
Abstract
Intestinal inflammation affects smooth muscle contractility contributing to altered motility, but changes to the individual smooth muscle cells are not well described. We used video microscopy to study the contractility of circular smooth muscle cells (CSMC) isolated from the rat mid-descending colon throughout the course of TNBS-induced colitis, measuring their shortening response to carbachol (CCh), 5-HT, histamine or high K+. In control CSMC, CCh caused a maximal shortening response of 28 (2%), similar to that for 5-HT of 27 (1%), but by day 4 of colitis, these responses were decreased by 35% and 37%, respectively. By day 36, all aspects of cholinergic contraction returned to control levels, while 5-HT-induced contraction remained significantly attenuated. In contrast, the contractile responses to histamine remained similar at all time points. K+-induced contraction was impaired only on day 4, and the maximal response remained substantially greater than CCh or 5-HT. Colitis caused a 121% increase in CSMC length by day 2 that persisted through day 36, independent evidence for phenotypic change. We conclude that impaired CSMC contractility at both the receptor and non-receptor levels contribute to altered smooth muscle function during colitis. Persistent changes in contractile response remained detectable after resolution of inflammation, and similar events may occur in post-enteritis syndromes seen in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammation of the intestine causes significant structural and functional modifications to the enteric nervous system and smooth muscle. In humans, as well as animal models of inflammatory bowel disease (IBD), inflammation results in a profound thickening of the intestinal wall. These structural changes can be at least partially attributed to alterations in the intestinal smooth muscle cells (SMC) since inflammation causes significant hyperplasia and hypertrophy [5, 13] as well as altered cellular protein content [6, 38]. More importantly, these changes represent long-lasting or even permanent alterations to the intestinal wall, which may contribute to post-inflammatory condition such as the irritable bowel syndrome (IBS).
Intestinal inflammation impairs agonist-induced contraction of the smooth muscle layer, observed experimentally in the intact organ as well as in tissue strips [7, 23, 25, 28]. The functional changes that occur during inflammation could be attributed either to extracellular factors that interact with the SMC to modify tissue function, or to intracellular modifications in the SMC that directly impair its contractility or a combination of extracellular and intracellular factors. The fact that inflammation causes changes to both the number and nature of the SMC [5, 6] suggests a cellular basis is responsible for this altered tissue function, but the actual source of the impaired motility has yet to be fully elucidated.
The use of isolated SMC is one way to better characterize the source of impaired tissue contractility, as this will reduce extracellular interactions to a minimum allowing for a more direct examination of cellular function. However, studies using this technique to explore the nature of altered contractility in the inflamed intestine have not identified a consistent underlying trend. For example, during TNBS-induced rabbit colitis, the impaired agonist-induced contractility observed in tissue strips was not detectable in the isolated circular SMC (CSMC) [10], and is thus supportive of the extracellular mediated hypothesis. In contrast, impaired tissue function observed during acetic acid-induced dog ileitis could be, at least partially, attributed to impaired CSMC function [33]. Moreels et al. [25] found that impaired agonist-induced longitudinal smooth muscle contractility observed during ileitis was not associated with impaired cellular function, while Akiho et al. [2] found altered contractility directly in the longitudinal SMC during ileitis, providing support for direct modifications to the SMC at the intracellular level.
Differences among animal models and inter-species variability may thus contribute to inconsistent outcomes. Further, the immunological profile of the inflammatory response may be an important determinant of these changes/alterations that occur in each model. For example, the cytokine profile of the inflammatory changes present in Crohn’s disease is Th-1 dominant, and insight into altered smooth muscle function might thus be drawn from TNBS-induced colitis in the rat, based on its similarly relatively severe, transmural inflammation and Th-1 profile [11, 29]. In contrast, the typically Th-2 predominant profile of parasitic infection (e.g., Trichinella spiralis) parallels the mucosal inflammation of ulcerative colitis in humans [32].
The diversity of neurotransmitters and other potential contractile agonists that are present within the inflamed intestinal wall adds further complexity to the understanding of altered contractility. However, much of the current literature has focused on the contractile response to acetylcholine (ACh) as the primary neurotransmitter of the gut, and few studies have evaluated contractility in response to other excitatory agonists. Other important contractile agonists such as serotonin (5-HT) and histamine use different intracellular pathways, and this raises the question of whether they are similarly or differentially affected during inflammation. Overall, this has yet to be determined. As well, the extent to which non-receptor mediated events contribute to altered contraction is also poorly understood. Finally, no studies have examined a protracted time-course, sufficient to identify the onset and resolution of inflammation-induced changes to CSMC function.
To address these questions, we have modified a recently developed cell isolation technique [39] to obtain a high yield of long, viable SMC that likely represents the in vivo status of the cell, and can thus optimally identify alterations in cellular contractility. We applied this technique to the model of TNBS-induced rat colitis, to characterize the effects of inflammation on the response of individual CSMC to the excitatory agonists carbachol (CCh), 5-HT, and histamine over the time course of inflammation from onset to overt recovery (as characterized by the absence of inflammation).
Materials and methods
TNBS-induced inflammation
Experiments were performed on adult male Sprague-Dawley rats (Charles River, QC, Canada). Animals were housed in pairs in microfilter-isolated cages with free access to food and water, except that food was removed 24 h before induction of colitis. All experimental procedures were approved by the local University Animal Care Committee. For experimental inflammation of the colon, rats were lightly anesthetized with halothane, and 500 μl of 200 mM trinitrobenzenesulfonic acid (TNBS; Fluka) dissolved in 50% ethanol was introduced into the colon 8 cm proximal to the anus using a PE-50 catheter [27]. During the first 4 days of recovery, rats were provided with salt/sugar solution (150 mM NaCl, 60 mM sucrose in water), and given an analgesic solution twice daily (Buprenex, 0.03 mg/kg). Control animals received either Buprenex similarly as above or no treatment.
Histology
Animals were sacrificed at various times after the initiation of colitis by cervical dislocation under halothane anesthesia, and the inflamed region of the mid-descending colon was removed. A small section of the involved colon was fixed in 10% neutral buffered formalin for 24 h before routine processing for paraffin sectioning. Histological sections (4 μm) were stained with hematoxylin and eosin for examination by a blinded observer according to the procedures described by Wallace and Keenan [37].
MPO activity was assessed as previously described [16, 17]. Briefly, snap-frozen intestinal segments were placed into a solution (1 ml/50 mg tissue) of hexadecyltrimethyl-ammonium bromide (0.5% wt/vol) in potassium phosphate buffer (50 mM, pH 6.0). Samples were homogenized on ice for 30 s, centrifuged for 30 min at 10,000 rpm at 4°C, and then incubated at 60°C for 2 h. After incubation, 100 μl of supernatant was added to 2.9 ml of 0.00005% H2O2 in an o-dianisidine dihydrochloride (Sigma, St. Louis, Mo., USA) solution in a cuvette, and the absorbance was measured at 460 nm. Results were expressed as units of MPO activity per 100 mg of tissue (wet weight), where 1 U of MPO activity is the amount of enzyme required to split 1 μmol H2O2/min at 25°C.
Isolation of circular smooth muscle cells
The protocol used for isolation of CSMC was similar to that previously described [39]. Briefly, the mid-descending colon was removed and placed in fresh Krebs’ solution (in mM: NaHCO3 25; NaCl 118; KCl 4.7; NaH2PO4 1; MgSO4 1.2; glucose 11; CaCl2 2.5; bubbled with 95%O2/5%CO2). The colon was opened along the longitudinal axis, pinned out in Krebs’ solution and the mucosa and sub-mucosa were removed by sharp dissection. Strips of circular smooth muscle (CSM) was carefully removed from the longitudinal smooth muscle layer and placed into HEPES physiological salt solution (HPSS)-digestion solution (in mM: NaCl 125; glucose 10; Na-HEPES 10; MgCl2 1; KCl 4; CaCl2 1; EDTA 0.25; taurine 10; pH 7.2) with papain (0.5 mg/ml; Sigma), BSA (1 mg/ml), dl-dithiothreitol (DTT) (1 μM; Sigma) and collagenase type-F (0.5 mg/ml; Sigma). The tissue was then incubated at 5°C for 2 h, room temperature for 20 min and finally 31°C for 10 min. The solution containing the CSM was then poured over a 200-μm nylon-mesh filter and thoroughly rinsed with HPSS-isolation solution (in mM: NaCl 112.5; KCl 5.5; KH2PO4 2; Na-HEPES 24; CaCl2 1.9; MgCl2 0.6; glucose 8; BME amino acids 40 μl/ml; soybean trypsin inhibitor 0.1 mg/ml; pH 7.4, 31°C) to remove excess enzyme solution. The CSM was re-suspended in HPSS-isolation solution and gently triturated to produce a suspension of individual CSMC.
Morphological evaluation of isolated CSMC
Immediately after obtaining a suspension of CSMC, phase contrast microscopy was used to quantify cells that were phase bright with membranes free of physical distortions and blebbing. Such cells were determined to be 100% viable by exclusion of trypan blue staining (data not shown). Overall, 90% of the isolated CSMC population demonstrated these characteristics.
To characterize the size and length of CSMC, freshly isolated cells were fixed with 1% acrolein (shown previously to preserve cell length [9]) and an aliquot of cell suspension was placed on a glass slide with cover slip. Cells were viewed with an inverted phase-contrast microscope (Olympus IMT-2), and images of 30 random CSMC were taken per slide using a digital imaging camera. Length measurements were obtained by manually tracing down the middle of each cell using an image-analysis program (ImagePro 5.0; Media Cybernetics, USA). Cell area was determined similarly by manually tracing the outline of the cell.
Contractility studies
For assessment of contractile response, CSMC were exposed to either CCh (0.1 pM–10 mM), 5-HT (0.1 pM–100 µM), histamine (0.1 pM–1 mM), or high K+ solution (80 mM) for 30 s and then fixed with acrolein at a final concentration of 1%. For each concentration of agonist used, the mean cell length was determined and expressed as a percentage of the initial resting length (cells fixed without agonist application), from animals at each of the different time points (days 0, 2, 4, 16 and 36). Since the receptor-mediated agonist-induced contraction on days 2 and 16 were not significantly different that observed on day 4 they were omitted from figures for clarity of presentation. In contrast, non-receptor mediated alterations show strikingly different trends on these days and therefore days 2 and 16 were included. All contractility experiments represent data from 4–10 animals at 30 cells per animal per concentration of agonist.
Typical assessment of agonist-induced contraction involves the evaluation of E max and pD2 (EC50) values between the different groups. However, with contractile responses where the Emax values are significantly decreased the pD2 values are artificially shifted to the left (appearing hyper-responsive) and thus are misleading. Therefore, in addition to the E max values, we report the EC100 values (calculated as the minimum agonist concentration required to elicit maximal contraction).
Assessment of individual cellular contractility
To ensure that the cells were in fact responsive to agonist-application, we evaluated the contractile response of individual CSMC. Briefly, aliquots of freshly isolated CSMC were placed in a 35 mm dish with inset glass cover-slips, allowed to settle for 10 min and viewed using an inverted phase-contrast microscope (Olympus IMT-2) connected to a closed circuit video camera. Individual CSMC were directly exposed to ACh (100 µM in Krebs’ solution) by pressure application via a puffer pipette (500 ms application at 10 psi through a 1-MΩ resistance tip) which was micro-manipulated to within 250 µm of the cell. Images of the cell were captured at 250-ms intervals for a total of 10 s so as to identify the resting length (cell length prior before ACh application) and maximal contractility of the cells isolated on days 0, 4 and 36. Negative controls consisted of pressure application of Krebs’ filled puffer pipettes, which failed to induce contraction at any time point (data not shown).
Statistical analysis
All data are expressed as mean±standard error of the mean (SEM). Statistical analysis was performed using one-way analysis of variance (ANOVA) with the student Newman-Keuls multiple comparison test or two-tailed Student’s t-test. For contractility experiments data was fitted using a standard curve fit equation with weight on SEM and maximal contraction was determined. P values of ≤0.05 were considered statistically significant.
Results
TNBS-induced inflammation of the mid-descending rat colon
Administration of TNBS caused colitis as previously described [31], with rats exhibiting weight loss and diarrhea. On days 2 and 4 post-TNBS, an overtly inflamed colon was present, frequently with adhesions. Only tissue from involved areas, which ranged from 1.5±4 cm of the mid-descending colon, was used in the contractility studies. By days 16 and 36, the acute effects of inflammation had resolved, but previously involved areas could be identified by mild adhesions and bowel wall thickening (a permanent result of inflammation-induced hypertrophy and hyperplasia). In all cases inflammation was identified using the independent criteria of weight loss, histological evaluation of transmural inflammation and MPO analysis.
Microscopic evaluation of TNBS-induced inflammation resulted in a transient increase in damage score with maximal inflammation present by day 4 post-TNBS and full resolution by day 36 (Fig. 1D). Figure 1B shows the typical microscopic appearance of the colon on day 4, with prominent inflammatory infiltrate. By day 16 post-TNBS there was nearly full resolution of the inflammation and by day 36 an overtly normal colon was apparent (Fig. 1C). In day 36 animals, previously inflamed regions could easily be identified by the thickening of the smooth muscle wall, which has previously been shown to be a result of inflammation-induced hypertrophy and hyperplasia [5, 6].

Depiction of onset and resolution of inflammation over the time course of TNBS-induced colitis in the rat. A–C; representative micrographs of haematoxylin- and eosin-stained cross sections of rat colon from control animals (A), day 4 of colitis (B) and day 36 (C). Note the mucosal damage as well as inflammatory infiltrate in both circular and longitudinal smooth muscle layers on day 4 (B; arrows) and adhesions (arrowhead). C The reversal of mucosal damage, absence of inflammatory infiltrate but persistent thickening of smooth muscle layers that was characteristic of tissue appearance by day 36. Scale bar for A–C: 200 μm. D Quantification of tissue damage during TNBS colitis, using the standard scoring system of Wallace and Keenan [37]. Damage score was maximal by day 4, with reversal to near control levels by day 16. Damage score was similar to control by day 36. E Measurement of myeloperoxidase (MPO) activity in colon over the time course of colitis. The MPO activity, primarily reflecting neutrophil influx, showed a rapid increase to maximal level by day 2, that declined by day 4 and thereafter. MPO was similar to control by day 16 and 36 (n=3 animals per treatment group; *P≤0.05 versus control)
Previous studies have demonstrated that TNBS resulted in a transient 115-fold increase in MPO activity that was maximal by day 1 [31]. This increased MPO persisted, albeit to a much less extent, through day 4 and by day 16 MPO levels had returned to control levels. Similar MPO results were obtained in our study (Fig. 1E). The MPO data in combination with histological evidence indicates that TNBS induces maximal inflammation by day 4 and this inflammation is fully resolved by day 36.
Inflammation increases the size of CMSC
Freshly isolated CSMC appeared phase-bright and free of membrane irregularities (Fig. 2). CSMC isolated from control animals were characteristically 126±3 µm (n=10 animals) in length, and this value was highly reproducible both within and among animals. However, colitis caused a rapid and lasting increase in resting cell length, which was 121% of control by day 2 and remained at 126% of control on day 36 post-TNBS (Fig. 3A). Analysis showed that CSMC area also increased in proportion with cell length, and reached 133% of control by day 16 (Figure 3B). There was no evidence of any reversal of these changes by day 36, suggesting that these changes were independent of the transient events of inflammation such as edema. We conclude that this increased size of CSMC is evidence of a significant and lasting cellular hypertrophy due to colitis.

Representative images showing the control and contracted appearances of circular smooth muscle cells (CSMC) isolated from the mid-descending colon of rats at day 0 (control), day 4 or day 36 after induction of colitis with TNBS. Live cells were viewed with phase contrast microscopy under control conditions (left column), and were re-photographed after maximal contraction with 100 µM CCh (right column), showing decreased cell length. Scale bar for all images: 50 µm

CSMC undergo hypertrophy during colitis. The resting length (A) and the cell area (B) of enzymatically isolated CSMC were determined from digital images obtained with phase contrast microscopy, and both parameters showed a rapid and lasting increase during colitis. Bars represent mean±SEM of 30 cells per animal, with 4–10 animals per time point; *P≤0.05 versus control
CSMC receptor-mediated contractility during inflammation
Examination of the effects of CCh on CSMC showed a dose-dependent contraction of the cells at all time points, and representative images of CSMC in both control and maximally contracted states are shown in Fig. 2. For cells isolated from control animals (n=7), the threshold for CCh-induced contraction was ~1 nM CCh with an EC100 of 100 µM, resulting in an E max of 28.6% shortening. For cells isolated from rats with colitis, there was no apparent modification to the threshold but there was a marked attenuation in the E max, which was reduced by 35% on day 4 (n=5; Fig. 4). However, upon resolution of the inflammation (day 36), there was full reversal of the impaired CCh-induced contractility with the percentage shortening being similar to control values (n=4; Fig. 4, inset).

Transient impairment of the contractile response of enzymatically isolated CSMC to a cholinergic agonist during colitis. The dose-dependent contraction of CSMC exposed to carbachol (CCh) was significantly impaired by day 4 post-TNBS (open circles) compared with control (filled circles), but returned to control levels by day 36 post-TNBS (filled triangles). Inset, expression of the shortening response to 100 µM CCh as percentage of control. Values represent mean±SEM of 30 cells per animal per concentration of CCh from 4 to 7 animals per time point; *P≤0.05 versus control, ‡ P≤0.05 for day 36 versus day 4
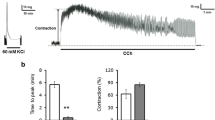
To verify our maximal contractile response, we evaluated the contractility of live individual CSMC. There was a consistent and highly reproducible response in CSMC isolated from control animals with a maximal shortening of 25.3±0.6% occurring 3.25 s following the initiation of contraction (n=5; Fig. 5). By day 4 post-TNBS there was a significant 24% decrease in the maximal ACh-induced contraction (n=4) and by day 36 there was complete recovery to all modifications in contractility (n=4).

Direct visualization of the contractile response of CSMC in response to local ACh application. Direct application of 100 µM ACh onto control CSMC (filled circles) via pressure application through a 1-MΩ resistant electrode resulted in a highly reproducible maximal contraction of 25.3±0.6% occurring 3.25 s after the initiation of contraction. By day 4 (open circles) this maximal contractile response was significantly reduced by 24%, which was fully reversed by day 36 (filled triangles). Values represent mean±SEM of 10 cells per animal from 4 to 5 animals; *P≤0.05 versus control
Application of 5-HT also caused a dose-dependent contraction of the CSMC, that in control animals was very similar to that seen with CCh. The threshold for response was ~1 nM with an EC100 of 10 µM, resulting in an E max of 27.2% shortening (Fig. 6). Furthermore, like the CCh-induced contraction, inflammation significantly reduced the maximal percentage contraction that could be elicited by 5-HT, with an E max that was impaired by 37% on day 4 (n=7), compared to controls (n=5). However, unlike CCh, the 5-HT-induced contraction did not recover upon resolution of inflammation, with the E max on day 36 (n=10) being only 67% that of control values (Fig. 6). In fact, the day 4 and day 36 contraction values remained similar at all 5-HT doses, showing the absence of recovery.

Lasting impairment of the contractile response of enzymatically isolated CSMC to 5-hydroxytryptamine (5-HT) following colitis. 5-HT dose-dependently increased the percentage contraction of CSMC isolated from all animals. In control CSMC (filled circles), maximal contraction caused a 27±1% shortening compared to basal resting length. By day 4 post-TNBS (open circles), the contractile response showed significant attenuation compared with control (filled circles) that was not reversed by day 36 post-TNBS (filled triangles). Inset, comparison of the contraction to 10 µM 5-HT expressed as percentage of control. Values represent mean±SEM of 30 cells per animal per concentration of 5-HT from 4 to 10 animals per time point; *P≤0.05 versus control
Previous studies have suggested that buprenorphine (the analgesic used during inflammation) has the potential to significantly alter gastrointestinal motility [3]. Therefore, we evaluated the impact of this analgesic on control CSMC. Administration of buprenorphine to control rats had no significant effects on control CSMC contractility or morphology as compared to littermate non-injected control rats (data not shown). Since the results were the same for both groups, all control data was pooled.
Application of histamine caused a dose-dependent contraction of control CSMC, resulting in an E max of 19.4% at 10 µM, significantly less than that caused by CCh or 5-HT. While the concentrations of histamine needed for threshold and maximal contraction were similar to those observed for CCh and 5-HT, histamine contrasted sharply in potency, causing only 69% and 71% of the maximal response elicited by CCh and 5-HT, respectively. Surprisingly, we found that during inflammation there were no significant modifications to the histamine-induced maximal contractile response at any time points studied (Fig. 7).

The contractile response of enzymatically isolated CSMC to histamine was not altered during colitis. While histamine produced a dose-dependent increase in the percent contraction in CSMC isolated from all animals, there were no significant differences in responses among CSMC from control (filled circles), day 4 (open circles) or day 36 (filled triangles) animals. Maximal contraction resulted in similar maximal shortening (inset). Values represent the mean±SEM of 30 cells per animal per concentration of histamine from 4 to 10 animals per time point
CSMC non-receptor-mediated contractility during inflammation
We assessed the capacity for non-receptor mediated contraction of CSMC through the addition of a HPSS containing 80 mM extracellular KCl (“high K+”). In control cells, high K+ resulted in maximal contraction of 29±2% (n=4; Fig. 8), which was similar to the maximum contraction observed with CCh and 5-HT. By day 4 post-TNBS, contractility elicited by high K+ (n=7) was significantly impaired, with the maximal contraction reduced to 73% of control values (Fig. 8). However, these changes in non-receptor mediated contraction were completely reversed by day 16, which was unlike the characteristics of CCh- and 5-HT-induced receptor mediated contraction (Fig. 8).

Non-receptor mediated contraction of enzymatically isolated CSMC is transiently impaired during colitis. The contraction of CSMC in response to high (80 mM) extracellular K+ was reduced during peak inflammation but became similar to control levels by day 16 post-TNBS and thereafter. The maximal contraction produced by high K+ was always substantially greater than that produced by receptor mediated stimulation, suggesting that both receptor and non-receptor mediated alterations exist during peak inflammation. Bars represent the mean±SEM of values from 30 cells per animal, with 4–8 animals per time point; *P≤0.05 control versus day 4; ‡P≤0.05 days 16 and 36 versus day 4
Comparison of the receptor/non-receptor-mediated contractility
We wished to compare the relative changes in receptor-mediated contraction with those initiated at a level downstream of the receptor (i.e., the high-K+ response). To study this, we used a contraction ratio, calculated as receptor/non-receptor mediated contraction (Table 1). In control animals, CCh and 5-HT both produced contraction ratios of nearly 1, suggesting that these agonists both have a similar ability to produce a high degree of contraction at the CSMC level. However, by day 4 post-TNBS there was a dramatic attenuation in contractility and while the impaired CSMC function appeared to be attributed to modifications in the non-receptor mediated contraction, analysis of the contraction ratios demonstrated the existence of both receptor and non-receptor mediated alterations, with CCh- and 5-HT-mediated contraction being 12% and 19% less than that to high K+, respectively (Table 1).
Discussion
In this animal model of intestinal inflammation, we have shown that a Th-1 dominant, transmural colitis can cause both structural and functional alterations at the level of the individual CSMC. In addition, we have identified differential impairment in CSMC contractility, which varied over the time course of inflammation and in response to various agonists (CCh, 5-HT and histamine). This represents alterations in the cellular responses to each of three major sources of contractile agonists—neuronal, enterochromaffin and inflammatory cells, respectively. In using multiple agonists and an extended time course, this work provides a more complete view of the effects of inflammation on contractility, extending our understanding of the effects of colitis beyond those identified in studies using CSM tissue strips. Overall, we conclude that alterations at the cellular level could be the major basis for the development of altered contractility of intestinal smooth muscle.
We observed that CCh and 5-HT both induced contraction of CSMC from the control colon that showed similar characteristics. For example, the agonist concentrations needed for the initiation and maximal contraction were similar, and both agonists produced identical maximal contraction. This was about 28% shortening from basal resting length, and was similar to that maximal ACh-induced contraction observed in other studies [18, 26, 33]. This shows that 5-HT has the potential to act directly on the SMC, as well as acting through 5-HT-stimulated neural pathways [14]. While current methods of detecting 5-HT containing neurons have failed to provide evidence of these motor neuron in the rat colon and therefore the CSMC may not encounter 5-HT directly under normal conditions, local 5-HT concentrations dramatically increase during inflammation [19]. In such cases, 5-HT is likely released from platelets, mast cells and enterochromaffin cells, providing an agonist capable of inducing strong contraction.
During the most severe phase of inflammation examined, the ability of the CSMC to contract to both 5-HT and CCh was significantly attenuated, with maximal contraction being reduced by approximately 35%. These results suggested that a major factor in the development of impaired tissue contractility is a diminished ability to respond that occurs at the level of the CSMC, and further, identifies the issue of reversibility as critical to the full resolution of colitis. While Myers et al. [28] showed that the impaired ACh-induced contractility in CSM tissue strips returned to control levels upon resolution of inflammation, this model used acetic acid-induced inflammation with a more superficial inflammatory response that is different from TNBS-induced transmural colitis. Nonetheless, we found that the impaired CCh-induced contraction seen in TNBS colitis did return to control levels, with no persisting indications of functional impairment. Thus, it is of particular interest that the impaired 5-HT-induced contractile response showed no tendency to return to control levels and remained indistinguishable from the suppressed contractility observed during peak inflammation.
In contrast to both CCh- and 5-HT-induced contraction, histamine produced a much weaker contractile response, resulting in a maximal contraction that was 28% less than that for CCh or 5-HT. This suggests that in the control state, histamine may not have a prominent role in contraction. However, unlike CCh- and 5-HT-induced contraction, the histamine-induced contractile response was not significantly influenced as a result of the TNBS-induced colitis. Since histamine is likely a major excitatory non-neuronal agonist present during inflammation, it may be that histamine-induced contraction assumes a more significant role in colitis, when neuronal sources of agonists are lost or damaged [8, 21, 31]. The fact that the histamine response was not impaired at any point of inflammation suggests that tachyphylaxis was either not an issue or that the cells somehow modified their receptor expression to counter the effects of tachyphylaxis. However, since the response to 5-HT remained diminished at day 36, a point where local inflammatory agonist concentrations would be back to control levels and therefore tachyphylaxis would not be an issue, and the histamine response remained unchanged, we feel that we can safely conclude that the modification in the 5-HT response are likely due to modifications at the receptor level.
We also looked for changes to non-receptor mediated contraction of CSMC, through the use of depolarizing concentrations of extracellular K+. In SMC from control colon, we found that high K+ caused a maximal contraction that was similar to that seen with maximal concentrations of CCh and 5-HT. However, this showed a trend to impairment by day 2 of colitis, and was significantly decreased by day 4 post-TNBS. However, these changes were reversed as early as day 16 post-TNBS, a time at which receptor mediated contraction remained significantly impaired.
While our results showed that colitis produced alterations at both the receptor and non-receptor level, Myers et al. [28] recently showed that acetic acid-induced colitis in the rat caused impaired contractility entirely at the post-receptor level. Certainly, the nature of altered smooth muscle contractility may be model-dependent, and this finding from a relatively superficial and acute colitis due to the application of acetic acid contrasts with our results in a transmural colitis model. In their study, the response to elevated K+ was substantially lower than the ACh-induced contraction, which suggests that the K+-induced contraction may not have been maximal. Indeed, we found that the contractile response initiated with 50 mM K+ solution was significantly lower than the response we obtained with 80 mM K+ (data not shown). In support, Depoortere et al. [10] identified both receptor and non-receptor components of impaired contraction during TNBS-induced colitis in the rabbit, although this was done using tissue strips.
The altered K+-induced contractility shows that the intracellular contractile apparatus does not function normally and this may be due to one or a combination of causes, such as impaired ion channel function [1, 20, 22]; altered contractile protein function [40]; altered Ca2+ handling properties [30, 34]; and/or changes in second messenger function [35]. Since inflammation results in extensive smooth muscle hypertrophy and hyperplasia [5, 6], it is possible that the observed alterations are a direct result of the remodeling of the CSMC. Supporting this, it was shown that proliferative SMC have fewer contractile proteins compared to contractile myocytes [15] and these proliferative SMC have a diminished response to ACh with an increased M2 receptor dependency [24, 33]. In our study, we found that inflammation caused distinct changes to receptor- and non-receptor-mediated contraction of CSMC, with unique time courses and patterns of reversal. Previous studies done in our lab have demonstrated that inflammation results in a significant loss of enteric neurons [31] as well as the importance of enteric neurons in the maintenance of cell phenotype [4]. Therefore, the alterations observed in this study may reflect transient changes to the CSMC phenotype due to cell growth and re-differentiation, as well as reversible changes due to the actions of inflammatory factors. However, we propose that it is the combination of these events, i.e., cell growth in the presence of inflammatory factors that is responsible for the development of the novel phenotype of CSMC that exhibits altered contractility. Clearly, further experiments are needed to clarify this.
Our results do not suggest the etiology of the sustained impairment of the 5-HT response, but one possible explanation is that inflammation alters the expression of receptors on the cell surface membrane. In partial support, it has been shown that inflammation causes a phenotypic switch in the expression of muscarinic receptors on dog colonic SMC [33]. Following this rationale, a similar phenotypic switch may occur that alters the proportions of 5-HT2A and 5-HT4 receptors, shown to be present on the SMC [18]. However, unlike muscarinic receptors, 5-HT4 receptors initiate relaxation via adenylate cyclase, and thus, any alterations to the 5-HT2A:5-HT4 receptor ratio could have profound effects on CSMC contractility. The impaired 5-HT-induced contractility that remained present in our CSMC following resolution of inflammation may represent a functional increase in 5-HT4 or a decrease in 5-HT2A receptors. If similar events were to occur in human intestinal inflammation, they might participate in the occurrence of post-inflammatory IBS, where 5-HT4 antagonist therapy has been shown beneficial [12]. However, much of this remains speculative and is the focus of ongoing investigation.
Several studies have demonstrated that receptor expression is sensitive to the effects of inflammation and that this is one way in which inflammation can modify the contractile response [33, 35]. For example, different receptor subtypes have different sensitivities for their agonist, and chronic stimulation could lead to the desensitization of one subtype and not the other [36]. In our study, the presence of an unchanged contractile response to histamine during colitis may indicate that this receptor type is more resistant to the deleterious effects of inflammation. Although the specifics of these events remain uncertain, it is clear that inflammation has selective actions on different receptor types, and suggests that the changes observed in our model may occur at the level of the receptors on the CSMC.
In summary, we have provided evidence that inflammation directly impairs the function of the individual CSMC, which likely contributes to altered motility observed at the organ level. This impaired CSMC function occurs at both receptor and non-receptor levels; however, the involvement of alterations downstream of the receptor were only significant during peak inflammation. The alterations in contraction are agonist specific, as impairments were observed for only CCh and 5-HT. While there was a degree of reversal of the impaired contraction following resolution of the inflammation, this too was agonist specific as only CCh-induced contractility returned to control levels, with 5-HT-mediated contraction still showing impairment by day 36 post-TNBS. Our experiments strongly support the hypothesis that changes at the level of the smooth muscle cell are a major contributing factor in the development of altered contractility, and hence altered intestinal motility.
References
Akbarali HI, Pothoulakis C, Castagliuolo I (2000) Altered ion channel activity in murine colonic smooth muscle myocytes in an experimental colitis model. Biochem Biophys Res Commun 275:637–642
Akiho H, Blennerhassett P, Deng Y, Collins SM (2002) Role of IL-4, IL-13, and STAT6 in inflammation-induced hypercontractility of murine smooth muscle cells. Am J Physiol 282:G226–G232
Bhounsule SA, D’Sa S, Fernandes N, D’Souza RD, Dhume VG (1996) Gastrointestinal actions of buprenorphine: are different receptors involved? Eur J Pharmacol 316:253–256
Blennerhassett MG, Lourenssen S (2000) Neural regulation of intestinal smooth muscle growth in vitro. Am J Physiol 279:G511–G519
Blennerhassett MG, Vignjevic P, Vermillion DL, Collins SM (1992) Inflammation causes hyperplasia and hypertrophy in smooth muscle of rat small intestine. Am J Physiol 262:G1041–G1046
Blennerhassett MG, Bovell FM, Lourenssen S, McHugh KM (1999) Characteristics of inflammation-induced hypertrophy of rat intestinal smooth muscle cell. Dig Dis Sci 44:1265–1272
Castro GA, Badial-Aceves F, Smith JW, Dudrick SJ, Weisbrodt NW (1976) Altered small bowel propulsion associated with parasitism. Gastroenterology 71:620–625
Collins SM (1996) The immunomodulation of enteric neuromuscular function: implications for motility and inflammatory disorders. Gastroenterology 111:1683–1699
Collins SM, Gardner JD (1982) Cholecystokinin-induced contraction of dispersed smooth muscle cells. Am J Physiol 243:G497–G504
Depoortere I, Van Assche G, Thijs T, Geboes K, Peeters TL (1999) Differential changes in ACh-, motilin-, substance P-, and K(+)-induced contractility in rabbit colitis. Am J Physiol 277:G61–G68
Elson CO, Sartor RB, Tennyson GS, Riddell RH (1995) Experimental models of inflammatory bowel disease. Gastroenterology 109:1344–1367
Farthing MJ (1998) New drugs in the management of the irritable bowel syndrome. Drugs 56:11–21
Gabella G (1975) Hypertrophy of intestinal smooth muscle. Cell Tissue Res 163:199–214
Grider JR (2003) Neurotransmitters mediating the intestinal peristaltic reflex in the mouse. J Pharmacol Exp Ther 307:460–467
Halayko AJ, Salari H, MA X, Stephens NL (1996) Markers of airway smooth muscle cell phenotype. Am J Physiol 270:L1040–L1051
Hogaboam CM, Jacobson K, Collins SM, Blennerhassett MG (1995) The selective beneficial effects of nitric oxide inhibition in experimental colitis. Am J Physiol 268:G673–G684
Krawisz JE, Sharon P, Stenson WF (1984) Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology 87:1344–1350
Kuemmerle JF, Martin DC, Murthy KS, Kellum JM, Grider JR, Makhlouf GM (1992) Coexistence of contractile and relaxant 5-hydroxytryptamine receptors coupled to distinct signaling pathways in intestinal muscle cells: convergence of the pathways on Ca2+ mobilization. Mol Pharmacol 42:1090–1096
Linden DR, Chen JX, Gershon MD, Sharkey KA, Mawe GM (2003) Serotonin availability is increased in mucosa of guinea pigs with TNBS-induced colitis. Am J Physiol 285:G207–G216
Liu X, Rusch NJ, Striessnig J, Sarna SK (2001) Down-regulation of L-type calcium channels in inflamed circular smooth muscle cells of the canine colon. Gastroenterology 120:480–489
Lourenssen S, Jeromin A, Roder J, Blennerhassett MG (2002) Intestinal inflammation modulates expression of the synaptic vesicle protein neuronal calcium sensor-1. Am J Physiol 282:G1097–G1104
Lu G, Mazet B, Sun C, Qian X, Johnson CP, Adams MB, Roman RJ, Sarna SK (1999) Inflammatory modulation of calcium-activated potassium channels in canine colonic circular smooth muscle cells. Gastroenterology 116:884–892
Martinolle JP, Garcia-Villar R, Fioramonti J, Bueno L (1997) Altered contractility of circular and longitudinal muscle in TNBS- inflamed guinea pig ileum. Am J Physiol 272:G1258–G1267
Mitchell RW, Halayko AJ, Kahraman S, Solway J, Wylam ME (2000) Selective restoration of calcium coupling to muscarinic M(3) receptors in contractile cultured airway myocytes. Am J Physiol 278:L1091–L1100
Moreels TG, De Man JG, Dick JM, Nieuwendijk RJ, De Winter BY, Lefebvre RA, Herman AG, Pelckmans PA (2001) Effect of TNBS-induced morphological changes on pharmacological contractility of the rat ileum. Eur J Pharmacol 423:211–222
Morini G, Kuemmerle JF, Impicciatore M, Grider JR, Makhlouf GM (1993) Coexistence of histamine H1 and H2 receptors coupled to distinct signal transduction pathways in isolated intestinal muscle cells. J Pharmacol Exp Ther 264:598–603
Morris GP, Beck PL, Herridge MS, Depew WT, Szewczuk MR, Wallace JL (1989) Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology 96:795–803
Myers BS, Martin JS, Dempsey DT, Parkman HP, Thomas RM, Ryan JP (1997) Acute experimental colitis decreases colonic circular smooth muscle contractility in rats. Am J Physiol 273:G928–G936
Parronchi P, Romagnani P, Annunziato F, Sampognaro S, Becchio A, Giannarini L, Maggi E, Pupilli C, Tonelli F, Romagnani S (1997) Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. Am J Pathol 150:823–832
Rich H, Sohn UD, Behar J, Kim N, Biancani P (1997) Experimental esophagitis affects intracellular calcium stores in the cat lower esophageal sphincter. Am J Physiol 272:G1523–G1529
Sanovic S, Lamb DP, Blennerhassett MG (1999) Damage to the enteric nervous system in experimental colitis. Am J Pathol 155:1051–1057
Sawa Y, Oshitani N, Adachi K, Higuchi K, Matsumoto T, Arakawa T (2003) Comprehensive analysis of intestinal cytokine messenger RNA profile by real-time quantitative polymerase chain reaction in patients with inflammatory bowel disease. Int J Mol Med 11:175–179
Shi XZ, Sarna SK (1999) Differential inflammatory modulation of canine ileal longitudinal and circular muscle cells. Am J Physiol 277:G341–G350
Shi XZ, Sarna SK (2000) Impairment of Ca(2+) mobilization in circular muscle cells of the inflamed colon. Am J Physiol 278:G234–G242
Sohn UD, Harnett KM, Cao W, Rich H, Kim N, Behar J, Biancani P (1997) Acute experimental esophagitis activates a second signal transduction pathway in cat smooth muscle from the lower esophageal sphincter. J Pharmacol Exp Ther 283:1293–1304
Takahara H, Fujimura M, Taniguchi S, Hayashi N, Nakamura T, Fujimiya M (2001) Changes in serotonin levels and 5-HT receptor activity in duodenum of streptozotocin-diabetic rats. Am J Physiol 281:G798–G808
Wallace JL, Keenan CM (1990) An orally active inhibitor of leukotriene synthesis accelerates healing in a rat model of colitis. Am J Physiol 258:G527–G534
Weisbrodt NW, Lai M, Bowers RL, Harari Y, Castro GA (1994) Structural and molecular changes in intestinal smooth muscle induced by Trichinella spiralis infection. Am J Physiol 266:G856–G862
Wells RW, Morris GP, Blennerhassett MG, Paterson WG (2003) Effects of acid-induced esophagitis on esophageal smooth muscle. Can J Physiol Pharmacol 81:451–458
Xie YN, Gerthoffer WT, Reddy SN, Cominelli F, Eysselein VE, Snape WJ Jr (1992) An abnormal rate of actin myosin cross-bridge cycling in colonic smooth muscle associated with experimental colitis. Am J Physiol 262:G921–G926
Acknowledgements
This work was supported by the Canadian Institutes of Health Research (CIHR). We thank the Canadian Digestive Health Foundation and CIHR for scholarship support (R.W.W.). The authors thank Dr. S. Lourenssen for help in preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wells, R.W., Blennerhassett, M.G. Persistent and selective effects of inflammation on smooth muscle cell contractility in rat colitis. Pflugers Arch - Eur J Physiol 448, 515–524 (2004). https://doi.org/10.1007/s00424-004-1286-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-004-1286-1