
Abstract
Dent’s disease is characterized by defective endocytosis in renal proximal tubules (PTs) and caused by mutations in the 2Cl−/H+ exchanger, CLC-5. However, the pathological role of endosomal acidification in endocytosis has recently come into question. To clarify the mechanism of pathogenesis for Dent’s disease, we examined the effects of a novel gating glutamate mutation, E211Q, on CLC-5 functions and endosomal acidification. In Xenopus oocytes, wild-type (WT) CLC-5 showed outward-rectifying currents that were inhibited by extracellular acidosis, but E211Q and an artificial pure Cl− channel mutant, E211A, showed linear currents that were insensitive to extracellular acidosis. Moreover, depolarizing pulse trains induced a robust reduction in the surface pH of oocytes expressing WT CLC-5 but not E211Q or E211A, indicating that the E211Q mutant functions as a pure Cl− channel similar to E211A. In HEK293 cells, E211A and E211Q stimulated endosomal acidification and hypotonicity-inducible vacuolar-type H+-ATPase (V-ATPase) activation at the plasma membrane. However, the stimulatory effects of these mutants were reduced compared with WT CLC-5. Furthermore, gene silencing experiments confirmed the functional coupling between V-ATPase and CLC-5 at the plasma membrane of isolated mouse PTs. These results reveal for the first time that the conversion of CLC-5 from a 2Cl−/H+ exchanger into a Cl− channel induces Dent’s disease in humans. In addition, defective endosomal acidification as a result of insufficient V-ATPase activation may still be important in the pathogenesis of Dent’s disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dent’s disease is an X-linked disorder that causes a dysfunction in renal proximal tubules (PTs) and is characterized by low-molecular-weight proteinuria (LMWP), hypercalciuria, nephrocalcinosis, nephrolithiasis, and renal failure [6, 8, 9, 48, 51]. The etiology of Dent’s disease has been associated with mutations in the CLCN5 gene that encodes the electrogenic 2Cl−/H+ exchanger CLC-5 [22, 31, 36, 56].
CLC-5 belongs to the CLC family [20] and is abundantly expressed in early endosomes of the PT, where it is co-localized with vacuolar-type H+-ATPase (V-ATPase) [14, 35]. Several lines of evidence indicate that CLC-5 plays an essential role in normal PT endocytosis by facilitating V-ATPase-mediated endosomal acidification [25]. Indeed, previous studies showed that two different CLC-5 knockout (KO) mice exhibited defective endosomal acidification and LMWP due to defective endocytosis [15, 16, 32, 49]. Therefore, a hypothesis was proposed that CLC-5 provides a Cl− shunt pathway required for maximal endosomal acidification by V-ATPase.
The identification of CLC-5 as a 2Cl−/H+ exchanger rather than a Cl− channel [31, 36, 56] has posed a challenge to the above hypothesis, because endosomal Cl− transport by the 2Cl−/H+ exchanger may waste energy by recycling H+ between endosomes and the cytoplasm. Nevertheless, simulation studies suggested that the 2Cl−/H+ exchange mode of CLC-7, rather than the simple Cl− conductance, would be more efficient in lysosomal acidification, and this is applicable to the situation of CLC-5 in endosomes [19, 50]. In contrast, Smith and Lippiat proposed that CLC-5 directly acidifies endosomes by transporting H+ into endosomes in exchange of Cl− [41].
In the 2Cl−/H+ exchanger CLC-ec1 of Escherichia coli, a glutamate residue in the selectivity filter (E148) acts not only as the outside Cl− gate but also as the extracellular H+ acceptor [10, 11]. Replacement of this conserved “gating glutamate,” which corresponds to E211 in CLC-5, with alanine converts 2Cl−/H+ exchangers to pure Cl− channels [1, 2, 31, 36]. Interestingly, in transgenic mice, the E211A mutation of CLC-5 acts as an artificial pure Cl− channel mutant and causes defective PT endocytosis similar to that of CLC-5 KO mice. Unlike CLC-5 KO mice, however, the E211A mice completely preserved normal endosomal acidification, suggesting that endosomal Cl− accumulation is more important for PT endocytosis than endosomal acidification [29].
While more than 150 mutations in CLCN5 have been identified [9, 24], mutations in the “gating glutamate” have not been reported to date. Notably, a recent genetic study of Japanese patients with typical Dent’s disease identified a previously unrecognized CLC-5 E211Q mutation [39]. Interestingly, the E148Q mutant in CLC-ec1, like the E148A mutant, was shown to behave as a pure Cl− channel. In this study, we aimed to clarify the mechanism of pathogenesis for Dent’s disease by analyzing the effects of the E211Q mutation on CLC-5 functions and endosomal acidification.
Methods
Patient description
The patient was a 7-year-old boy who presented with high urinary β2-microglobulin (37,580 μg/L; normal range <230 μg/L), hypercalciuria (calcium/creatinine 0.313 mg/mg; normal range <0.2 mg/mg), and renal calcification without renal failure. The genetic analysis identified a codon change c.631G>C resulting in the CLC-5 E211Q mutation [39].
Animals
All animal procedures including sacrifice were approved by the University of Tokyo Ethics Committee for Animal Experiments and conformed to the guidelines for animal experiments of the University of Tokyo.
Molecular modeling of CLC-5
A three-dimensional model of the human CLC-5 transmembrane domains was constructed as previously reported [52] based on X-ray crystallographic data for CLC-ec1 [10, 11]. This model utilized the optimization of the molecular structure by energy minimization and molecular dynamic simulation. Molecular modeling and analysis were performed using a package for molecular structure analyses, Molecular Operating Environment (MOE 2011.10, Chemical Computing Group Inc., Québec, Canada http://www.chemcomp.com/). The E211Q and E211A mutant models were also constructed by the same manner.
Preparation of CLC-5-expressing constructs
Hemagglutinin (HA)-tagged human wild-type (WT) CLC-5 subcloned into pTLN [13] and pcDNA3.1 vectors were used for expression in Xenopus laevis oocytes and HEK293 cells, respectively. Both of the vectors were kindly provided by Dr. T. J. Jentsch (Leibniz-Institut für Molekulare Pharmakologie, Berlin, Germany). To introduce mutations, the QuickChange II XL Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA) was used and the complete cDNA sequences were verified by DNA sequencing.
Expression in oocytes
The mMESSAGE mMACHINE high-yield Capped RNA Transcription kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to synthesize capped cRNAs from the CLC-5 constructs. Female X. laevis were anesthetized before surgery by immersion in 0.2 % tricaine methanesulfonate (MS-222, Sigma-Aldrich, St. Louis, MO, USA) solution for 30 min. Oocytes were surgically collected from ovaries, treated with collagenase as described [17, 40, 47], and injected with 5 ng of each cRNA, while the frogs were kept in separate tanks for 2 days after the suture and put back in the aquarium. Electrophysiological experiments were performed 3–4 days after cRNA injection.
Voltage clamp in oocytes
An oocyte was placed in a perfusion chamber and superfused with nominally HCO3 −-free ND96 solution (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 5 mM HEPES) that was adjusted to pH 7.4. To measure the CLC-5 current, a two-electrode voltage clamp method consisting of 20 mV steps from −80 to +80 mV during 100 ms from a folding potential of −25 mV was performed with a model OC-725C oocyte clamp (Warner Instruments, Hamden, CT, USA) controlled by the Clampex module of pCLAMP software (Molecular Devices, Sunnyvale, CA, USA). Using the same voltage clamp method, we measured the CLC-5 current in perfusates of various pH (5.4, 6.4, 7.4, and 8.4) to examine its sensitivity to extracellular acidosis. For pH 6.4 and 5.4 solutions, 5 mM HEPES was replaced by 5 mM 2-(N-morpholino)-ethanesulfonic acid (MES). To exclude a contribution of any leaky endogenous currents, a reduction in current amplitude to the replacement of 80 mM Cl− in ND96 by equivalent amounts of I− was routinely confirmed [46].
Surface pH measurement in oocytes
An oocyte was voltage clamped and the surface pH of the oocyte was measured by pH-sensitive and reference microelectrodes connected to a Duo773 high-input impedance differential electrometer (WPI, Sarasota, FL, USA). Tips of pH-sensitive microelectrodes were siliconized with dichlorodimethylsilane vapor (Serva, Heidelberg, Germany) and filled with the ionophore cocktail (95293, Sigma-Aldrich) as described [21, 38], and only electrodes responding with a slope larger than 50 mV per pH unit were used. For these measurements, HEPES concentration in perfusates was reduced to 0.5 mM as described [31].
Expression in HEK293 cells
HEK293 cells were transfected with CLC-5 constructs using Lipofectamine 2000 (Thermo Fisher Scientific). To facilitate the expression of CLC-5 constructs, Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal calf serum and 5 mM sodium butyrate was used [7]. Measurements of cell and endosomal pH, immunoblotting, or immunofluorescence analysis were performed 48 h after transfection.
Measurement of V-ATPase activity in HEK293 cells
HEPES-buffered solution (127 mM NaCl, 5 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 1 mM Na2SO4, 25 mM HEPES, and 5.5 mM glucose) was adjusted to pH 7.4. In isotonic Na+-free solution, Na+ was replaced by equivalent amounts of N-methyl-d-glucamine. In hypotonic Na+-free solution, the osmolality was reduced to 210 mOsm [33]. In NH4Cl solution, 30 mM NaCl in HEPES-buffered solution was replaced by equimolar amounts of NH4Cl. Cellular pH was measured by detecting fluorescence emissions from the pH dye acetoxymethylester of bis(carboxyethyl)carboxyfluorescein (BCECF/AM; Doujin, Kumamoto, Japan) with a photometry system (OSP-10; Olympus, Tokyo, Japan) [17, 47, 53]. To induce intracellular acidification, cells were first pulsed with 30 mM NH4Cl for 3 min. Afterwards, the solution was exchanged for isotonic Na+-free solution followed by hypotonic Na+-free solution, which was previously shown to activate V-ATPase [3, 33]. Intracellular pH was calibrated by incubating the cells with solutions containing 120 mM KCl, 20 mM NaCl, and 10 μM nigericin adjusted to pH 6.5 to 7.5 [54].
Measurement of endosomal pH in HEK293 cells
To measure endosomal pH, we used a ratiometric biosensor comprised of the pH-sensitive GFP mutant pHluorin fused to the vesicle-associated membrane protein VAMP2, kindly provided by Dr. J. D. Lippiat (University of Leeds, Leeds, UK) [26, 41]. HEK293 cells were cotransfected with VAMP2-pHluorin and CLC-5 constructs. A Leica TCS SP5II confocal microscope (Leica Microsystems, Wetzlar, Germany) was used to excite VAMP2-pHluorin at 405 and 488 nm. Emissions were measured through a long-pass filter covering 505 to 696 nm. Endosomal pH was determined in two or three vesicles in a single cell across ≥20 cells that were transfected with CLC-5 constructs or a blank vector control. To convert the 405-/488-nm fluorescence ratio to pH, in situ pH calibration was performed as described [43].
Sample preparation, cell surface biotinylation, and immunoblotting
HEK293 cells were grown on a 10-cm culture dish, transfected with the CLC-5 constructs, collected, lysed with the lysis buffer (Tris/pH 8.0, 150 mM NaCl, 1 % Nonidet P-40, 0.5 % sodium deoxycholate, 0.1 % SDS, and 1 mM PMSF), and subjected to immunoblotting of total cellular fractions. To examine the effect of hypotonicity on plasma membrane V-ATPase expression, HEK293 cells transfected with each CLC-5 construct were superfused with 30 mM NH4Cl solution for 3 min, isotonic Na+-free solution for 90 s, and hypotonic Na+-free solution (210 mOsm) for 1 min at 37 °C. Thereafter, cell surface biotinylation was performed using the Pierce Cell Surface Protein Isolation Kit (Thermo Scientific) as described [47, 53] and samples were subjected to biotinylation western blotting. Immunoblotting of total cellular fractions or cell surface fractions was performed by using antibodies against HA (Roche Diagnostics, Meyland, France), B2 subunit of V-ATPase (Santa Cruz Biotechnology, Dallas, TX, USA), β-actin (Merck Millipore), or Na/K pump α1 (Merck Millipore, Darmstadt, Germany).
Immunofluorescence
HEK293 cells were cotransfected with CLC-5 constructs and VAMP2-pHluorin and grown on polylysine-coated coverslips. After fixation with 4 % paraformaldehyde in PBS, cells were permeabilized with 0.1 % Triton X-100 (Sigma-Aldrich) and stained with a rabbit polyclonal anti-HA antibody (BETHYL, Montgomery, TX, USA) followed by an Alexa Fluor 546 goat anti-rabbit IgG (Molecular Probes) secondary antibody. The Leica TCS SP5II confocal microscope was used to visualize VAMP2-pHluorin (488 nm) and CLC-5 constructs (543 nm).
Measurement of luminal Na+/H+ exchanger (NHE) and V-ATPase activities in isolated mouse PTs
PTs were isolated from 6-week-old C57BL/6 mice which were sacrificed by intraperitoneal injection of excessive amounts of pentobarbital and attached to a glass coverslip coated with Cell-Tak glue (Corning, NY, USA). A capillary glass was used to expose the luminal surface as described for isolated mouse distal tubules [37]. To measure luminal V-ATPase and NHE activities, intracellular pH was monitored with an inverted fluorescence microscope equipped with MetaFluor 7.7 software (Molecular Devices, Sunnyvale, CA, USA) similar to the measurements of V-ATPase activity in HEK293 cells. In brief, isolated and split-opened mouse PTs in the chamber were first loaded with BCECF/AM in HEPES-buffered solution for 5 to 10 min, and then the perfusate was switched to isotonic Na+-free solution. After intracellular acidification was achieved by the NHE activities, the solution was replaced with hypotonic Na+-free solution to activate plasma membrane V-ATPase. The V-ATPase activity was determined by the rates of cellular pH recovery that was observed both in isotonic and hypotonic Na+-free solutions. For the calculation of the activity, the cellular pH change during the initial 30 s was measured.
siRNA treatment in isolated mouse PTs
Freshly isolated mouse PTs were treated with siRNA for the V-ATPase B2 subunit, CLC-5, or scrambled negative control (all from Santa Cruz Biotechnology, Dallas, TX, USA) using Lipofectamine 2000 and were incubated overnight in DMEM supplemented with 10 % FBS as previously reported [27].
RNA extraction and quantitative PCR analysis
Total RNA was extracted from isolated mouse PTs by using Isogen II (Nippon Gene, Tokyo, Japan), and cDNA was synthesized with the cDNA Synthesis Kit (Takara, Tokyo, Japan) as previously described [27]. Quantitative PCR was performed with TaqMan Gene Expression Master Mix (Applied Biosystems, Foster City, CA, USA), TaqMan Gene Expression Assays (Mm00431987_ml for V-ATPase B2, Mm00443851_ml for CLC-5, and Mm00607939_sl for β-actin; all from Applied Biosystems), and sequence detection system (7500 Fast Real-time PCR System; Applied Biosystems). The expression level was quantified relative to the abundance of β-actin cDNA.
Statistical analysis
The data were presented as the mean ± SEM. Unpaired Student’s t test or ANOVA with Bonferroni’s correction were used to determine significant differences as appropriate. Statistical significance was set at p < 0.05.
Results
Functional analysis of E211Q in Xenopus oocytes
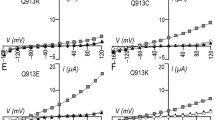
We first examined the functional properties of the CLC-5 E211Q mutant. Figure 1a shows the current–voltage relationships of oocytes expressing WT CLC-5, the E211Q mutant, or the artificial E211A mutant. WT CLC-5 showed strongly outwardly rectifying currents as previously reported [13, 46]. In contrast, the pure Cl− channel E211A mutant showed nearly linear currents [31, 36]. Notably, the E211Q mutant also showed linear currents similar to that of E211A. Although extracellular acidosis significantly reduced the WT currents, E211A and E211Q mutants had no reduction in currents as shown in Fig. 1b. These results suggest that the E211Q mutant behaves as a pure Cl− channel.

Electrophysiological properties of WT and CLC-5 mutants. a Steady-state current–voltage relationships of oocytes expressing WT CLC-5 (n = 8), E211A (n = 9), and E211Q mutants (n = 6). b Current sensitivity to changes in external pH. Currents at 80 mV were monitored for each oocyte expressing WT CLC-5 (n = 6), the E211A mutant (n = 7), or the E211Q mutant (n = 7) (*p < 0.05 versus pH 7.4). Each data point is normalized to the current value at pH 7.4. c A protocol of each single depolarizing pulse. d A representative CLC-5 current elicited by a single depolarizing pulse. e Changes in surface pH of oocytes in response to repetitive depolarizing pulse trains measured with pH-sensitive and reference microelectrodes
To further characterize the properties of the E211Q mutant, we examined changes in the surface pH of oocytes in response to trains of depolarizing pulses (Fig. 1c). As shown in Fig. 1d, e, WT CLC-5 elicited an immediate and reversible reduction in surface pH by 0.31 ± 0.04 (n = 4) in response to the pulse trains, consistent with its function as a 2Cl−/H+ exchanger [31]. In contrast, either the E211A mutant (n = 7) or the E211Q (n = 7) mutant failed to elicit changes in surface pH in response to the pulse trains. These results confirmed that the E211Q mutation, like E211A, converts CLC-5 into the pure Cl− channel.
Effects of E211Q and E211A mutations on CLC-5 gaiting structure
To gain insight into molecular mechanism underlying the mutation-based changes in CLC-5 function, we also performed simulation analysis on the gating structure. Transmembrane domain molecular structures of WT CLC-5, E211Q, and E211A mutant were predicted based on X-ray crystallographic data of CLC-ec1 [10, 11].
Figure 2a shows that carboxyl group of E211 in WT CLC-5 was fixed by hydrogen bonds to main-chain NH groups of E211, G212, and I456. The homologous E148 in CLC-ec1 has also similar interaction to neighboring main-chain [23]. Figure 2b illustrates the structural effects of the E211Q mutation. In the case of Q211, the side-chain direction was changed, and only hydrogen bond to Q211 main-chain NH group remained. The NH2 group of Q211 would break the hydrogen bonds to G212 and I456, and its partially positive charge weakly interacted to a neighboring chloride ion. This “open conformation,” as also observed in CLC-ec1 E148Q mutation [23], is similar to the structure of a temporary protonated E211 in the theoretical model of human CLC-5 proton transformation [55]. Figure 2c also illustrates the structural effects of the E211A mutation. In this model, A211 main-chain was shifted away from F455-I456 due to its small side-chain. Thus, the conformational shift of neighboring side-chains (including F455, I456, and Y558) in the E211A mutant was significantly larger enough to form slightly wider “open conformation” than that in the E211Q mutant. The similar main-chain shift was also observed in the CLC-ec1 E148A mutation [28]. Unlike the E211Q mutant, the E211A mutant failed to directly interact with a chloride ion due to the lack of NH2 group.

Predicted molecular structures of CLC-5 constructs. Transmembrane domain molecular structure of WT human CLC-5, E211Q, and E211A were predicted on X-ray crystallographic data of CLC-ec1 and shown in a, b, and c, respectively. E211 (WT), Q211 (mutant), and A211 (mutant) are colored in yellow, light blue, and deep green. Oxygen, nitrogen, and chloride are colored in red, blue, and light green, respectively. Hydrogen atoms are not described. Theoretical van del Waals surface of chloride ions are described in light green. Hydrogen bonds and distances related to side-chains of E211 and Q211 are shown by green dashed lines with the lengths (Å)
These structural analyses confirmed that the protonation of E211 played a key role in the gating mechanism for Cl− transport [30, 55]. Furthermore, E211Q and E211A mutations were expected to induce similar changes in the gating structure, resulting in an “open conformation” that allowed for free Cl− transport even in the absence of E211 protonation.
Effects of CLC-5 constructs on V-ATPase in HEK293 cells
We next examined the impact of WT and mutant CLC-5 constructs on plasma membrane V-ATPase activity in HEK293 cells. Cellular pH recovery from acid load was monitored in the absence of Na+, and V-ATPase was activated by hypotonicity as previously reported [3, 33]. As shown in Fig. 3a, superfusion with isotonic Na+-free solution after NH4Cl pulse decreased cellular pH to approximately 6.8. While the subsequent hypotonic Na+-free solution had a negligible effect on pH recovery in cells transfected with the control vector alone, WT CLC-5 induced a rapid pH recovery that was completely inhibited by 200 nM bafilomycin, an effect that is consistent with the activation of V-ATPase [3, 33]. Both of the E211A and E211Q mutants also activated V-ATPase, but the magnitude of V-ATPase activation was reduced compared with WT CLC-5 (Fig. 3b). Western blot analysis confirmed that E211A levels were comparable to that of WT CLC-5, and E211Q levels were higher than WT CLC-5 as shown in Fig. 3c, d. This indicates that the reduced activation of V-ATPase by E211A and E211Q mutants was not due to a difference in protein expression levels of these mutants.

Effects of CLC-5 constructs on V-ATPase in HEK293 cells. a Intracellular pH tracings of WT or the control vector are shown. Note that WT, but not vector, exhibited a rapid cell pH recovery in hypotonic Na+-free solution. b V-ATPase activity determined by the rates of intracellular pH recovery in hypotonic Na+-free solution (*p < 0.05 versus vector, and # p < 0.05 versus WT). c Total cellular expression of CLC-5 constructs and β-actin in HEK293 cells. d Intensity data for CLC-5 constructs/β-actin semi-quantified by densitometry and normalized to the average value of WT (n = 5 for each construct) (# p < 0.05 versus WT)
We also examined the surface expression of the V-ATPase B2 subunit. Figure 4a shows that the CLC-5 constructs had no effect on the surface expression of endogenous V-ATPase. In addition, Fig. 4b, c shows that incubation with hypotonic Na+-free solution did not impact the surface expression of V-ATPase. These results indicate that the difference in surface expression of V-ATPase cannot account for the difference in the degree of V-ATPase activation by CLC-5 constructs. These data also confirmed that hypotonicity stimulates V-ATPase activity without altering V-ATPase surface expression as previously reported [3, 33].

Surface expression of endogenous V-ATPase B2 subunit in HEK293 cells. a Biotinylation western blotting in isotonic Na+-free solution. Intensity data for V-ATPase B2 subunit/Na+-K+ ATPase were semi-quantified by densitometry and normalized to the average value of the vector (n = 4 for each construct). b Biotinylation western blotting in hypotonic Na+-free solution. Intensity data for V-ATPase B2 subunit/Na-K ATPase were semi-quantified by densitometry and normalized to the average value of the vector (n = 4 for each construct). c Effects of hypotonicity on surface expression of B2 subunit. Open bars represent data in isotonic solution, and closed bars represent data in hypotonic solution (n = 4 for each construct)
Effects of CLC-5 constructs on endosomal acidification in HEK293 cells
We next examined the effects of CLC-5 constructs on endosomal acidification in HEK293 cells by using VAMP2-pHluorin [41]. Figure 5a illustrates the localization of CLC-5 and VAMP2-pHluorin in HEK293 cells transfected with each CLC-5 construct. As can be seen in these images, WT CLC-5, E211A, and E211Q showed similar intracellular localization in endosomes and at the plasma membrane. Figure 5b shows that while all of the CLC-5 constructs significantly reduced endosomal pH, the magnitude of acidification by E211A and E211Q was reduced compared with WT. Given that bafilomycin abolished the effects of the CLC-5 constructs on endosomal acidification (Fig. 5c), CLC-5-induced endosomal acidification reflected V-ATPase activity at the endosome.

Effects of CLC-5 constructs on endosomal pH. a Co-localization of HA-CLC-5 constructs and VAMP2-pHluorin. Confocal images of HEK293 cells showed that each CLC-5 construct (red) and VAMP-2 pHluorin (green) localized in endosomes and also at the plasma membrane. Scale bars = 10 μm. b Endosomal pH measured by the ratiometric VAMP2-pHluorin analysis. (*p < 0.05 versus Vector and # p < 0.05 versus WT). c Endosomal pH in the presence of 200 nM bafilomycin
Effects of CLC-5 on plasma membrane V-ATPase in isolated mouse PTs
While the activation of endosomal V-ATPase by CLC-5 has been well established [14, 42], the activation of plasma membrane V-ATPase by CLC-5 is rather unexpected. Therefore, we examined whether CLC-5 was required for the plasma membrane V-ATPase activation in isolated mouse PTs.
Figure 6a shows an isolated and split-opened mouse PT. In this preparation, luminal NHE activity was reliably measured (Fig. 6b, c). Moreover, Fig. 6d, e shows that hypotonicity activated bafilomycin-sensitive V-ATPase also in isolated mouse PTs. As shown in Fig. 7, luminal NHE and V-ATPase activities were largely preserved after overnight incubation with control small interference RNA (siRNA), consistent with the functional preservation of cultured PTs through the duration of the experiments [27].

Activities of luminal transporters in freshly isolated and split-opened mouse PTs. a An image of an isolated and split-opened mouse PT. It was attached to a glass coverslip by a tissue adhesive, and one side was split by a glass pipet (shown in a yellow circle). a Scale bar = 10 μm. b Intracellular pH tracings of lumen-open and lumen-closed mouse PTs in response to Na+ removal. Lumen-open PTs exhibited a faster and greater pH decrease by Na+ removal, which reflected the higher NHE activity. c NHE activity determined by the rates of cellular pH decrease in response to Na+ removal. Open and closed bars indicate data from lumen-open and lumen-closed PTs, respectively (n = 6 for each and *p < 0.05 versus lumen-closed). d Hypotonicity-inducible luminal V-ATPase activation in lumen-open mouse PTs. e Bafilomycin-sensitive V-ATPase activity in lumen-open mouse PTs. Open and closed bars indicate data in isotonic (n = 6) and hypotonic (n = 7) Na+-free solution, respectively (*p < 0.05 versus isotonic)

Preserved activities of luminal transporters in isolated mouse PTs after overnight incubation with control small interference RNA (siRNA). a NHE activity of freshly isolated PTs (day 0) and PTs treated with control siRNA (day 1). Open and closed bars indicate data from day 0 and day 1, respectively (n = 6 for each and *p < 0.05 versus day 0). b Plasma membrane V-ATPase activity of freshly isolated PTs (day 0) and PTs treated with control siRNA (day 1), activated by hypotonic solution. Open and closed bars indicate data from day 0 and day 1, respectively (n = 7 for day 0, n = 6 for day 1, and *p = 0.046 versus day 0). c Plasma membrane V-ATPase activity of freshly isolated PTs (day 0) and PTs treated with control siRNA (day 1) in isotonic solution. Open and closed bars indicate data from day 0 and day 1, respectively (n = 6 for each and *p < 0.05 versus day 0)
Figure 8a, b shows that treatment with siRNA against V-ATPase B2 subunit or CLC-5 effectively and selectively suppressed the mRNA expression of B2 subunit or CLC-5, respectively. Consistent with the predominant role of the B2 subunit in V-ATPase in PTs [4], siRNA against B2 markedly suppressed hypotonicity-induced V-ATPase activity in PTs and, moreover, siRNA against CLC-5 also largely suppressed V-ATPase activity as shown in Fig. 8c, d. These results indicate that CLC-5 is required for plasma membrane V-ATPase activity not only in cultured cells but also in intact PTs.

V-ATPase activity in isolated mouse PTs requires CLC-5. Isolated mouse PTs were treated with scrambled siRNA (control), siRNA against V-ATPase B2 subunit (anti-B2), or siRNA against CLC-5 (anti-CLC-5). a mRNA expression level of V-ATPase B2 subunit (n = 4). Each data point is normalized to the average value of control. b mRNA expression level of CLC-5 (n = 4). Each data point is normalized to the average value of control. c Intracellular pH tracings of siRNA-treated PTs. Note that cell pH recovery in hypotonic Na+-free solution was markedly suppressed by anti-B2 or anti-CLC-5 siRNA. d Summary data for V-ATPase activity (8 for control, n = 7 for V-ATPase anti-B2 subunit, n = 8 for anti-CLC-5 and *p < 0.05 versus control)
Discussion
We performed the functional characterization of the “gating glutamate” mutation E211Q in CLC-5 identified in a typical Dent’s disease patient. In Xenopus oocytes, WT CLC-5 showed outward-rectifying currents that were inhibited by extracellular acidosis. On the other hand, E211Q, like the artificial mutant E211A, displayed linear currents that were insensitive to extracellular acidosis. While WT CLC-5 induced a robust decrease in surface pH in response to the depolarizing pulses, E211Q and E211A failed to induce such changes in surface pH. These results indicate that the E211Q mutant behaves as a Cl− channel similar to the E211A mutant. Simulation analysis based on the structure model of CLC-5 further supports this conclusion. Previously, the E211A mutant was shown to cause defective endocytosis in PTs of transgenic mice [29]. This study is the first to show that the E211Q mutation also converts the functional mode of CLC-5 from a 2Cl−/H+ exchanger into a Cl− channel, which leads to the pathogenesis of Dent’s disease in humans. Traditionally, endosomal acidification via functional coupling between CLC-5 and V-ATPase has been considered to be essential for normal endocytosis [25]. However, endosomal acidification of vesicles purified from PTs was reported to be completely preserved even in E211A mice. This seemed to suggest that Cl− accumulation might be critical for normal endocytosis rather than endosomal acidification [29, 50]. Nevertheless, our present study indicates that defective endosomal acidification due to insufficient V-ATPase activation still plays an important role in the pathogenesis of Dent’s disease.
In HEK293 cells, we observed that both of the E211Q and E211A mutants caused a moderate reduction in endosomal pH. However, endosomal acidification by these mutants was significantly less than that by WT CLC-5, in contrast to previous findings in E211A mice. Methodological differences may account for this discrepancy. For example, significant acidification could still be detected in endosomal vesicles prepared from mouse PT fractions even in the complete absence of CLC-5 [15]. In contrast, basal V-ATPase activity was negligible in HEK293 cells, lacking the endogenous CLC-5 activity [41]. Accordingly, the difference in endosomal acidification by the CLC-5 constructs as detected in the present study might have been missed in vesicles prepared from the E211A mice [29]. Some compensatory mechanisms might have also emerged in the gene-targeted mice. While the involvement of cell type-specific factors cannot be excluded, lack of basal V-ATPase activity in HEK293 cells may support the functional coupling of V-ATPase and CLC-5 at the plasma membrane as will be discussed below. On the other hand, our study confirms the model that the Cl− shunt pathway can facilitate endosomal acidification by V-ATPase [25, 34]. Although this observation contradicts the idea of direct endosomal acidification by CLC-5 [41], theoretical considerations also put the direct acidification theory in doubt [44, 45].
The previous simulation studies on lysosomal acidification support the advantage of the 2Cl−/H+ exchange mode. Thus, CLC-7 as an antiporter provides more negative intravesicular potential than a pure Cl− conductor, resulting in the electrochemical environments that are more favorable for V-ATPase [19, 50]. Such indirect activation of V-ATPase is one possible explanation for the more efficient endosomal acidification by WT CLC-5 than by the E211Q and E211A mutants. However, the present study raised another intriguing possibility that the 2Cl−/H+ exchange mode of WT CLC-5 directly activates V-ATPase.
At usual inside negative cell membrane potentials, the simple linear Cl− channel should provide a negative charge shunt by exporting Cl− from inside cells, leading to the acceleration of H+ extrusion. Conversely, the outwardly rectifying 2Cl−/H+ exchanger should be nearly inactive at this potential according to the current–voltage relationship (Fig. 1a) and would induce no changes in electrochemical gradients for H+. Nevertheless, WT actually activated plasma membrane V-ATPase in HEK293 cells more efficiently than the E211Q and E211A mutants. Moreover, silencing of CLC-5 by siRNA in isolated mouse PTs significantly reduced plasma membrane V-ATPase activity without changing the expression level of the B2 subunit. These results support the view that tight functional coupling between V-ATPase and CLC-5 exists also in the plasma membrane.
How did WT CLC-5 activate plasma membrane V-ATPase more efficiently than the Cl− channel mutants? The answer to this question is currently unknown, but V-ATPase is known to have multiple functions. For example, V-ATPase may be critical for endosomal pH-sensing mechanism by interacting with the small GTPase Afr6 and ARNO [18]. Moreover, a multi-protein complex including V-ATPase senses amino acids in lysosomes and thereby activates target of rapamycin (mTOR) kinase [57], which may in turn regulate megalin-mediated PT endocytosis [12]. It is therefore possible that the 2Cl−/H+ exchange mode of CLC-5 induces the maximal plasma membrane V-ATPase activation by recruiting unknown cellular factors and/or modifying V-ATPase functions. A previous study using immortalized rat renal proximal tubule cells also supports this view [5]. Hypothetical models for the functional coupling between V-ATPase and CLC-5 in both endosomes and plasma membrane were summarized in Fig. 9. On the other hand, it is unclear whether our present in vitro data directly corresponds to in vivo condition where many compensatory functions will happen. To clarify this issue, future studies on E211Q knock-in mice or PT cells from the patient carrying the E211Q mutation will be required.

Hypothetical models for the functional coupling between V-ATPase and CLC-5 in endosomes and plasma membrane. a A schematic model of endocytosis and localization of CLC-5 and V-ATPase in a renal proximal tubular cell. CLC-5 expressed in early endosomes with V-ATPase is involved in normal endocytosis, while it is also expressed in the plasma membrane with V-ATPase. Mutations in the coding gene CLCN5 cause Dent’s disease that is characterized primarily by low-molecular-weight proteinuria (LMWP). b Proposed models for roles of CLC-5 in endosome. In indirect acidification model, CLC-5 activates V-ATPase indirectly by functioning as a Cl− shunt pathway. Although the results of our present study supports this model, we cannot exclude that Cl− accumulation into endosome rather than Cl− shunting is more important in the same model. In direct acidification model, on the other hand, CLC-5 functions in the opposite direction as a Cl− exit pathway. c Functional coupling of V-ATPase and CLC-5 in the plasma membrane. CLC-5 activates V-ATPase in the plasma membrane via unknown mechanisms that require the 2Cl−/H+ exchange activity. This type of functional coupling between V-ATPase and CLC-5 is presumed to exist even in endosomes
In summary, our present study revealed that the conversion of CLC-5 2Cl−/H+ exchanger into the pure Cl− channel indeed induces Dent’s disease in humans. We found that the 2Cl−/H+ exchange mode of CLC-5 is required for the maximal V-ATPase activation in both endosome and plasma membrane. While we cannot exclude the potential involvement of endosomal Cl− accumulation, our data support the critical role of defective endosomal acidification due to insufficient V-ATPase activation in the pathogenesis of Dent’s disease.
References
Accardi A, Kolmakova-Partensky L, Williams C, Miller C (2004) Ionic currents mediated by a prokaryotic homologue of CLC Cl− channels. J Gen Physiol 123:109–19. doi:10.1085/jgp.200308935
Accardi A, Miller C (2004) Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature 427:803–7. doi:10.1038/nature02314
Amlal H, Goel A, Soleimani M (1998) Activation of H+-ATPase by hypotonicity: a novel regulatory mechanism for H+ secretion in IMCD cells. Am J Physiol 275:F487–501
Cao X, Yang Q, Qin J, Zhao S, Li X, Fan J, Chen W, Zhou Y, Mao H, Yu X (2012) V-ATPase promotes transforming growth factor-beta-induced epithelial-mesenchymal transition of rat proximal tubular epithelial cells. Am J Physiol Renal Physiol 302:F1121–32. doi:10.1152/ajprenal.00278.2011
Carraro-Lacroix LR, Lessa LM, Bezerra CN, Pessoa TD, Souza-Menezes J, Morales MM, Girardi AC, Malnic G (2010) Role of CFTR and ClC-5 in modulating vacuolar H+-ATPase activity in kidney proximal tubule. Cell Physiol Biochem 26:563–76. doi:10.1159/000322324
Claverie-Martin F, Ramos-Trujillo E, Garcia-Nieto V (2011) Dent’s disease: clinical features and molecular basis. Pediatr Nephrol 26:693–704. doi:10.1007/s00467-010-1657-0
D’Antonio C, Molinski S, Ahmadi S, Huan LJ, Wellhauser L, Bear CE (2013) Conformational defects underlie proteasomal degradation of Dent’s disease-causing mutants of ClC-5. Biochem J 452:391–400. doi:10.1042/BJ20121848
Dent CE, Friedman M (1964) Hypercalcuric rickets associated with renal tubular damage. Arch Dis Child 39:240–9
Devuyst O, Thakker RV (2010) Dent’s disease. Orphanet J Rare Dis 5:28. doi:10.1186/1750-1172-5-28
Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R (2002) X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 415:287–94. doi:10.1038/415287a
Dutzler R, Campbell EB, MacKinnon R (2003) Gating the selectivity filter in ClC chloride channels. Science 300:108–12. doi:10.1126/science.1082708
Gleixner EM, Canaud G, Hermle T, Guida MC, Kretz O, Helmstadter M, Huber TB, Eimer S, Terzi F, Simons M (2014) V-ATPase/mTOR signaling regulates megalin-mediated apical endocytosis. Cell Rep 8:10–9. doi:10.1016/j.celrep.2014.05.035
Grand T, Mordasini D, L’Hoste S, Pennaforte T, Genete M, Biyeyeme MJ, Vargas-Poussou R, Blanchard A, Teulon J, Lourdel S (2009) Novel CLCN5 mutations in patients with Dent’s disease result in altered ion currents or impaired exchanger processing. Kidney Int 76:999–1005. doi:10.1038/ki.2009.305
Gunther W, Luchow A, Cluzeaud F, Vandewalle A, Jentsch TJ (1998) ClC-5, the chloride channel mutated in Dent’s disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc Natl Acad Sci U S A 95:8075–80
Gunther W, Piwon N, Jentsch TJ (2003) The ClC-5 chloride channel knock-out mouse—an animal model for Dent’s disease. Pflugers Arch 445:456–62. doi:10.1007/s00424-002-0950-6
Hara-Chikuma M, Wang Y, Guggino SE, Guggino WB, Verkman AS (2005) Impaired acidification in early endosomes of ClC-5 deficient proximal tubule. Biochem Biophys Res Commun 329:941–6. doi:10.1016/j.bbrc.2005.02.060
Horita S, Yamada H, Inatomi J, Moriyama N, Sekine T, Igarashi T, Endo Y, Dasouki M, Ekim M, Al-Gazali L, Shimadzu M, Seki G, Fujita T (2005) Functional analysis of NBC1 mutants associated with proximal renal tubular acidosis and ocular abnormalities. J Am Soc Nephrol 16:2270–8. doi:10.1681/ASN.2004080667
Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada GH, Bourgoin S, Casanova J, Wildeman A, Bechoua S, Ausiello DA, Brown D, Marshansky V (2006) V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat Cell Biol 8:124–36. doi:10.1038/ncb1348
Ishida Y, Nayak S, Mindell JA, Grabe M (2013) A model of lysosomal pH regulation. J Gen Physiol 141:705–20. doi:10.1085/jgp.201210930
Jentsch TJ, Steinmeyer K, Schwarz G (1990) Primary structure of Torpedo marmorata chloride channel isolated by expression cloning in Xenopus oocytes. Nature 348:510–4. doi:10.1038/348510a0
Kondo Y, Fromter E (1987) Axial heterogeneity of sodium-bicarbonate cotransport in proximal straight tubule of rabbit kidney. Pflugers Arch 410:481–6
Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, Harding B, Bolino A, Devoto M, Goodyer P, Rigden SP, Wrong O, Jentsch TJ, Craig IW, Thakker RV (1996) A common molecular basis for three inherited kidney stone diseases. Nature 379:445–9. doi:10.1038/379445a0
Lobet S, Dutzler R (2006) Ion-binding properties of the ClC chloride selectivity filter. EMBO J 25:24–33. doi:10.1038/sj.emboj.7600909
Lourdel S, Grand T, Burgos J, Gonzalez W, Sepulveda FV, Teulon J (2012) ClC-5 mutations associated with Dent’s disease: a major role of the dimer interface. Pflugers Arch 463:247–56. doi:10.1007/s00424-011-1052-0
Mellman I, Fuchs R, Helenius A (1986) Acidification of the endocytic and exocytic pathways. Annu Rev Biochem 55:663–700. doi:10.1146/annurev.bi.55.070186.003311
Miesenbock G, De Angelis DA, Rothman JE (1998) Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394:192–5. doi:10.1038/28190
Nakamura M, Yamazaki O, Shirai A, Horita S, Satoh N, Suzuki M, Hamasaki Y, Noiri E, Kume H, Enomoto Y, Homma Y, Seki G (2015) Preserved Na/HCO3 cotransporter sensitivity to insulin may promote hypertension in metabolic syndrome. Kidney Int 87:535–42. doi:10.1038/ki.2014.351
Nguitragool W, Miller C (2006) Uncoupling of a CLC Cl−/H+ exchange transporter by polyatomic anions. J Mol Biol 362:682–90. doi:10.1016/j.jmb.2006.07.006
Novarino G, Weinert S, Rickheit G, Jentsch TJ (2010) Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science 328:1398–401. doi:10.1126/science.1188070
Picollo A, Malvezzi M, Accardi A (2010) Proton block of the CLC-5 Cl−/H+ exchanger. J Gen Physiol 135:653–9. doi:10.1085/jgp.201010428
Picollo A, Pusch M (2005) Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 436:420–3. doi:10.1038/nature03720
Piwon N, Gunther W, Schwake M, Bosl MR, Jentsch TJ (2000) ClC-5 Cl−-channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature 408:369–73. doi:10.1038/35042597
Rahmati N, Kunzelmann K, Xu J, Barone S, Sirianant L, De Zeeuw CI, Soleimani M (2013) Slc26a11 is prominently expressed in the brain and functions as a chloride channel: expression in Purkinje cells and stimulation of V H+-ATPase. Pflugers Arch 465:1583–97. doi:10.1007/s00424-013-1300-6
Sabolic I, Burckhardt G (1986) Characteristics of the proton pump in rat renal cortical endocytotic vesicles. Am J Physiol 250:F817–26
Sakamoto H, Sado Y, Naito I, Kwon TH, Inoue S, Endo K, Kawasaki M, Uchida S, Nielsen S, Sasaki S, Marumo F (1999) Cellular and subcellular immunolocalization of ClC-5 channel in mouse kidney: colocalization with H+-ATPase. Am J Physiol 277:F957–65
Scheel O, Zdebik AA, Lourdel S, Jentsch TJ (2005) Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 436:424–7. doi:10.1038/nature03860
Schodel J, Klanke B, Weidemann A, Buchholz B, Bernhardt W, Bertog M, Amann K, Korbmacher C, Wiesener M, Warnecke C, Kurtz A, Eckardt KU, Willam C (2009) HIF-prolyl hydroxylases in the rat kidney: physiologic expression patterns and regulation in acute kidney injury. Am J Pathol 174:1663–74. doi:10.2353/ajpath.2009.080687
Seki G, Fromter E (1990) The chloride/base exchanger in the basolateral cell membrane of rabbit renal proximal tubule S3 segment requires bicarbonate to operate. Pflugers Arch 417:37–41
Sekine T, Komoda F, Miura K, Takita J, Shimadzu M, Matsuyama T, Ashida A, Igarashi T (2014) Japanese Dent disease has a wider clinical spectrum than Dent disease in Europe/USA: genetic and clinical studies of 86 unrelated patients with low-molecular-weight proteinuria. Nephrol Dial Transplant 29:376–84. doi:10.1093/ndt/gft394
Shirakabe K, Priori G, Yamada H, Ando H, Horita S, Fujita T, Fujimoto I, Mizutani A, Seki G, Mikoshiba K (2006) IRBIT, an inositol 1,4,5-trisphosphate receptor-binding protein, specifically binds to and activates pancreas-type Na+/HCO3 − cotransporter 1 (pNBC1). Proc Natl Acad Sci U S A 103:9542–7. doi:10.1073/pnas.0602250103
Smith AJ, Lippiat JD (2010) Direct endosomal acidification by the outwardly rectifying CLC-5 Cl−/H+ exchanger. J Physiol 588:2033–45. doi:10.1113/jphysiol.2010.188540
Smith AJ, Reed AA, Loh NY, Thakker RV, Lippiat JD (2009) Characterization of Dent’s disease mutations of CLC-5 reveals a correlation between functional and cell biological consequences and protein structure. Am J Physiol Renal Physiol 296:F390–7. doi:10.1152/ajprenal.90526.2008
Sonawane ND, Thiagarajah JR, Verkman AS (2002) Chloride concentration in endosomes measured using a ratioable fluorescent Cl− indicator: evidence for chloride accumulation during acidification. J Biol Chem 277:5506–13. doi:10.1074/jbc.M110818200
Stauber T, Jentsch TJ (2013) Chloride in vesicular trafficking and function. Annu Rev Physiol 75:453–77. doi:10.1146/annurev-physiol-030212-183702
Stauber T, Weinert S, Jentsch TJ (2012) Cell biology and physiology of CLC chloride channels and transporters. Compr Physiol 2:1701–44. doi:10.1002/cphy.c110038
Steinmeyer K, Schwappach B, Bens M, Vandewalle A, Jentsch TJ (1995) Cloning and functional expression of rat CLC-5, a chloride channel related to kidney disease. J Biol Chem 270:31172–7
Suzuki M, Vaisbich MH, Yamada H, Horita S, Li Y, Sekine T, Moriyama N, Igarashi T, Endo Y, Cardoso TP, de Sa LC, Koch VH, Seki G, Fujita T (2008) Functional analysis of a novel missense NBC1 mutation and of other mutations causing proximal renal tubular acidosis. Pflugers Arch 455:583–93. doi:10.1007/s00424-007-0319-y
Thakker RV (2000) Pathogenesis of Dent’s disease and related syndromes of X-linked nephrolithiasis. Kidney Int 57:787–93. doi:10.1046/j.1523-1755.2000.00916.x
Wang SS, Devuyst O, Courtoy PJ, Wang XT, Wang H, Wang Y, Thakker RV, Guggino S, Guggino WB (2000) Mice lacking renal chloride channel, CLC-5, are a model for Dent’s disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum Mol Genet 9:2937–45
Weinert S, Jabs S, Supanchart C, Schweizer M, Gimber N, Richter M, Rademann J, Stauber T, Kornak U, Jentsch TJ (2010) Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl- accumulation. Science 328:1401–3. doi:10.1126/science.1188072
Wrong OM, Norden AG, Feest TG (1994) Dent’s disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM 87:473–93
Wu F, Roche P, Christie PT, Loh NY, Reed AA, Esnouf RM, Thakker RV (2003) Modeling study of human renal chloride channel (hCLC-5) mutations suggests a structural-functional relationship. Kidney Int 63:1426–32. doi:10.1046/j.1523-1755.2003.00859.x
Yamazaki O, Yamada H, Suzuki M, Horita S, Shirai A, Nakamura M, Satoh N, Fujita T, Seki G (2013) Identification of dominant negative effect of L522P mutation in the electrogenic Na+-HCO3 − cotransporter NBCe1. Pflugers Arch 465:1281–91. doi:10.1007/s00424-013-1277-1
Yamazaki O, Yamada H, Suzuki M, Horita S, Shirai A, Nakamura M, Seki G, Fujita T (2011) Functional characterization of nonsynonymous single nucleotide polymorphisms in the electrogenic Na+-HCO3 − cotransporter NBCe1A. Pflugers Arch 461:249–59. doi:10.1007/s00424-010-0918-x
Zifarelli G, De Stefano S, Zanardi I, Pusch M (2012) On the mechanism of gating charge movement of ClC-5, a human Cl−/H+ antiporter. Biophys J 102:2060–9. doi:10.1016/j.bpj.2012.03.067
Zifarelli G, Pusch M (2009) Conversion of the 2 Cl−/1 H+ antiporter ClC-5 in a NO3 −/H+ antiporter by a single point mutation. EMBO J 28:175–82. doi:10.1038/emboj.2008.284
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 334:678–83. doi:10.1126/science.1207056
Acknowledgments
This study was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Daisuke Yamamoto deceased.
Parts of this paper were taken from the thesis written in English by Nobuhiko Satoh. The title of the thesis, which is in Japanese, is as follows: “CLC-5の2Cl/H交換輸送機能はV-ATPaseを介する効率的エンドゾーム酸性化に必要である”. The summary (in Japanese) of the thesis is accessible at http://repository.dl.itc.u-tokyo.ac.jp/index_e.html.
Rights and permissions
About this article

Cite this article
Satoh, N., Yamada, H., Yamazaki, O. et al. A pure chloride channel mutant of CLC-5 causes Dent’s disease via insufficient V-ATPase activation. Pflugers Arch - Eur J Physiol 468, 1183–1196 (2016). https://doi.org/10.1007/s00424-016-1808-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-016-1808-7