
Abstract
Purpose
To evaluate ophthalmological and molecular findings in eight patients with a clinical diagnosis of neurofibromatosis type 2 (NF2). New pathological mutations are described and variability in the ophthalmic phenotype and NF2 allelic heterogeneity are discussed.
Methods
Eye examination was performed in eight NF2 patients, and it included the measurement of the visual acuity, biomicroscopy, dilated fundus examination, color fundus photography, infrared photography, and spectral domain optical coherence tomography (SD-OCT). Molecular analysis was performed with whole-exome sequencing using DNA derived from peripheral blood mononuclear cells from each individual.
Results
Ophthalmological features were present in all patients, ranging from subtle retinal alterations identified only using SD-OCT to severe ocular damage present at birth. Six mutations were observed: two patients with stop codon mutation as shown on table 1 and result section, three patients with frameshift mutation as shown on table 1 and result section. Three novel mutations were found among them.
Conclusions
It is a descriptive study of a rare disease, with poor previous literature. Clinical and genetic data are shown, reviving the need to further studies to clarify the genotype-phenotype correlations in NF2.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurofibromatosis is a group of three genetic conditions caused by mutations in different genes [1] as follows: neurofibromatosis 1 (NF1) is caused by mutations that inactivate the neurofibromin gene on chromosome 17q [2]; neurofibromatosis 2 (NF2) is caused by inactivating mutations of the NF2 gene on chromosome 22q [3, 4]; and schwannomatosis is caused by germline mutations in SMARCB1 and LZTR1 genes [5].
Neurofibromatosis type 2 (NF2) is characterized by bilateral vestibular schwannomas, multiple central and peripheral nervous system tumors, and ocular manifestations [6]. Its estimated incidence lies between 1 in 33,000 [7] live births and 1 in 87,410 live births [8]. NF2 has a heterogeneous presentation and the disease severity ranges widely, from a single tumor developed later in life to multiple tumors developed in the first decade. There is also a diverse landscape of disease-causing NF2 mutations, including nonsense mutations, splice site mutations, and missense mutations [9]. Genotype-phenotype correlations in NF2 have been proposed, with nonsense and frameshift mutations being associated with the most severe clinical picture. Correlations between truncating mutations and ocular findings have also been observed [10, 11].
Neurofibromatosis type 2 (NF2) is a monogenic autosomal dominant disease, caused by mutations on 22a12.2 of NF2 gene. The NF2 gene is a tumor suppressor gene that has 17 exons and codes for a protein termed Merlin or neurofibromin 2. This protein is homologous to ezrin/radixin/moesin (ERM) family actin-binding proteins and serves as a linker between plasma membrane proteins and the actin [12]. Neurofibromin 2 is broadly expressed in vertebrate nervous tissues and in neuroepithelial compartments of the developing mouse eye. The loss of mouse neurofibromin 2 causes hyperplasia in these compartments, replicating the ocular abnormalities seen in human NF2 patients [12].
Ocular abnormalities associated with NF2 are cataract, retinal pigment epithelial hamartomas, retinal hamartomas, epiretinal membranes (ERM), and paralytic strabismus [13,14,15]. In children with a severe phenotype, ophthalmological alterations such as strabismus, cataract, ERM, and retinal hamartomas usually are present before the typical neurological symptoms, and their recognition can help to reduce the delay in diagnosis of pediatric patients with NF2.
Optical coherence tomography (OCT) is a noninvasive imaging test that uses infrared light waves to produce high-resolution cross-section images of the retina. In the last years, the introduction of OCT in clinical practice provided the discovery of new NF2 specific alterations such as flame-shaped ERM, retinal tufts, and retinal microhamartomas [16]. It is still poorly understood how the nature of the NF2 mutation affects the ocular involvement. The discovery of these subtle and specific retinal alterations and their phenotype-genotype correlation could serve in the direction of an appropriate medical treatment and monitoring and can help to better understand pathogenesis in NF2.
The present study aims to evaluate ophthalmological and molecular findings in eight patients with a clinical diagnosis of NF2. New mutations are described and variability in the ophthalmic phenotype and NF2 allelic heterogeneity are discussed.
Patients and methods
The study was approved by the ethics committee of Federal University of Minas Gerais, Brazil, and followed the principles of the Declaration of Helsinki. All patients received written and oral information about the study, and written consent was obtained from all subjects.
The subjects selected for the study were patients diagnosed with NF2 who were under the care of the neurofibromatosis outpatient reference center of Federal University of Minas Gerais, Brazil. The studied group included eight patients, five male and three females, with a mean age of 25 years (range 14 to 50 years). Patients 1, 2, 3, 4, 5, and 6 were included in a previous study [16]. All patients fulfilled the Manchester criteria for NF2. All patients were unrelated, and at least one first-degree relative of each patient received a complete ophthalmological exam, including spectral domain OCT (SD_OCT). Age and sex-matched controls also received an ophthalmological exam. There was no history of consanguinity.
Eye examination was performed in 16 NF2 eyes, and it included the measurement of the visual acuity and biomicroscopy before and after pupillary dilatation, dilated fundus examination, color fundus photography, infrared photography, fundus autofluorescence, and spectral domain optical coherence tomography (SD-OCT). SD-OCT was accomplished in 15 NF2 eyes; in one eye, OCT scans were not performed, due to a dense subcapsular lens opacity. SD-OCT examinations were performed by means of Spectralis HRA + OCT (Heidelberg Engineering, Heidelberg, Germany).
Molecular analysis was performed with whole-exome sequencing using DNA derived from peripheral blood mononuclear cells from each individual. DNA was extracted from peripheral white blood cells in a high salt method using a QIAamp Blood DNA Mini Kit (Qiagen®, Milano, Italy) according to the manufacturer’s instructions. Capture of exons was performed using Nextera Rapid Capture Mendelics Custom Panel V2 followed by new sequencing generation with Illumina Hiseq (HiSeq paired readings 2 × 75 pb) and alignment with human genome reference (hg19) using Burrows-Wheeler Aligner software. Genotyping of variants with genome analysis toolkit was performed according to the best practices at Broad’s Institute. Annotation and prediction of pathogenicity of variants using the in-house software for mutation prediction (Mendelics Abracadabra®) and checked in the Human Genome Mutation Database and ClinVar, associated with frequency control population, were performed.
The homology modeling server SWISS-MODEL [17] was used to predict the tertiary structure of wild NF2 protein and the protein product from the three new variants found.
The incidence of cataract, retinal abnormalities, and other ocular involvement was analyzed. The relationship between specific ocular findings and different types of mutations of the NF2 gene were pointed and analyzed.
Results
The results are summarized in Table 1. All patients presented with bilateral vestibular schwannomas (BVS).
The overall delay between the onset of symptoms and diagnosis of NF2 averaged 7 years. Of the eight patients, six had an early onset of NF2 (< 20 years), and the delay between the first symptoms and diagnosis in this group was an average of 9.3 years (range 2 to 18 years) (Table 1). In half of the patients with early onset, the initial symptoms were ophthalmological disorders (two patients presented with strabismus and one patient with cataract) and the mean delay in diagnosis in these patients was 11 years. Late onset of NF2 (> 20 years) was observed in two of eight patients, and their NF2 diagnosis was based on the discovery of BVS just a few months after the beginning of the first symptoms. The initial symptoms of these two patients were inferior limb paresis and facial pain.
Ophthalmological features were present in all patients and range widely from subtle retinal alterations, identified only by SD-OCT, to severe ocular involvement present at birth.
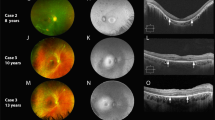
Cataracts were noted in five of eight patients (four with early onset and one with late onset). Cataracts were visually impacting in three eyes of three patients (Fig. 1a, b). One patient in the early-onset group had chronic-appearing retinal detachment and retinal hamartoma in the right eye. Epiretinal membranes were noticed in all five patients in the early-onset group (Fig. 1c, d); four patients had thick ERM with a flame-shaped appearance on SD-OCT scans, and one patient had subtle ERM with fine undulations on the inner retinal surface. Retinal hamartomas were noted in four patients in the early-onset group. In two patients, hamartomas were subclinical, detected only by SD-OCT (Fig. 2d, e). Choroidal nodules, similar to those described in NF1 patients, were found in three patients in the early-onset group (Fig. 3a, b). All patients with epiretinal membrane, retinal hamartomas, and choroidal nodules were in the early-onset group and presented with truncating mutations. Tufts of retinal internal layers detected exclusively by SD-OCT (Fig. 2a–c) were found in four patients; two patients had early-onset NF2 and two had late-onset NF2. Paralytic strabismus was noted in three patients in the early-onset group.

a Anterior segment photography of case 1 showing typical subcapsular and cortical cataract. b Retinography of case 6 showing a retinal hamartoma. c Retinography of case 5 showing a typical NF2 ERM. d SD-OCT of case 5 showing the ERM with the characteristic flame-shaped appearance

a SD-OCT of case 8 showing a peripheral retina tuft. b SD-OCT of case 5 showing a small retinal tuft in the macular region. c SD-OCT of case 2 showing an evident retinal tuft in parafoveal region. d SD-OCT of case 7 showing a small retinal hamartoma evident only by SD-OCT. e SD-OCT of case 8 showing an intraretinal retinal hamartoma

a Infrared imaging and SD-OCT of case 4 showing a choroidal nodule. b Infrared imaging and SD-OCT of case 8 showing a choroidal nodule
No first-degree relative presented with NF2 specific ocular findings, and the only ophthalmological alteration found in some of them was low ametropias.
Molecular findings
Molecular analysis was performed with whole-exome sequencing. In the studied group, we found three categories of truncating mutations: two patients with premature stop codon (nonsense) mutations, three patients with frameshift mutations, and one patient with splice site mutation.
All three novel mutations found were frameshift mutations that caused a change of reading frame and from this point leading to premature interruption of protein translation. The two patients with nonsense mutations presented with a C-T transition, one at position 196 and the other one at position 165. They presented with widely different phenotypes.
No germline mutations were detected in two patients with late-onset NF2. Those patients presented with retinal alterations, but they were subtle and appear only in SD-OCT. The ophthalmological features and molecular findings of the eight NF2 patients are summarized in Table 1.
In a comparison of wild-type NF2 protein structure, tridimensional modeling of new frameshift mutation revealed conformation changes in protein structure in all three variants (Fig. 4).

Structure modeling of wild-type and frameshift mutations of NF2 gene with SWISS-MODEL. a 3D model of wild-type NF2 protein. b 3D model of variant NF2 chr22:30.064.367- 30.064.368 AG > A p.Ala313Pro fs*9 of patient 4. c Protein structure of variant chr22:30.032.802 -30.032.809 CTGGTTCT > C p.Trp60Leu fs*9 of patient 8. d 3D model of variant chr22:30.057.260 30.057.268 AACAGACTG > A p.Arg249Pro fs*9 of patient 6.
Discussion
To date, over 400 mutations in the NF2 gene have been described (HGMD, Human Gene Disease Mutation Database, http://www.hgmd.cf.ac.uk/, accessed on 07 September 2018). The majority of these mutations were truncating mutations, leading to a smaller and probably nonfunctional protein product. The most common mutations in the NF2 gene are nonsense mutations and small deletions [9, 18].
About 25% of NF2 patients have a mosaic disease, in which only a proportion of cells contain the mutated NF2 gene, frequently resulting in cases with a mild presentation. The initiating mutation occurs after conception, leading to two separate cell lineages. The proportion of cells affected depends on how early in development the mutation occurs [9, 19, 20]. Two patients in our series had late-onset NF2 and besides fulfilling the clinical criteria, no mutations were found on them. We suppose they might have a mosaic pattern.
Different from previous studies [10, 11], at this sample, we observed ocular features in a 100% of the sample. The main difference between our group of patients and previous reports relays on retinal alterations found in patients with a mild phenotype and no germline mutations. To our knowledge, no retinal changes have been reported in patients without germline mutation. The retinal alterations found in our study were tufts of retinal internal layers identifiable only using SD-OCT. The differences in findings might be a technological issue since the use of SD-OCT was not usually reported. It is possible that with the inclusion of SD-OCT in clinical practice, these subtle findings will be reported more frequently. Retinal tufts were first described by Chan C on histopathology exam of three patients with severe NF2 [21]. Retinal tuft identifiable by SD-OCT was first described recently in a previous report involving patients 1, 2, 3, 4, and 5 of the present study [16].
The relationship between choroidal nodules and NF1 has been well established. Choroidal findings are so prevalent in NF1 patients that some authors suggested that they should be included in the diagnostic criterion of NF1 [22, 23]. Unlike NF1, there is little information about choroidal changes in NF2 [16]. Choroidal nodule in an NF2 patient was first described in a previous study including patient 4 [16]. In our series, patients who presented with ophthalmological features as the first sign of NF2 also show choroidal nodules in near-infrared imaging (Fig. 3). Two of these patients had frameshift mutations and one patient had exon 2 splice site mutation (Table 1). These findings are in accordance with previous reports associating constitutional splice site mutations in exons 1–5 to more severe disease. Exons 2 and 3 seem to be required for self-association of the amino terminal of the NF2 protein [6]. Frameshift mutations are also associated with more severe disease and retinal alterations in patients with NF2 [9, 10].
In both patients with nonsense C to T substitution, a stop codon was inserted resulting in a shorter protein. Clinically, one patient presented with multiple tumors in very young age, paralytic strabismus, cataract, subtle ERM, and retinal hamartoma whereas the other presented with early-onset NF2 and flame-shaped ERM.
Retinal hamartomas, epiretinal membranes, and retinal tufts are part of a wide array of ocular abnormalities that occur in NF2. These retinal lesions are thought to result from developmental defects involving glial cells and the vascular epithelium; consequently, it has been suggested that they represent different faces of the same pathological process [16, 21, 24,25,26,27]. Until now, little has been reported on the ophthalmologic course in patients with NF2. It has been suggested that most NF2-specific ophthalmologic findings remain stable during the course of illness [14]. Long-term follow-up of retinal alterations with SD-OCT may improve comprehension of pathology and relationship between these lesions.
In conclusion, although this study has case-series limitations, including the small number of patients in a descriptive study, NF2 is a rare disease and there are only a few previous reports about the ocular findings. This study shed light and adds new information on both clinical and genetic features in NF2. Ocular alterations and the associated genetic features can pave for additional studies on the genotype-phenotype correlations in NF2.
References
NIH conference statement (1990) Neurofibromatosis 1 (von Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis). Ann Intern Med 113:39–52
Seizinger BR, Rouleau GA, Ozelius LJ et al (1987) Genetic linkage of von Recklinghausen neurofibromatosis to the nerve growth factor receptor gene. Cell 49:589–594
Roleau G, Seizinger BR, Ozelius LG et al (1987) Genetic linkage analysis of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22. Nature 329:246–248
Cooper J, Giancotti FG (2014) Molecular insights into NF2/Merlin tumor suppressor function. FEBS Lett 16:2743–2752
Rodrigues LO, Batista PB, Goloni-Bertollo EM et al (2014) Neurofibromatosis part 1 – diagnosis and differential diagnosis. Arq Neuropsiquiatr 72:241–250
Basser ME, Kuramoto L, Woods R et al (2005) The location of constitutional neurofibromatosis 2 (NF2) splice site mutations is associated with the severity of NF2. J Med Genet 42:540–546
Evans DG, Howard E, Giblin C et al (2010) Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet 152A:327–332
Antinheimo J, Sankila R, Carpén O, Pukkala E, Sainio M, Jääskeläinen J (2000) Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology 54:71–76
Evans DGR (2009) Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis 4:16
Feucht M, Kluwe L, Mautner VF, Richard G (2008) Correlation of nonsense and frameshift mutations with severity of retinal abnormalities in neurofibromatosis 2. Arch Ophthalmol 126:1376–1380
Parry DM, MacCollin MM, Kaiser-Kupfer MI et al (1996) Germ-line mutations in the neurofibromatosis 2 gene: correlations with disease severity and retinal abnormalities. Am J Hum Genet 59:529–539
Moon KH, Kim HT, Lee D, Rao MB, Levine EM, Lim DS, Kim JW (2018) Differential expression of NF2 in neuroepithelial compartments is necessary for mammalian eye development. Dev Cell 44:13–28
Kaye LD, Rothner AD, Beauchamp GR, Meyers SM, Estes ML (1992) Ocular findings associated with neurofibromatosis type II. Ophthalmology 99:1424–1429
Bosch MM, Boltshauser E, Harpes P, Landau K (2006) Ophthalmologic findings and a long-term course in patients with neurofibromatosis type 2. Am J Ophthalmol 141:1068–1077
Feucht M, Griffiths B, Niemüller I, Haase W, Richard G, Mautner VF (2008) Neurofibromatosis 2 leads to higher incidence of strabismological and neuro-ophthalmological disorders. Acta Ophthalmol 86:882–886
Waisberg V, Rodrigues LO, Nehemy MB, Frassom M, De Miranda DM (2016) Spectral-domain optical coherence tomography findings in neurofibromatosis type 2. Inves Ophthalmol Vis Sci 57:OCT262–OCT267
Schwede T (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31(13):3381–3385
Ahronowitz I, Xin W, Kiely R, Sims K, Maccollin M, Nunes FP (2007) Mutational spectrum of the NF2 gene: a meta-analysis of 12 years of research and diagnostic laboratory findings. Hum Mutat 28:1–12
Kluwe L, Mautner V, Heinrich B et al (2003) Molecular study of frequency of mosaicism in neurofibromatosis 2 patients with bilateral vestibular schwannomas. J Med Genet 40:109–114
Kluwe L, Mautner VF (1998) Mosaicism in sporadic neurofibromatosis 2 patients. Hum Mol Genet 7:2051–2055
Chan C, Koch CA, Kaiser-Kupfer MI et al (2002) Loss of heterozygosity for the NF gene in retinal and optic nerve lesions of patients with neurofibromatosis 2. J Pathol 198:14–20
Viola F, Villani E, Natacci F et al (2012) Choroidal abnormalities detected by near-infrared reflectance imaging as a new diagnostic criterion for neurofibromatosis 1. Ophthalmology 119:369–375
Moramarco A, Giustini S, Nofroni I, Mallone F, Miraglia E, Lacovino C, Calvieri S, Lambiase A (2018) Near-infrared imaging: an in vivo, non-invasive diagnostic tool in neurofibromatosis type 1. Graefes Arch Clin Exp Ophthalmol 256:307–311
Landau K, Yasargil GM (1993) Ocular fundus in neurofibromatosis type 2. Br J Ophthalmol 77:646–649
Meyers SM, Gutman FA, Kaye LD, Rothner AD (1995) Retinal changes associated with neurofibromatosis 2. Trans Am Ophthalmol Soc 93:245–257
Sisk RA, Berrocal AM, Schefler AC, Dubovy SR, Bauer MS (2010) Epirretinal membranes indicate a severe phenotype of neurofibromatosis type 2. Retina 30:51–58
McLaughlin ME, Pepin SM, MacCollin MM, Choopong P, Lessell S (2007) Ocular pathologic findings of neurofibromatosis type 2. Arch Ophthalmol 125:389–394
Acknowledgments
This research was partially supported by Federal University of Minas Gerais, Brazil.
Funding
This study was partially funded by FAPEMIG (Grupos Emergentes).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
All procedures performed in the study involving human participants were in accordance with ethical standards of the institutional committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Waisberg, V., Rodrigues, L.O.C., Nehemy, M.B. et al. Ocular alterations, molecular findings, and three novel pathological mutations in a series of NF2 patients. Graefes Arch Clin Exp Ophthalmol 257, 1453–1458 (2019). https://doi.org/10.1007/s00417-019-04348-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-019-04348-5