zeiss, Jena, Germany) and kept at 37 °C. The bath was perfused continuously with Ringer solution (145 mM NaCl, 0.4 mM KH2PO4, 1.6 mM K2HPO4, 6 mM d-glucose, 1 mM MgCl2, and 1.3 mM calcium gluconate, pH 7.4) at the rate of 5 ml/min. For fast whole cell patch clamping, pipettes were filled with intracellular-like “physiological” solution containing 30 mM KCl, 95 mM potassium gluconate, 1.2 mM NaH2PO4, 4.8 mM Na2HPO4, 1 mM EGTA, 0.758 mM calcium gluconate, 1.034 mM MgCl2, 5 mM d-glucose, and 3 mM ATP (pH 7.2), with an input resistance of 2–4 MΩ. For the given Cl− concentrations, the Nernst potential E Cl − was −39.7 mV. In some experiments, a potassium-free pipette filling solution was used in which potassium was replaced by NMDG+. The access conductance was measured continuously and was 70–140 nS. The junction potentials occurring at the bath reference electrode were small as we used a flowing 3.5 M KCl electrode and continuous bath perfusion. Currents (voltage clamp) and voltages (current clamp) were recorded using a patch clamp amplifier (EPC 7, List Medical Electronics, Darmstadt, Germany), the LIH1600 interface, and PULSE software (HEKA, Lambrecht, Germany) as well as Chart software (ADInstruments, Spechbach, Germany). Data were stored continuously on a computer hard disc and were analyzed using the PULSE software. In regular intervals, membrane voltages (V c) were clamped in steps of 10 mV from −50 to +50 mV. Membrane conductance G m was calculated from the measured current (I) and V c values according to Ohm’s law.
Intracellular pH studies
Changes in intracellular pH (pHi) were monitored using the acetoxymethyl ester of the pH-sensitive fluorescent dye 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF-AM) as described [2]. HEK-293 cells were grown to semi-confluence on coverslips and then transfected with either Slc26a11 cDNA or empty vector (mock transfected). Forty-eight hours after transfection, cells were incubated in the presence of 5 μM BCECF in a solution consisting of 115 mM NaCl, 25 mM Na-gluconate, 0.8 mM K2HPO4, 0.2 mM KH2PO4, 1 mM CaCl2, 1 mM MgCl2, 4 mM KCl, 10 mM HEPES, and 5 mM glucose at pH 7.4 and gassed with 100 % O2. To measure the pHi, each coverslip was positioned diagonally in a cuvette that was placed in a thermostatically controlled holding chamber (37 °C) in a Deltascan dual-excitation spectrofluorometer (PTI, double-beam fluorometer; Photon Technology International, South Brunswick, NJ). The monolayer was then perfused with the above solution to obtain the baseline pHi. The fluorescence ratio at excitation wavelengths of 500 and 450 nm was utilized to determine the pHi values in the experimental groups by comparison to the calibration curve that was generated by KCl/nigericin technique. The emission wavelength was recorded at 525 nm. Calibration curves were established daily by incubating the BCECF-loaded cells with 3.3 μM nigericin in a medium containing (in millimolar) 120 KCl, 1 CaCl2, 1 MgCl2, 0.8 K2HPO4, 0.2 KH2PO4, and 10 HEPES and adjusted at various pH values with Tris-buffered solution. F 500/F 450 was found to be linearly related to pHi over the pH range of 7.40–6.30 (y = 2.1x − 12.5; r = 0.997). The intracellular acid load was induced by the NH3/NH4 + pulse technique [2]. The H+-ATPase activity was determined as the initial rate of a sodium-independent, concanamycin A sensitive pHi recovery (dpHi/dt, pH/min) from an acid load induced by NH3/NH4 + in the presence of a Na+-free, hypotonic (210 mOsm/l) solution. The Na-free solution was prepared by replacing Na−Cl with TMA−Cl (tetramethylammonium chloride) and Na-gluconate with NMDG-gluconate when necessary. The dpHi/dt was calculated by fitting to a linear equation the first 3 min of the time course of pHi recovery. Correlation coefficients for these linear fits averaged 0.986 ± 0.003. In addition to Na and chloride (or TMA and Cl for Na-free experiments), all solutions contained (in millimolar per liter) 0.8 K2HPO4, 0.2 KH2PO4, 1 CaCl2, and 1 MgCl2.
Antibodies and immunoblot analysis
A peptide corresponding to amino acids 274–287 (TRDNKTISFSEMVQC) of mouse KBAT (GenBank accession number AF345196) was synthesized and used for antibody generation in two rabbits. The specificity of KBAT antibody was recently demonstrated [52]. For Western blot analysis, microsomal membrane proteins were prepared from cerebellum, resolved by SDS-PAGE (30 μg/lane), and transferred to nitrocellulose membrane. The membrane was blocked with 5 % milk proteins and then incubated for 6 h with 20 μl of KBAT-immune serum diluted at 1:600. The secondary antibody was a donkey anti-rabbit IgG conjugated to horseradish peroxidase (Pierce). The site of antigen–antibody complex formation on the nitrocellulose membranes was visualized using chemiluminescence method (SuperSignal Substrate, Pierce) and captured on a light-sensitive imaging film (Kodak). For V H+-ATPase, a monoclonal antibody to the E 31 kD subunit was used [6].
Immunohistochemistry studies
Mice were perfused through the left ventricle with 4 % paraformaldehyde. Brains were removed, postfixed in the same fixative for 1 h at room temperature, and then placed in 10 % sucrose at 4 °C overnight. Brains were embedded in gelatin and then protected in 30 % sucrose at 4 °C overnight. Sections at 40 μm thickness were cut with a freezing microtome and washed with Tris-buffered saline (TBS, pH 7.6). Sections were permeabilized and blocked in TBS containing 0.4 % Triton X-100 and 10 % normal horse serum at room temperature for 1 h. Rabbit polyclonal Slc26a11 (KBAT) antibodies (1:500) were applied to the sections at 4 °C for 48 h. Thereafter, the sections were incubated in avidin–biotin complex at room temperature for 1.5 h. Slc26a11 (KBAT) antibodies were labeled by goat anti-rabbit antibodies at 1:200 dilution for 1.5 h. Immunostaining was revealed with 0.6 % diaminobenzidine dissolved in 0.05 M Tris–HCl (pH 7.6) containing 0.03 % H2O2. Nonspecific labeling was controlled by using purified pre-immune serum and pre-adsorbed immune serum at the same dilution.
Immunofluorescent studies
Mice were perfused and postfixed the same as in immunohistochemical experiments. Sections were incubated in 10 mM Na+-citrate at 80 °C for 2 h. For single immunofluorescence labeling, sections were permeabilized and blocked in TBS containing 0.4 % Triton X-100 and 10 % normal horse serum at room temperature for 1 h. Rabbit polyclonal Slc26a11 (KBAT) antibodies (1:200) were applied to the sections at 4 °C for 48 h. For double immunofluorescence labeling, sections were pretreated with pepsin for 20 min. After blocking with 10 % normal horse serum, they were incubated with donkey anti-mouse Fab fragments (1:200) for 2 h (Jackson ImmunoResearch Laboratories, Inc). KBAT and H+-ATPase (E subunit) antibodies were used for colocalization studies. The dilutions for KBAT and H+-ATPase antibodies were 1/200 and 1/50, respectively. For visualization, green (FITC) and red (Cy3) fluorescent conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc) were used for KBAT and H+-ATPase. Sections were mounted and images were acquired on a Zeiss LSM 700 laser confocal scanning microscope.
Materials
[32P]dCTP was purchased from New England Nuclear (Boston, MA). Nitrocellulose filters and other chemicals including valinomycin were purchased from Sigma (St. Louis, MO). A RadPrime DNA labeling kit was purchased from GIBCO-BRL. BCECF was from Molecular Probes. A mMESSAGE mMACHINE Kit was purchased from Ambion (Austin, TX).
Statistical analysis
The results for patch clamp experiments or pHi studies are presented as means ± SE. Statistical significance between various experimental groups was determined by ANOVA or Student's unpaired t test whenever applicable. P <0.05 was considered significant. All experiments were performed at least three times.
Results
Expression of Slc26a11 (KBAT)
To assess the expression levels of KBAT in the brain, RT-PCR and Northern and Western blot analyses were performed. Figure 1a shows an ethidium bromide staining of RT-PCR experiment demonstrating the expression of KBAT in whole brain, cerebellum, and cerebral cortex. Northern blot analysis showed that KBAT is abundantly expressed in the cerebellum. The expression of KBAT in the cerebral cortex was significantly less compared to that in the cerebellum (p < 0.05, n = 3 separate animals). The expression of KBAT in kidney is shown for comparison (Fig. 1b). Western blot analysis was performed on microsomes isolated from mouse cerebellum. KBAT was detected as a ∼72-kDa band in cerebellum. The labeling of the 72-kDa band was completely blocked in the presence of pre-adsorbed serum (Fig. 1c).

Slc26a11 (KBAT) expression and distribution in mouse brain. a Expression of KBAT by RT-PCR. A representative ethidium bromide staining of agarose gel demonstrates a PCR product of expected size (1,200 bp) in whole brain, cerebellum, and cerebral cortex. b Northern hybridization of KBAT in brain sections. KBAT is abundantly expressed in cerebellum with lower levels in cerebral cortex. c Western blot analysis of KBAT in cerebellum. Microsomal membranes from cerebellum were resolved by SDS-PAGE and blotted against KBAT antibody. KBAT appears as ∼72 kDa band in cerebellar microsomes. The experiments for Northern and Western blots have been performed at least two times
Immunohistochemical staining and regional localization
The purpose of the next series of experiments was to ascertain the cellular distribution and subcellular localization of KBAT in the brain. Toward this end, immunohistochemical staining with KBAT antibody was performed on brain sections according to the “Experimental procedures.” Initial experiments indicated abundant level of KBAT expression in the cerebellum with moderate levels in the hippocampus. Considerable expression of KBAT was also detected in substantia nigra reticularis, superior colliculus, olfactory bulb, and mammillary body (see detailed expression results in Table 1). As shown in Fig. 2a (sagittal sections), b (coronal sections), the labeling of KBAT in the cerebellum was predominantly detected in Purkinje cells, with both the cell body and dendrites displaying abundant expression (left panels: low magnification; right panels: high magnification). KBAT labeling in granule cells of the cerebellum was lower compared to Purkinje cells. The immunolabeling with the purified pre-immune sera (Fig. 2c, right and left panels) or the pre-adsorbed antibody (Fig. 3c, right and left panels) did not reveal any labeling in the cerebellum or other brain sections, indicating the specificity of the labeling with KBAT antibody. The localization of KBAT in the cerebellum was also studied by immunofluorescent staining (“Experimental procedures”). As indicated, Purkinje cells displayed the highest expression levels of KBAT, with specific labeling detected in the dendrite (yellow arrow), plasma membrane (blue arrow), and the cytoplasm (red arrow) of cell bodies (Fig. 2d, left and right panels). The expression levels of KBAT in granule cells were lower compared to those in Purkinje cells.

Expression of KBAT in the cerebellum. a Immunohistochemical staining with KBAT antibodies in sagittal sections of the cerebellum. Specific labeling with KBAT antibodies was detected in cell bodies and dendrites of Purkinje cells. Arrow in right shows a cell body of Purkinje cell (pc). Right panel: high magnification; left panel: low magnification (the scale bars indicate 500 μm in right panel and 50 μm in left panel). b Immunohistochemical staining with KBAT antibodies in coronal sections of the cerebellum. Similar to Fig. 1a (above), KBAT antibodies detect high expression in Purkinje cells. Right panel: high magnification; left panel: low magnification (the scale bars indicate 500 μm in right panel and 50 μm in left panel). c Immunostaining with purified pre-immune or pre-adsorbed sera (scale bars 500 μm). Immunostaining with either purified pre-immune serum (right image) or pre-adsorbed immune serum (left image) failed to detect any labeling in the cerebellum or any other regions in the brain, indicating the specificity of the staining with KBAT antibody in (a) and (b). d Immunoflourescent staining with KBAT antibody in cerebellum. Slc26a11 (KBAT) is detected in both the plasma membrane and the cytoplasmic compartment of Purkinje cells (blue arrows show the membrane labeling and red arrows point to the cytoplasmic labeling; yellow arrows point to the dendritic labeling of KBAT). Hb habenula

Expression of KBAT in the hippocampus, olfactory bulb, cerebral cortex, and substantia nigra. a Immunohistochemical staining with KBAT antibodies in coronal sections of the substantia nigra. KBAT antibodies show intermediate to high expression levels in substantia nigra reticularis. Expression of KBAT in zonal layer and superficial gray layer of superior colliculus as well as in mammillary body is shown for comparison. b Immunohistochemical staining with KBAT antibodies in coronal sections of the hippocampus. Specific labeling with KBAT antibodies was detected in pyramidal cells and lacunosum molecular layer of the hippocampus. The scale bars (500 μm in right panel and 250 μm in left image) are depicted. c The immunostaining with pre-adsorbed immune serum. The labeling with KBAT antibodies was specific, as pre-adsorbed immune serum failed to detect any labeling in substantia nigra and hippocampus or any other regions in the brain. Scale bars 500 μm. d The immunostaining with KBAT antibodies in coronal sections of the olfactory bulb and cerebral cortex. Intermediate to high level of KBAT labeling was observed in glomerular (Gl) and external plexiform (EPI) layers as well as anterior nucleus (AON) of the olfactory bulb. In the cerebral cortex, moderate to high labeling was detected in layers II, III, and V. Scale bars 250 μm. Hip hippocampus; Zo zonal layer of the superior colliculus; SuG superficial gray layer, superior colliculus; SNR substantia nigra, reticular part; Mb mammillary body; Cx cerebral cortex; Py pyramidal cell layer; LMol lacunosum molecular layer; DG dentate gyrus; Gl glomeruli of olfactory bulb; EPI external plexiform layer, olfactory bulb; AON anterior olfactory nucleus; II, III, IV, V, VI layers 2, 3, 4, 5, and 6
Immunohistochemical staining also revealed high expression levels of KBAT in the stratum lacunosum-moleculare and pyramidal layers of the hippocampus (Fig. 3b) and glomerular layer and anterior olfactory nucleus of the olfactory bulb (Fig. 3d). In addition, intermediate to high expression levels were observed in substantia nigra reticularis, as well as zonal layer (Zo) and superficial gray layer (SuG) of superior colliculus (Fig. 3a). Immunostaining with the pre-adsorbed immune serum did not detect any significant labeling (Fig. 3c).
Slc26a11 (KBAT) functions as a chloride channel
KBAT has been shown to transport chloride and mediate Cl−/HCO3 − exchange [52]. Two other members of SLC26 family, namely SLC26A7 and SLC26A9, can function in both Cl−/HCO3 − exchange and chloride channel modes [8, 10, 16, 51, 53]. There are no studies that have directly examined the ability of KBAT to function either as a chloride channel or an electrogenic chloride transporter. In the next series of experiments, we examined the functional identity of KBAT by patch clamp studies in HEK 293 cells transfected with KBAT cDNA.
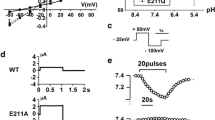
As shown in Fig. 4a, KBAT increases baseline whole cell currents without any stimulation, suggesting that it is already active under baseline conditions. Summation of whole cell conductance in Slc26a11 or mock-transfected cells is shown in Fig. 4b. I–V curves show a significant increase (p < 0.05) in whole cell currents in KBAT-transfected cells compared to that in mock-transfected cells (Fig. 4c). The results reflect the summation of all experiments, with no experiment being excluded. The affinity of Slc26a11 for chloride, bromide, and iodide was examined by replacing extracellular chloride with bromide (Br−) or iodide (I−). The results (I/V relationship) are depicted in Fig. 4d and indicate that although the permeability for I− was slightly larger than that for Cl− and Br−, the channel conducts the latter anions better than I−. The reduction in extracellular Cl− to 32 mM (Fig. 4e) or 5 mM (Fig. 4f) led to shifts in the reversal potential by 9 and 15 mV, respectively, which is less than the expected shifts for a pure Cl− conductance, suggesting possible contribution of ions other than Cl−. Performing the anion replacement (5Cl−) experiment with a potassium-free pipette solution shifted the current voltage relationship more significantly by about 36 mV, clearly indicating the presence of a Cl− permeable conductance in KBAT-expressing HEK293 cells (Fig. 4g). Finally, we reduced extracellular NaCl from 145 to 29 mM by replacing NaCl with NMDG-gluconate, which strongly reduced the whole cell current but shifted the reversal potential by 12 mV (Fig. 4h). Using the Goldman–Hodgkin–Katz equation, we found a permeability ratio of Cl−/Na+ of 1.6. The results indicate that Slc26a11 may also be capable of functioning as an electrogenic chloride transporter, with the ability to carry a cation such as sodium. The currents are shown as raw data, and no subtraction or correction has been used. Extracellular Cl− removal had no significant effect on mock-transfected cells (data not shown).

Expression of KBAT induces a Cl− conductance in HEK293 cells. a Original recordings of whole cell currents measured in mock-transfected and Slc26a11 (KBAT)-overexpressing HEK293 cells. Cells were voltage clamped from −50 to +50 mV in steps of 10 mV. b Summary of the calculated whole cell conductance in Slc26a11 expressing HEK293 and mock-transfected cells. c I/V relationship of Slc26a11-induced whole cell currents. d I/V relationships of Slc26a11-induced whole cell currents: effect of replacement of extracellular Cl− by I− or Br−. The calculated conductances were G Cl −, 35 nS, G I −, 21 nS, \( {G}_{{\mathrm{Br}}^{-}} \), 40 nS. e Reduction of extracellular Cl− to 32 mM shifted the I/V curve by 9 mV (E Cl − = 0 mV, expected shift for pure G Cl −, 39.7 mV). f Reduction of extracellular Cl− to 5 mM shifted the I/V curve by 15 mV. (E Cl − = +48 mV, expected shift for pure G Cl −, 87.7 mV). g Anion replacement (5Cl−) with a potassium-free pipette solution shifted the current voltage relationship by 36 mV. (E Cl − = +48 mV, expected shift for pure G Cl −, 87.7 mV). h Reduction of extracellular NaCl from 145 to 29 mM by replacing NaCl with NMDG-gluconate inhibited whole cell currents and shifted the reversal potential by 12 mV (E Cl − = −2.5 mV). Permeability ratio of Cl−/Na+, 1.6 (Goldman–Hodgkin–Katz equation). Mean ± SEM (number of experiments). Number sign indicates significant difference when compared to mock (unpaired t test; p < 0.05). Asterisk indicates significant difference when compared to 5Cl− (paired t test; p < 0.05)
Slc26a11 (KBAT) is active under basal conditions and is not regulated by forskolin, ionomycin, or co-expression with CFTR
Our results demonstrate that about 28 % of the SLC26A11 current is inhibited by either 10 μM GlyH or 5 μM CFTRinh (Fig. 5a). The presence of 20 μM CFTRinh blocked the SLC26A11-mediated current by 39 % (data not shown). Our results further demonstrated that there was almost no inhibitory effect of any Cl− inhibitor on KBAT except for application of GlyH-101 and CFTRinh172, which induced a slight inhibition (Fig. 5a). CFTR is a PKA/cAMP-activated chloride channel, which is widely distributed in various brain regions, including the cerebellum [24]. The inhibitory effect of GlyH-101 and CFTRinh172, two known inhibitors of CFTR, on KBAT-mediated current raises the possibility that KBAT is likely activated by CFTR. We therefore tested the effect of cAMP on KBAT activity by co-transfecting the cells with KBAT and/or CFTR and repeating the experiments in the absence or presence of increased intracellular cAMP. We found that increasing intracellular cAMP levels by 10 μM, forskolin did not further increase KBAT-generated currents (Fig. 5b). In cells co-expressing KBAT together with CFTR, forskolin activated CFTR and this CFTR current was additive with the KBAT currents. No evidence was found for an additional activation of KBAT during stimulation of CFTR (Fig. 5b). Lastly, we examined the effect of increased intracellular calcium on KBAT activity by adding 1 μM ionomycin to HEK293 cells transfected with KBAT cDNA. However, ionomycin did not affect KBAT-mediated current in patch clamp studies (Fig. 5b). Lastly, we examined the effect of increased intracellular calcium on KBAT activity by adding 1 μM ionomycin to HEK293 cells transfected with KBAT cDNA. Increasing the intracellular calcium did not affect KBAT-mediated current in patch clamp studies (Fig. 5c). For comparison, ionomycin treatment resulted in a significant activation of whole cell currents in HEK293 cells overexpressing the Ca2+-activated Cl− channel anoctamin 1 [54] (data not shown). A total of 288 out of 294 cells transfected with anoctamin 1 responded with the activation of a Ca2+-dependent Cl− channel by 1 μM ionomycin, consistent with a transfection rate of ∼98 % in HEK293 cells. In the last series of patch clamp experiments, we examined the effect of co-expression with CFTR on activation of KBAT and the impact of luminal acidic pH on KBAT-mediated Cl− current (Fig. 5d). The results indicate that the co-expression with CFTR, irrespective of cAMP presence or luminal acidity, does not activate Slc26a11-mediated Cl− current in HEK293 cells.

Effect of inhibitors, agonists, CFTR, and low pH on KBAT whole cell currents. a Summary of whole cell conductances in mock-transfected and Slc26a11-overexpressing HEK293 cells and effects of Cl− channel inhibitors GlyH-101 (GlyH; 10 μM), CFTRinh-172 (CFTR inh ; 5 μM), and 4,4′-diisothiocyano-2,2′-stilbene disulfonic acid (DIDS; 100 μM). b Summary of the whole cell conductances obtained from mock-transfected and Slc26a11-overexpressing HEK293 cells and effects of stimulation with IBMX (100 μM) and forskolin (2 μM) (I/F) or ionomycin (Iono; 1 μM). c Summary of the whole cell conductances obtained from CFTR- or CFTR/Slc26a11-co-expressing cells and effects of stimulation with I/F in the absence or presence of GlyH or CFTRinh. d Whole cell conductances obtained from cells expressing Slc26a11 and CFTR or co-expressing Slc26a11 and CFTR and effects of I/F and low pH (pH 6.0). Mean ± SEM (number of experiments). Number sign indicates significant difference when compared to mock (unpaired t test; p < 0.05). Asterisk indicates significant activation by I/F or inhibition by channel blockers (paired t test; p < 0.05). Section sign indicates significant inhibition by blockers after I/F (paired t test; p < 0.05)
Slc26a11 (KBAT) expression in the plasma membrane of cells in cerebellum and cerebral cortex
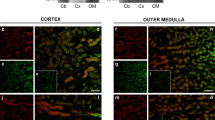
The purpose of the next series of experiments was to examine the plasma membrane localization of Slc26a11 in both the cerebellar and non-cerebellar neurons. The localization of KBAT in Purkinje cells was first studied using both red and green fluorescent secondary antibodies. The high-magnification images in Fig. 6(A) (top panel) demonstrate that Slc26a11 is predominantly localized to the plasma membrane in Purkinje cells, with some cells displaying cytoplasmic expression as well (Fig. 6(A-a) in red and Fig. 6(A-b) in green). The membrane localization of Slc26a11 (KBAT) in the cerebral cortex is shown in Fig. 6(A-c) (in green) and Fig. 6(A-d, A-e) (in red). The images clearly depict predominant plasma membrane localization and some intracellular distribution of Slc26a11 in pyramidal cells in the cerebral cortex (Fig. 6(A-e)). Whether the intracellulary distributed Slc26a11 represents a recycling pool which is utilized for trafficking to or from the plasma membrane remains speculative.

Slc26a11 (KBAT) expression in the plasma membrane of cells in cerebellum and the cerebral cortex: colocalization with H+-ATPase in Purkinje cells. A, top panel: Slc26a11 is predominantly localized to the plasma membrane in Purkinje cells, with certain cells displaying cytoplasmic expression as well (A-a in red, A-b in green). B, bottom panel: The images in A-c (in green) and A-d and A-e (in red) clearly demonstrate predominant plasma membrane localization of Slc26a11 in pyramidal cells in the cerebral cortex. Some cells also display intracellular expression of Slc26a11. B Localization of H+-ATPase in Purkinje cells (B-a). Labeling with H+-ATPase antibodies +/− DAPI counterstain demonstrate a granular pattern prominently present in the plasma membrane in Purkinje cells. The labeling with H+-ATPase antibodies was absent in the nucleus but was detected in submembranous compartment (B-a, left and right panels). Double immunofluorescence labeling of H+-ATPase and Slc26a11 in Purkinje cells (B-b). The double immunofluorescence labeling studies show that both KBAT (Slc26a11) (green) and V H+-ATPase (red) are detected in the plasma membrane of Purkinje cells. Slc26a11 (KBAT) shows more intracellular abundance relative to H+-ATPase in Purkinje cells
KBAT and V H+-ATPase localization in the plasma membrane of Purkinje cells
Recent studies have indicated the expression of H+-ATPase subunits in Purkinje cells as well as in other neurons [25, 31, 57]. In the last series of experiments, we sought to determine the localization of KBAT (Slc26a11) in relation to H+-ATPase in Purkinje cells. We first examined the expression of H+-ATPase by immunofluorescence labeling in Purkinje cells using DAPI as a nuclear counterstain. The H+-ATPase fluorescent was not present as a discrete line, but rather had a granular appearance in Purkinje cells (Fig. 6(B-a), left and right panels). These results support the localization of H+-ATPase to the plasma membrane and possibly some submembranous compartments. Figure 6(B-b; top and bottom panels) is a double immunofluorescence labeling and shows that both KBAT (Slc26a11) (green) and V H+-ATPase (red) are detected in the plasma membrane of Purkinje cells. As evident, Slc26a11 (KBAT) shows more intracellular abundance relative to H+-ATPase in Purkinje cells.
KBAT stimulates acid transport via V H+-ATPase in cultured cells
Several known chloride channels have been shown to regulate H+-ATPase activity in either the plasma membrane or endomembrane of intracellular organelles. We next examined the effect of Slc26a11 (KBAT) on H+-ATPase activity in HEK 293 cells. Toward this end, HEK 293 cells were transiently transfected with KBAT cDNA, and plasma membrane H+-ATPase activity was assayed 48 h later by the pH fluorescence dye BCECF, as the rate of sodium independent, concanamycin A sensitive intracellular pH recovery from an acid load (dpHi/dt) which was calculated by fitting to a linear equation the first 3 min of the time course of pHi recovery. The concentration of concanamycin A was 200 nM. Figure 7a depicts representative pHi tracings in KBAT-transfected (lower panel) and mock-transfected cells (top panel) and indicates a faster recovery from intracellular acidosis in Slc26a11-transfected cells. The intracellular pH recovery was completely inhibited in the presence of concanamycin A, at 200 nM, in both control (mock transfected) and Slc26a11-expressing cells (Fig. 7a, the linear traces in the presence of concanamycin A are overlaid on pHi tracings recorded in the absence of any inhibitors). These results indicate that the Na-independent pHi recovery from acidosis is mediated via V H+-ATPase. Summation of the results in Fig. 7b demonstrates that KBAT stimulated Na-independent pHi recovery from acidosis by ∼120 % (n = 5 in each group; p < 0.02 vs. mock-transfected cells).

Functional interaction of Slc26a11 (KBAT) and H+-ATPase. a Intracellular pH studies. Cultured HEK293 cells (mock transfected) were acid loaded with NH4 + pulse in the presence of Na-free (TMA)-Cl solution. Switching from the isotonic, Na-free solution to a hypotonic, Na-free solution resulted in a Na-independent, intracellular pH (pHi) recovery from intracellular acidosis (top panel). KBAT-transfected cells showed a more robust recovery from intracellular acidosis relative to mock-transfected cells (right panel). The near linear traces in the presence of concanamycin A are overlaid on pHi tracings recorded in the absence of any inhibitors (top and bottom panels) and show the complete inhibition of pHi recovery by concanamycin A. b Intracellular pH studies: results summation. Summation of recovery rates from acidosis from six separate coverslips showed that H+-ATPase-mediated pHi recovery from intracellular acidosis was significantly increased in KBAT-transfected cells
Discussion
The SLC26 (human)/Slc26 (mouse) gene family encodes ten distinct isoforms capable of transporting a wide variety of anions. They are expressed in various tissues and organs, with some paralogs displaying a specific tissue and cellular and/or subcellular expression pattern [17, 18, 29, 35, 40, 41, 46, 51, 52]. Several SLC26A members function as chloride/bicarbonate exchangers. These include SLC26A3 (DRA), SLC26A4 (pendrin), SLC26A6 (PAT1), SLC26A7, and SLC26A9. SLC26A7 and SLC26A9 can also function as chloride channels [10, 16, 27, 53].
In the present studies, Slc26a11 expression was examined by Northern and Western blot analysis and by immunohistochemical staining in brain sections. The highest expression of Slc26a11 was detected in Purkinje cells of the cerebellum. Several other regions of the brain were found to express intermediate to high expression levels, including pyramidal and lacunosum molecular cell layers of the hippocampus, glomerular layer of olfactory bulb, layers II, III, and V of the cerebral cortex, Zo and SuG layers of superior colliculus, interpeduncular nuclei, and substantia nigra (Table 1). At the subcellular level, KBAT was detected both in the cytoplasm and at the plasma membrane, raising the possibility that it might be regulated by trafficking to and from the plasma membrane.
To investigate the functional identity of Slc26a11, patch clamp studies in HEK293 cells transfected with Slc26a11 cDNA were performed. Our data indicated that Slc26a11 is a constitutively active Cl− channel, is not regulated by cAMP or intracellular calcium, and conducts Cl− and Br− better than I− (Figs. 4 and 5). These aspects of KBAT functional profile—being active at baseline state and not affected by cAMP or calcium—make this channel distinct from several other Cl− channels such as CFTR or calcium-activated chloride channels, which are not active under baseline conditions and need cAMP or calcium, respectively, to be activated. It is therefore possible that KBAT may play an important role in baseline membrane potential regulation and/or intra- and extracellular ion homeostasis. It is worth mentioning that in addition to KBAT-mediated Cl− conductance, other conductances, particularly K+ conductances, could potentially contribute to the overall conductance and the stabilization of membrane voltage.
Our experiments indicate that decreasing the extracellular Na+ and Cl− from 145 to 29 mmol/l NaCl resulted in a significant reduction in whole cell current, with a mild shift (∼12 mV) in the reversal potential. These results indicate that SLC26A11 may also be capable of functioning as an electrogenic chloride transporter, with the ability to carry a cation such as sodium. These aspects of SLC26A11 are very similar to those of SLC26A9 which has been shown to function as a chloride channel, an electrogenic Cl−/HCO3 − exchanger, and an electrogenic Na-coupled Cl− transporter [8, 10, 16, 53].
It is plausible that KBAT-mediated chloride current could regulate electrical activity in neurons by regulating the intracellular chloride concentration and/or the magnitude of membrane hyperpolarization. In support of this latter possibility, several studies have demonstrated that the opening of anion channels, in particular Cl− channels, lead to anion influx and hyperpolarization, with subsequent inhibition of electrical activity in neurons [14]. In fact, such functions have also been described for Purkinje cells [14]. Our assessment of the role of Slc26a11 (KBAT) in the mediation of chloride conductance in Purkinje cells is based on both the specific localization studies in Purkinje cells (Figs. 1, 2, and 3) and the functional properties that were ascertained in cultured cells transfected with the KBAT cDNA (Figs. 4 and 5). Thus, any extrapolation regarding Slc26a11 contribution to Purkinje cells current is at best indirect and requires verification in native cells.
Given the fact that Slc26a11 is obviously trafficked to the cell membrane, where it produces a Cl− conductance, we want to emphasize that all cellular components that are required for the generation of such a current are intact in HEK293 cells. From earlier studies and the present report, it is also clear that HEK293 cells possess all necessary components to regulate/activate ion channels by Ca2+, Ca2+-dependent kinases, cAMP, or other common kinases. Whether there are factors or proteins acting as accessory subunits to Slc26a11 in Purkinje cells that are missing in HEK293 cells cannot be excluded.
Disrupting the Kcc2 KCl cotransporter in Purkinje cells increases [Cl−]i twofold and, as a consequence, the reduced Cl− gradient nearly abolishes GABA-induced hyperpolarization in these cells. Since some forms of motor learning and consolidation are affected in Purkinje cell-specific knockouts of Kcc2 [39], it will be interesting to find out whether knockouts of KBAT show similar aberrations in their behavior. Such disrupting effects on procedural memory formation may come about by corrupting the spatiotemporal patterns of simple spike activity of Purkinje cells [9, 41]. These activity patterns of Purkinje cells are largely determined by the feed-forward inhibition from the molecular layer interneurons [50]. Indeed, ablation of synaptic inhibition of Purkinje cells by deleting GABAA receptors (PC-Δγ2 mice) leads to altered regularity of Purkinje cell firing patterns as well as learning and consolidation deficits.
Inhibitory circuits mediated by Cl− channels are also shown to have vital roles in enhancing network performance in other brain regions, including the cerebral cortex and hippocampus. It is known that inhibitory circuits in the hippocampus are important for synaptic integration and action potential generation in pyramidal cells, which are necessary for encoding declarative memories [32]. Thus, identification of new chloride channels and their functions could shed new light on the fundamental mechanisms involved in plasticity of neuronal networks and the necessity of related inhibitory circuits in various regions of the brain.
In nerve cells, changes in intracellular pH can affect neuronal excitability subsequent to their effects on ion channel regulation [44]. Further, neuronal activity can affect intracellular pH. It is therefore highly plausible that regulation of acid extruding pathways in neurons would be critical for homeostasis. Lastly, neurons by virtue of their high metabolic rates may be susceptible to injury if exposed to increased amount of acid load, which can happen during anoxia or ischemia. Therefore, pH regulation of neurons is likely to be crucial in both physiological and pathophysiological conditions. As such, KBAT may play an important role in intracellular pH regulation by modifying H+-ATPase activity.
Several chloride channels [45–48] have been shown to activate vacuolar H+-ATPases (V-ATPases) that are ATP-dependent proton pumps translocating H+ across the plasma membranes of certain specialized cells such as renal epithelial cells and osteoclasts [1, 5, 36, 55] and are also essential for acid transport across the endomembrane of subcellular organelles, such as lysosomes and endosomes [20, 26, 37].
The localization of KBAT and V H+-ATPase to the plasma membrane of Purkinje cells raises interesting possibilities. The first one is that KBAT, via its chloride current, could play an important role in regulating the intracellular chloride concentration and chloride gradient across the neuronal plasma membrane and in intracellular compartments. As such, KBAT may play a role in neuronal excitability or the regulation of presynaptic inhibition by ligand-gated (GABA-R or glycine) chloride channels. The second possibility is that KBAT, via activation of H+-ATPase, could play an important role in the concentration of neurotransmitters in synaptic vesicles in Purkinje cells. It has been demonstrated that the accumulation of neurotransmitters in synaptic vesicles is in large part regulated by specific vesicular transporters that couple with the electrochemical proton gradient generated by V-ATPase [22, 23].
In conclusion, our results demonstrate the identification of a novel chloride channel in the brain, with a most prominent expression in cerebellar Purkinje cells. This channel, known as Slc26a11 and designated as KBAT, is constitutively active and is not regulated by cAMP or calcium. KBAT is also capable of functioning as an electrogenic chloride transporter. KBAT colocalizes with V H+-ATPase in plasma membrane and the cytoplasmic compartments in Purkinje cells and can regulate acid translocation via H+-ATPase in cultured cells. We propose that KBAT plays a role in neuronal excitability or the regulation of presynaptic inhibition by ligand-gated (GABA-R or glycine) chloride channels and is likely involved in H+-ATPase-mediated accumulation of neurotransmitters in synaptic vesicles.
References
Alper SL, Natale J, Gluck S, Lodish HF, Brown D (1989) Subtypes of intercalated cells in rat kidney collecting duct defined by antibodies against erythroid band 3 and renal vacuolar H+-ATPase. Proc Natl Acad Sci U S A 86(14):5429–5433
Amlal H, Goel A, Soleimani M (1998) Activation of H+-ATPase by hypotonicity: a novel regulatory mechanism for H+ secretion in IMCD cells. Am J Physiol 275(4 Pt 2):F487–F501
Bellemer A, Hirata T, Romero MF, Koelle MR (2011) Two types of chloride transporters are required for GABA(A) receptor-mediated inhibition in C. elegans. EMBO J 30(9):1852–1863
Berend K, van Hulsteijn LH, Gans RO (2012) Chloride: the queen of electrolytes? Eur J Intern Med 23(3):203–211
Blake-Palmer KG, Karet FE (2009) Cellular physiology of the renal H+ ATPase. Curr Opin Nephrol Hypertens 18(5):433–438, Review
Brown D, Hirsch S, Gluck S (1988) Localization of a proton-pumping ATPase in rat kidney. J Clin Invest 82(6):2114–2126
Burge JA, Hanna MG (2012) Novel insights into the pathomechanisms of skeletal muscle channelopathies. Curr Neurol Neurosci Rep 12(1):62–69, Review
Chang MH, Plata C, Zandi-Nejad K, Sindić A, Sussman CR, Mercado A, Broumand V, Raghuram V, Mount DB, Romero MF (2009) Slc26a9—anion exchanger, channel and Na+ transporter. J Membr Biol 228(3):125–140
De Zeeuw C, Hoebeek F, Bosman L et al (2011) Spatiotemporal firing patterns in the cerebellum. Nat Rev Neurosci 12:327–344
Dorwart MR, Shcheynikov N, Wang Y, Stippec S, Muallem S (2007) SLC26A9 is a Cl− channel regulated by the WNK kinases. J Physiol 584:333–345
Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED (1997) Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 17:411–422
Hoglund P, Haila S, Socha J, Tomaszewski L, Saarialho-Kere U, Karjalainen-Lindsberg M-L, Airola K, Holmberg C, de la Chapelle A, Kere J (1996) Mutations of the down-regulated in adenoma (DRA) gene cause congenital chloride diarrhea. Nat Genet 14:316–319
Jentsch TJ, Stein V, Weinreich F, Zdebik AA (2002) Molecular structure and physiological function of chloride channels. Physiol Rev 82(2):503–568
Karlsson U, Druzin M, Johansson S (2011) Cl− concentration changes and desensitization of GABAA and glycine receptors. J Gen Physiol 138:609–626
Kasper D, Planells-Cases R, Fuhrmann JC, Olaf S, Oliver Z, Klaus R, Schmitt A, Mallorie P, Robert S, Michaela S, Uwe K, Jentsch TJ (2005) Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J 24(5):1079–1091
Kim KH, Shcheynikov N, Wang Y, Muallem S (2005) SLC26A7 is a Cl− channel regulated by intracellular pH. J Biol Chem 280:6463–6470
Lohi H, Kujala M, Kerkela E, Saarialho-Kere U, Kestila M, Kere J (2000) Mapping of five new putative anion transporter genes in human and characterization of SLC26A6, a candidate gene for pancreatic anion exchanger. Genomics 70:102–112
Lohi H, Kujala M, Makela S, Lehtonen E, Kestila M, Saarialho-Kere U, Markovich D, Kere J (2002) Functional characterization of three novel tissue-specific anion exchangers SLC26A7, -A8, and -A9. J Biol Chem 277:14246–14254
Lynch J (2003) Molecular structure and function of the glycine receptor chloride channel. J Physiol Rev 84:1051–1095
Marshansky V, Futai M (2008) The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol 20(4):415–426, Review
Martins JR, Faria D, Kongsuphol P, Reisch B, Schreiber R, Kunzelmann K (2011) Anoctamin 6 is an essential component of the outwardly rectifying chloride channel. Proc Natl Acad Sci U S A 108(44):18168–18172
Moriyama Y, Maeda M, Futai M (1992) The role of V-ATPase in neuronal and endocrine systems. J Exp Biol 172:171–178
Moriyama Y, Tsai HL, Futai M (1993) Energy-dependent accumulation of neuron blockers causes selective inhibition of neurotransmitter uptake by brain synaptic vesicles. Arch Biochem Biophys 305:278–281
Mulberg A, Resta L, Windner E et al (1994) Expression and localization of the cystic fibrosis transmembrane conductance regulator mRNA and its protein in rat brain. J Clin Invest 96:646–652
Murata Y, Sun-Wada GH, Yoshimizu T, Yamamoto A, Wada Y, Futai M (2002) Differential localization of the vacuolar H+ pump with G subunit isoforms (G1 and G2) in mouse neurons. J Biol Chem 277(39):36296–36303
Nelson N, Perzov N, Cohen A, Hagai K, Padler V, Nelson H (2000) The cellular biology of proton-motive force generation by V-ATPases. J Exp Biol 203(Pt 1):89–95, Review
Ohana E, Yang D, Shcheynikov N, Muallem S (2009) Diverse transport modes by the solute carrier 26 family of anion transporters. J Physiol 587(Pt 10):2179–2185, Review
Okada Y, Sato K, Numata T (2009) Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J Physiol 587(Pt 10):2141–2149, Review
Petrovic S, Ju X, Barone S, Seidler U, Alper SL, Lohi H, Kere J, Soleimani M (2003) Identification of a basolateral Cl-/HCO3- exchanger specific to gastric parietal cells. Am J Physiol Gastrointest Liver Physiol 284(6):G1093–G1103
Plans V, Rickheit G, Jentsch TJ (2009) Physiological roles of CLC Cl(−)/H (+) exchangers in renal proximal tubules. Pflugers Arch 458(1):23–37, Review
Poëa-Guyon S, Amar M, Fossier P, Morel N (2006) Alternative splicing controls neuronal expression of v-ATPase subunit a1 and sorting to nerve terminals. J Biol Chem 281(25):17164–17172
Pouille F, Scanziani M (2001) Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293:1159–1163
Ratte´ S, Prescott S (2011) ClC-2 channels regulate neuronal excitability, not intracellular chloride levels. J Neurosci 31(44):15838–15843
Rivera C, Voipio J, Kaila K (2005) Two developmental switches in GABAergic signaling: the K+-Cl− cotransporter KCC2 and carbonic anhydrase CAVII. J Physiol 562(1):27–36
Romero MF, Chang MH, Plata C, Zandi-Nejad K, Mercado A, Broumand V, Sussman CR, Mount DB (2006) Physiology of electrogenic SLC26 paralogues. Novartis Found Symp 273:126–138
Saroussi S, Nelson N (2009) Vacuolar H(+)-ATPase-an enzyme for all seasons. Pflugers Arch 457(3):581–587, Review
Saroussi S, Nelson N (2009) The little we know on the structure and machinery of V-ATPase. J Exp Biol 12:1604–1610, Review
Schweinfest CW, Spyropoulos DD, Henderson KW, Kim JH, Chapman JM, Barone S, Worrell RT, Wang Z, Soleimani M (2006) Slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. J Biol Chem 281:37962–37971
Seja P, Schonewille M, Spitzmaul G et al (2012) Raising cytosolic Cl− in cerebellar granule cells affects their excitability and vestibulo-ocular learning. EMBO I 31:1217–1230
Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, Burnham CE (2001) Pendrin: an apical Cl-/OH-/HCO3- exchanger in the kidney cortex. Am J Physiol Ren Physiol 280:F356–F364
Soleimani M, Xu J (2006) SLC26 chloride/base exchangers in the kidney in health and disease. Semin Nephrol 26(5):375–385
Stadler H, Tsukita S (1984) Synaptic vesicles contain an ATP-dependent proton pump and show 'knob-like' protrusions on their surface. EMBO J 3:3333–3337
Strauss O, Neussert R, Müller C, Milenkovic VM (2012) A potential cytosolic function of bestrophin-1. Adv Exp Med Biol 723:603–610, Review
Takahashi KI, Copenhagen DR (1996) Modulation of neuronal function by intracellular pH. Neurosci Res 24(2):109–116
Tornberg J, Voikar V, Savilahti H et al (2005) Behavioral phenotype of hypomorphic KCC2-deficient mice. J Eur J NeuroSci 21:1327–1337
Vincourt JB, Jullien D, Amalric F, Girard JP (2003) Molecular and functional characterization of SLC26A11, a sodium-independent sulfate transporter from high endothelial venules. FASEB J 17:890–892
Walcott BP, Kahle KT, Simard JM (2012) Novel treatment targets for cerebral edema. Neurotherapeutics 9(1):65–72, Review
Wellhauser L, D'Antonio C, Bear CE (2010) ClC transporters: discoveries and challenges in defining the mechanisms underlying function and regulation of ClC-5. Pflugers Arch 460(2):543–557, Review
Weyler R, Yurko-Mauro K, Rubenstein R et al (1999) CFTR is functionally active in GnRH-expressing GT1-7 hypothalamic neurons. Am J Physiol Cell Physiol 277:C563–C571
Wulff P, Schonewille M, Renzi M, Viltono L, Sassoè-Pognetto M, Badura A, Gao Z, Hoebeek FE, van Dorp S, Wisden W, Farrant M, De Zeeuw CI (2009) Synaptic inhibition of Purkinje cells mediates consolidation of vestibulo-cerebellar motor learning. Nat Neurosci 12(8):1042–1049
Xu J, Song P, Nakamura S, Miller M, Barone S, Alper SL, Riederer B, Bonhagen J, Arend LJ, Amlal H, Seidler U, Soleimani M (2009) Deletion of the chloride transporter slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J Biol Chem 284(43):29470–29479
Xu J, Barone S, Li H, Holiday S, Zahedi K, Soleimani M (2011) Slc26a11, a chloride transporter, localizes with the vacuolar H(+)-ATPase of A-intercalated cells of the kidney. Kidney Int 80(9):926–937
Xu J, Song P, Miller ML, Borgese F, Barone S, Riederer B, Wang Z, Alper SL, Forte JG, Shull GE, Ehrenfeld J, Seideler U, Soleimani M (2008) Deletion of the chloride transporter Slc26a9 causes loss of tubulovesicles in parietal cells and impairs acid secretion in the stomach. Proc Natl Acad Sci U S A 105(46):17955–17960
Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U (2008) TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455(7217):1210–1215
Yao G, Feng H, Cai Y, Qi W, Kong K (2007) Characterization of vacuolar-ATPase and selective inhibition of vacuolar-H(+)-ATPase in osteoclasts. Biochem Biophys Res Commun 357(4):821–827, Review
Young A, Chu D (1990) Distribution of GABAA and GABAB receptors in mammalian brain: potential targets for drug development. J Drug Dev Res 21:161–167
Zhang Z, Nguyen KT, Barrett EF, David G (2010) Vesicular ATPase inserted into the plasma membrane of motor terminals by exocytosis alkalinizes cytosolic pH and facilitates endocytosis. Neuron 68(6):1097–1108
Acknowledgments
The authors express appreciation to M. Rutteman and E. Haasdijk from Erasmus MC for their contribution and to Dr. D. Jaarsma from Erasmus and Dr. Masato Nakafuku, Professor of Pediatrics, Division of Developmental Biology, Cincinnati Children's Hospital Research Foundation, for their helpful discussions. These studies were supported by the Dutch Organization for Medical Sciences (ZonMw; CIDZ), DFG SFB699A7 (KK), National Institute of Health R56DK62809 (MS), Life Sciences (ALW; CIDZ), Senter (Neuro-Basic; CIDZ), Merit Review Award from the Department of Veterans Administration (MS), Prinses Beatrix Fonds (CIDZ), ERCadvanced, CEREBNET and C7 programs of the European Community (CIDZ), and funds from US Renal Care (MS) and Center on Genetics of Transport at University of Cincinnati (MS).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Rahmati, N., Kunzelmann, K., Xu, J. et al. Slc26a11 is prominently expressed in the brain and functions as a chloride channel: expression in Purkinje cells and stimulation of V H+-ATPase. Pflugers Arch - Eur J Physiol 465, 1583–1597 (2013). https://doi.org/10.1007/s00424-013-1300-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-013-1300-6