
Abstract
Estimating the time since death is a very important aspect in forensic sciences which is pursued by a variety of methods. The most precise method to determine the postmortem interval (PMI) is the temperature method which is based on the decrease of the body core temperature from 37 °C. However, this method is only useful in the early postmortem phase (~0–36 h). The aim of the present work is to develop an accurate method for PMI determination beyond this present limit. For this purpose, we used sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), Western blotting, and casein zymography to analyze the time course of degradation of selected proteins and calpain activity in porcine biceps femoris muscle until 240 h postmortem (hpm). Our results demonstrate that titin, nebulin, desmin, cardiac troponin T, and SERCA1 degraded in a regular and predictable fashion in all samples investigated. Similarly, both the native calpain 1 and calpain 2 bands disintegrate into two bands subsequently. This degradation behavior identifies muscular proteins and enzymes as promising substrates for future molecular-based PMI determination technologies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The delimitation of the postmortem interval (PMI) is a very important aspect in forensic sciences. To determine the PMI as exactly as possible aids to reveal the circumstances of death and therefore has legal relevance in appropriate cases. To date, there are numerous approaches that deal with different physical, chemical, or biological aspects of tissue and/or organ decomposition. The most precise method to determine the PMI is the temperature method which is based on the decrease of the body core temperature from 37 °C. However, this method is only useful in the early postmortem phase (~0–36 h postmortem, hpm) [1]. Additional methods for PMI delimitation in this phase involve supravital reactions of tissue [2], such as electrical stimulation of eyelid muscle [3, 4] and pharmacological excitability of the iris [3, 4]. Further applied methods include the analysis of vitreous potassium and hypoxantine [5], rigor and livor mortis [6], and analyses of biological clock genes [7]. The late phase (from ~120 hpm onward) is relatively well characterized by forensic entomology [8]. More recently, the late phase was characterized by postmortem changes of the microbial community in the abdomen and on the skin in mice [9] or degradation of tooth pulp RNA and colorimetric analysis of pulp decomposition [10]. In contrast to the early and the late postmortem phases, there is a lack of confidential methods for PMI delimitation in the intermediate phase between approximately 36 and 120 hpm. Several approaches have been described to close this knowledge gap, such as quantitative analyses of DNA degradation [11] or changes of cartilage structure and function [12]. However, these procedures have major weaknesses regarding their practicability and accuracy.
Skeletal muscle appears to be a viable target tissue for PMI analysis because it is the most abundant tissue in the body and has a much greater delay in postmortem change compared to kidney, pancreas, liver, and heart [13]. Thus, degradation of muscle tissue is especially interesting for analysis of intermediate and late PMI. Several studies have addressed this issue in the rodent model. Degeneration of muscle has been characterized at ultrastructural [14] as well as on protein level [15–17]. Remarkably, it was also shown that human skeletal muscle undergoes a highly active transcriptional, and possibly translational phase during the initial 46 hpm [18].
Postmortem degradation of skeletal muscle has also been intensively studied in the context of meat tenderness and storage. Studies using domestic animals have shown that particularly the degradation of two large cytoskeletal proteins, titin and nebulin, improves meat tenderness [19–25]. Other cytoskeletal proteins that have been suggested to play a role in meat tenderization are desmin [26], troponin T [23], and filamin [24]. All these proteins have different rates of degradation. Some proteins have been shown to reproducibly degenerate into the same degradation products, even in a variety of species. The native form of titin (also known as T1), for example, very early degrades into a degradation product called T2 in pigs [23, 27], cattle [20, 24, 25], rabbits [28, 29], and chickens [22, 26].
It is known that the degradation of such proteins depends upon the activity of Ca2+-dependent calpains, a family of proteolytic enzymes [21, 30]. In particular, the family members calpain 1 and calpain 2 (formerly referred to as μ-calpain and m-calpain, respectively) have been demonstrated to increase meat tenderness by their proteolytic activity. Calpain 1 and 2 are activated upon an increase in the intracellular Ca2+ concentration [31] which occurs when the integrity of the sarcolemma is compromised after death. More recently, it was suggested that calpain 1 is the main isoform responsible for protein degradation at low temperatures while calpain 2 is activated at higher temperatures [32].
Based on the knowledge gained from studies investigating the degradation of specific muscle proteins in domestic animals, we aim to develop an accurate method for PMI delimitation, especially in the intermediate postmortem phase. We also intend to gain additional information about early and late postmortem changes to supplement or eventually substitute inaccurate or inefficient existing methods. For this purpose, we analyzed the time course of degradation of selected proteins in porcine biceps femoris muscle until 240 h postmortem. The degradation behavior of titin, nebulin, α-actinin, tropomyosin, desmin, cardia troponin T (cTnT), and SERCA1 was assessed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) or Western blotting. Casein zymography was employed to determine the activities of calpain 1 and calpain 2. The results are intended to identify proteins which are promising substrates for future molecular-based PMI determination technologies.
Materials and methods
Animals and sampling
Three male pigs (commercial crossbreed animals, German Large White × German Landrace, 3 months old) were used for this study. The animals were killed according to standard procedures (captive bolt stunning and exsanguination). Both hind limbs were separated by dissection from each animal and the first samples taken from each leg (approximately 30 min after death = 0 h samples). The legs were stored separately under standardized laboratory conditions (temperature, 21 ± 1 °C; humidity, 35 ± 5 %), and muscle tissue from the belly of the musculus biceps femoris was sampled at pre-defined points of time within a period of 10 days pm (6, 12,18, 24, 36, 48, 60, 72, 84, 96, 108, 120, 144, 168, 192, 216, and 240 h). For sampling, an incision was made through the skin and the underlying fascia using a surgical scalpel, and a 2 × 2 × 2 cm piece of muscle tissue was removed in a depth of 2 cm. Minimum distance between sampling sites was 3 cm. Excised muscle pieces were subdivided in samples of approximately 100 mg. Samples were snap frozen and stored in liquid nitrogen until further use. Frozen samples were homogenized by cryogenic grinding and subsequent sonication (three bursts at maximum power). RIPA buffer containing protease inhibitor cocktail (SIGMA) was used as protein extraction buffer for SDS-PAGE and western blot. For casein zymography, an extraction buffer containing 50 mM Tris, 5 mM EDTA, and 10 mM 3-mercaptopropane-1,2-diol was used. The homogenate was centrifuged at 1000×g for 6 min and supernatant was removed and stored at −20 °C (SDS-PAGE and western blot) or liquid nitrogen (zymography) until further use. Protein concentration was measured using Pierce BCA-Assay Kit (Thermo Fisher Scientific Inc.).
SDS-PAGE
SDS-PAGE was performed according to Laemmli [33]. For the detection of α-actinin, tropomyosin, desmin, cTnT, and SERCA1 10 % polyacrylamide gels (acrylamide/N,N’-bis-methylene acrylamide = 37.5:1, 0.1 % SDS, 0.05 % TEMED, 0.05 % APS, and 375 mM Tris HCl, pH 8.8) were used as resolving gels. Five percent polyacrylamide gels (acrylamide/N,N’-bis-methylene acrylamide = 37.5:1, 0.1 % SDS, 0.125 % TEMED, 0.075 % APS, and 125 mM Tris HCl, pH 6.8) were used as stacking gels. Total protein (20–40 μg) was diluted in 20 μl Aqua bidest/sample buffer (40 % glycerine, 10 % mercaptoethanol, 0.04 % bromphenol blue, and 250 mM Tris HCl pH 6.75) = 3:1. Samples were then denatured at 90 °C for 5 min and inserted into the gel wells. Electrophoresis was run at a constant voltage of 150 V until the dye front reached the bottom of the gel (approximately 1.5 h). The running buffer contained 25 mM Tris, 195 mM glycine, 2 mM EDTA, and 0.1 % SDS. Gels were transferred onto polyvinylidine fluoride (PVDF) membranes in transfer buffer (192 mM glycine, 20 % methanol, and 25 mM Tris) at a constant current of 250 mA for 75 min.
Five percent polyacrylamide gels (acrylamide/N,N’-bis-methylene acrylamide = 100:1, 0.1 % SDS, 2 mM EDTA, 0.067 % TEMED, 0.1 % APS, and 200 mM Tris HCl, pH 8.0) were used for the detection titin of and nebulin, without stacking gels. Samples were diluted to 80 μg total protein diluted in 20 μl A. bidest/sample buffer = 3:1, heated at 90 °C for 5 min and inserted into the gel wells. Electrophoresis was run at a constant current of 10 mA for approximately 22 h. Running buffer contained 25 mM Tris, 192 mM glycine, 2 mM EDTA, 0.1 % mercaptoethanol, and 0.1 % SDS.
After electrophoresis, titin and nebulin gels were stained in Coomassie dye (0.1 % Coomassie Brilliant Blue R250, 50 % methanol, and 10 % glacial acidic acid) for 2 h and thoroughly destained in the same solution without Coomassie dye.
Western blotting
All blots were blocked for 1 h in TTBS (150 mM NaCl, 0.05 % Tween, and 25 mM Tris, pH 7.5) including 1 % bovine serum albumin (BSA). The following primary antisera were used: α-actinin (N-19, Santa Cruz, 1:500), tropomyosin (CH1, DSHB, 1:250), mouse monoclonal anti-desmin (DE-R-11, Santa Cruz, 1:8000), mouse monoclonal anti-cardiac troponin T (CT3, DSHB, 1:250), goat polyclonal anti-SERCA1 (N-19, Santa Cruz, 1:400), mouse monoclonal anti-titin (9D10, Developmental Studies Hybridoma Bank (DSHB), 1:250), and anti-nebulin (N-19, Santa Cruz, 1:250). Secondary antibodies applied were HRP conjugated polyclonal goat anti-mouse (DAKO, 1:10000) and HRP conjugated polyclonal rabbit anti-goat (DAKO, 1:10000). All primary and secondary antibodies were diluted in 1 % BSA solution in TTBS.
After each antibody application, the membranes were extensively washed in TTBS. Antibody binding was visualized by 5-min incubation in chemiluminescence substrate (SuperSignal West Pico) and consecutive exposure to autoradiography film (Amersham Hyperfilm ECL).
Casein zymography
Casein zymography was performed according to Raser [34] with slight modifications. Polyacrylamide gels (12.5 %; acrylamide/N,N’-bis-methylene acrylamide = 37.5:1, 0.1 % TEMED, 0.05 % APS and 375 mM Tris HCl, pH 8.8) were copolymerized with 0.1 % casein and used as resolving gels. Polyacrylamide gels (5 %; acrylamide/N,N’-bis-methylene acrylamide = 37.5:1, 0.125 % TEMED, 0.075 % APS, and 330 mM Tris HCl, pH 6.8) were used as stacking gels. Sample supernatant was diluted 1:1 in sample buffer (25 % glycerol, 0.1 % bromphenol blue. and 62.5 mM Tris HCl, pH 6.8). This was again diluted 1:1 in A. bidest and inserted into the gel well without denaturation. The running buffer contained 25 mM Tris, 192 mM glycine, and 1 mM EDTA. After a 15-min prerun at 75 V and 4 °C, the samples were inserted into the gel wells and electrophoresis was run for approximately 6 h at 75 V and 4 °C.
The gels were briefly rinsed with A. bidest, transferred into incubation buffer with varying CaCl2 levels (4 mM, 400 μM, 40 μM CaCl2, 0.1 % 3-mercapto-1,2-propanediol, and 50 mM Tris HCl, pH 7.5), and incubated overnight (12–18 h at room temperature). Subsequently, the gels were stained in Coomassie dye (0.1 % Coomassie Brilliant Blue R250, 50 % methanol, and 10 % acidic acid) for 2 h and thoroughly destained in the same solution without Coomassie dye.
Statistical analyses
The intensity of all protein bands was measured using Gel Analysis Tools in ImageJ (1.48 v NIH, National Institutes of Health, USA). Data were further converted into binary information (presence vs. absence of bands), and abundance of bands per time point was used for statistical analysis. For survival analysis, the Kaplan-Meier estimator was used to determine the statistic mean point of time as well as the 95 % confidence interval (CI) of all qualitative band changes. Statistical analysis was performed using SPSS 21 (IBM, USA).
Results
High molecular weight proteins
In a first experimental series, we aimed to investigate whether the presence and/or size of certain muscle proteins changes within the first 10 days pm. For this purpose, we used SDS-PAGE on 5 % gels to analyze the proteins with the highest molecular masses present in the muscle biopsies. Seven different protein bands were clearly detected over the investigated time course (Fig. 1a). In all six legs examined, these bands revealed similar patterns of location in the gel and similar temporal appearances/disappearances. The bands conform to those of large proteins that have been identified in various mammals [29, 35]. It is known that the proteins between 600 and 900 kDa correspond to nebulin [26, 35]. Proteins larger than 900 kDa (up to 4 MDa) represent titin isoforms and titin derivatives [29]. Using monoclonal titin and nebulin antibodies, we were able to identify the two bands with the largest proteins as titin 1 and 2 (T1 at 0 hpm and T2 at 24 hpm) and two bands as nebulin 1 and 2 (N1 at approximately 900 kDa and N2 at about 700 kDa) (Fig. 1b). An additional band between T1 and N1 most likely corresponds to a 1200-kDa titin derivate, which is a well-known degeneration product of titin [24, 28]. Two bands that appear only after 72 hpm most likely represent titin degradation products [20] and are therefore further referred to as titin dp1 and titin dp2.

Titin and nebulin degradation during postmortem storage until 240 h. a Coomassie-stained 5 % acrylamide gels of a porcine musculus biceps femoris muscle sample reveals the presence of native and/or degraded forms of titin and nebulin at various time points postmortem. b Western blots (WB) using anti-titin and anti-nebulin antibodies demonstrate the identities of bands at 0 and 24 hpm. T1 = native form of titin, T2 = titin derivative, N1 = nebulin 1, N2 = nebulin 2, dp = degradation product
Results demonstrate that at 0 hpm (hours postmortem), only four bands were present (T1, the 1200-kDa titin derivate, N1, and N2) (Fig. 1a). A statistical analysis of the binary outcome (presence/absence) of the native titin band T1 using the Kaplan-Meier estimator demonstrates that this protein was present until 14.0 ± 2.0 hpm (Fig. 2a and Table 1). The confidence interval indicates that the T1 band disappeared to 95 % between 10.1 and 17.9 hpm. The first major titin degradation product T2 was present from 16.0 ± 2.0 hpm onward (Figs. 1a, 2a, Table 1). The appearance of this band occurred in a close time window such that this band statistically emerged between 12.1 and 19.9 hpm. The 1200-kDa titin derivative was only detectable for a short period, with slightly varying vanishing times between samples (Fig. 1a). The mean disappearance time was at 13.2 ± 2.2 hpm; 95 % vanished between 8.8 and 17.6 hpm (Fig. 2a and Table 1). At the approximately same position as the 1200-kDa titin form, an additional degradation product of titin, titin dp1, appeared on average at 69.6 ± 7.0 hpm (Figs. 1a and 2a and Table 1). Compared to T1, T2, and 1200-kDa titin forms, there was a large temporal variance in appearance so that the 95 % confidence interval is between 55.9 and 83.3 hpm. The titin degradation product titin dp2 has a molecular weight of approximately 800 kDa and emerged on average at 202.0 ± 18.4 hpm (Figs. 1a and 2a and Table 1). Also, for this peptide, there was a longer period of appearance which ranged between 165.9 and 238.1 hpm. Titin dp1 and titin dp2 were both present until the end of the investigated period of time.

Changes in protein band appearance over 240 hpm. a Titin, b nebulin, c desmin, d cardiac troponin T, e SERCA1, f calpain 1 and 2. Gray bars indicate the 95 % CI where individual changes happen. Vertical lines and error bars within the gray bars mark the mean and SE. Black bars stand for the presence of a band, white bars for the absence. The survival analysis was performed using the Kaplan-Meier estimator
The nebulin band N1 was detectable until 52.8 ± 6.1 hpm (Figs. 1a and 2a and Table 1). Similar to the native titin band T1, the time frame of disappearance was rather short, so that 95 % disappeared between 40.8 ± 64.8 hpm. N2 was detected in all legs until 84.0 ± 10.7 hpm and it significantly disappeared between 63.0 and 105.0 hpm (Figs. 1a and 2a and Table 1).
Degradation of α-actinin, tropomyosin, desmin, troponin T, and SERCA1
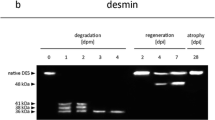
Western blot analysis using α-actinin, tropomyosin, desmin, cardiac troponin T (cTnT), and SERCA1 antibodies were performed to examine whether these muscle proteins also go through regular decay processes. These analyzes were initially performed for 0, 120, and 240 hpm samples. The results revealed that tropomyosin and α-actinin bands were present throughout the sampling period and no degradation products could be identified within the first 240 hpm (Fig. 3a, b). By contrast, analyses of desmin, cTnT, and SERCA1 expression patterns demonstrated that these proteins degraded in a predictable fashion in all samples investigated. A more detailed analysis of desmin showed that the native protein at about 53 kDa was at least present until 224.0 ± 13.3 hpm (Fig. 3c and Table 1). The Kaplan-Meier estimate indicates that this band was significantly present until 198.0 hpm (Fig. 2c). Within the sampling period, several desmin degradation products were detected with molecular weights between 35 and 50 kDa (Fig. 3c). The most prominent band above 35 kDa, further referred to as desmin dp1, appeared at 45.0 ± 10.0 hpm (95 % CI between 25.4 and 64.6 hpm) and sometimes disappeared again together with the native band. The mean appearance of the 35 kDa desmin band (desmin dp2) was at 35.0 ± 6.6 hpm (95 % CI between 22.0 and 48.0 hpm) (Fig. 2c, Table 1). This band remained detectable throughout the sampling period in all samples (Fig. 3c).

Degradation behavior of α-actinin, tropomyosin, desmin, cTnT, and SERCA1 at various time points postmortem (0–240 hpm). Western blot analysis of a porcine musculus biceps femoris muscle sample shows that α-actinin (a) and tropomyosin (b) were present throughout the investigated period and no degradation products were found. By contrast, desmin (c), cTnT (d), and SERCA1 (e) degraded in a predictable fashion in all muscle samples. dp = degradation product
The native cTnT band of approximately 40 kDa remained stable almost throughout the investigated period (Fig. 3d). It was present until 232.0 ± 4.6 hpm (Fig. 2d and Table 1). Analysis further reveals that two split products of cTnT regularly appeared in the gels. The first degradation product cTnT dp1 has a molecular weight of about 37 kDa and appeared at 120.0 ± 8.5 hpm (95 % CI at 103.4–136.6 hpm). Only slightly later, the degradation product cTnT dp2 with a molecular mass of approximately 35 kDa emerged at 132.0 ± 6.9 hpm (95 % CI between 118.4 and 145.6 hpm; Figs. 2d and 3d and Table 1).
Western blot analysis using an anti-SERCA1 antibody revealed that this protein appears as a double band at 0 hpm (Fig. 3e). This doublet was at least present until 192 hpm and vanished on average at 230.4 ± 6.1 hpm (Fig. 2e and Table 1). From 168 hpm onward, several degradation products of various sizes between approximately 35 and 100 kDa were detected. Two of these products were present at high intensities in all legs. The degradation product 1 (SERCA1 dp1) has a molecular weight of approximately 60 kDa and appeared at 199.2 ± 31.3 hpm. SERCA1 dp2 with a molecular mass of approximately 50 kDa emerged even later at 230.4 ± 8.6 hpm. Evaluation of the 95 % CI of appearance demonstrates that SERCA1 dp1 and SERCA1 dp2 are not present before 137.9 and 213.6 hpm, respectively (Figs. 2e and 3e and Table 1).
Calpains
A native PAGE on casein copolymerized gels with subsequent zymography was performed to analyze the activity of calpains, the major proteases that are known to be responsible for degradation of skeletal muscle proteins [36]. Upon activation by 4 mM and 400 μM CaCl2, two bands of different sizes were detected in all samples at 0 hpm (Fig. 4a, b). When CaCl2 concentration in the incubation buffer is reduced to 40 μM, only the upper band containing the slower migrating protein remained detectable. No bands were detected when using an incubation buffer without CaCl2 (Fig. 4b). From these experiments, it can be inferred that the bands most likely represent μM- and mM-Ca2+ concentration dependent proteases and therefore could be identified as calpain 1 and calpain 2. Within the sampling period, calpain 1 (represented by the top band) vanished earlier than calpain 2 (Fig. 4a). On average, the native calpain 1 band was present with decreasing intensity until 86.4 ± 2.4 hpm (Table 1 and Fig. 2f). There was a short temporal variation in the disappearance of this band so that 95 % of the changes from presence to absence occurred between 81.7 and 91.1 hpm. At 19.2 ± 1.2 hpm, an additional band appeared underneath the original band. This band is likely to be the autolyzed form of calpain 1. This band also appeared in a narrow time frame so that 95 % of all changes occurred within 16.8 and 21.6 hpm. In most cases, the autolyzed calpain 1 was active throughout the investigated period. The calpain 2 band was detected with decreasing intensity until 144.0 ± 18.2 hpm (Table 1 and Fig. 2f). Evaluation of the 95 % CI reveals a significant change from presence to absence at 108.4–179.6 hpm. Similar to calpain 1, an additional band appeared when the intensity of the calpain 2 band decreased markedly at 88.8 ± 13.3 hpm (Fig. 4a). This protein form arose in a wider time frame between 62.6 and 115.0 hpm.

Zymogram shows different calpain activities at different time points postmortem. The native calpain 1 band disintegrates into two bands between 18 and 36 hpm. In contrast, it takes until 72 hpm that the same occurs for calpain 2. A similar difference became evident for the final disappearance of the native calpain 1 and 2 bands which lasts until 84 and 168 hpm, respectively. Activities of calpains 1 and 2 are dependent upon calcium concentrations. The use of different concentrations of calcium demonstrates that calpain 1 is already active at 40 μM CaCl2 (b), while calpain 2 activity is only present at concentrations of 400 μM (lower band in (c)) and 4 mM CaCl2 (lower band in (d)). Without calcium in the incubation buffer, no activity is detected (a). Note that at 4 μM CaCl2, the degraded calpain 1 is also active
Discussion
The results of the present study demonstrate that analyses of skeletal muscle protein degradation are very promising tools for the delimitation of the postmortem interval (PMI). In general, we could demonstrate that the degradation of the investigated proteins occurred with a high regularity at different times postmortem. Some proteins degraded very early (e.g., T1 and N1), and others degraded late (e.g., desmin and SERCA1) or never (α-actinin, tropomyosin) within the first 10 days postmortem. In addition, split products of certain proteins regularly appeared within the gels at certain points of time.
The protein changes detected are of varying significance for PMI delimitation because some changes happen within a wider time range (e.g., appearance of titin dp2 and autolyzed calpain 2 or disappearance of native calpain 1 and calpain 2) while others were found to be limited to a time frame of only a couple of hours (e.g., degradation of T1 and 1200-kDa unit, and appearance of T2 and autolyzed calpain 1). Used in combination, the described patterns of appearance/disappearance of all investigated proteins and their degradation products characterize certain time points and thus allow a more precise delimitation of PMI at the intermediate and early late postmortem phase.
The present results of protein degeneration in porcine muscle are very much in agreement with published data from other species. Analysis of native titin (T1) degradation reveals high similarities with data derived from cattle [20, 24, 25, 37], rabbits [28, 29], and chickens [22, 26]. This includes the appearance of similar degeneration products, such as T2 (at approximately 2400 kDa) and the 1200-kDa titin polypeptide, and a similar order of temporal appearance/disappearance of these proteins. However, a direct comparison is not possible due to different temperature regimes used in the present study (21 °C) and the above mentioned studies (−1.5 °C to 4 °C). Thus, it can be expected that rates of protein degeneration found in this study are much higher than described in the literature [38]. T1, for example, vanished at 14.0 ± 2.0 hpm but was present for 3 days in bovine muscle stored at 2–4 °C [20]. Hardly any degradation of T1 was found until 168 hpm in porcine muscle stored at 4 °C [23]. At a lower temperature of −1.5 °C, a complete degradation of T1 into T2 occurred only after 7 days postmortem in bovine muscle [24]. This degradation was also shown to depend on the pH value. In addition to the degradation products T2 and 1200-kDa unit, the SDS-PAGE gel analysis performed in the present study further identified a multiple number of titin degradation products that appeared in the 10-day period. Some of these, e.g., the titin degradation products 1 and 2, were found very regularly and may also be of great interest for future PMI delimitation studies.
In contrast to titin, we were unable to detect any smaller degradation products of nebulin. Thus, degradation of both nebulins N1 and N2 is considerably later than degradation of native titin. A comparison with other studies reveals that this degradation sequence occurs in various species. This includes bovine and ovine muscle, in which nebulin degradation under cooler storage conditions (at 2–4 °C) occurred at 6 and 7 days postmortem, respectively [19, 39]. Only in chicken, a complete degradation of nebulin was shown to occur before 48 hpm [26].
The postmortem degradation of desmin has also been well characterized in a number of different species. Similar to our results, native desmin is present for several days postmortem in muscles of cattle [21, 40], pigs [41], lambs [42], and chickens [26]. Also in accordance with these studies, degradation of desmin involves the regular appearance of several split products between 35 and 50 kDa from 1–2 days postmortem onward. Such products were shown to develop after incubation of muscle with calpain 1 and calpain 2 [43].
To our knowledge, no data are present for the degradation behavior of SERCA1 and cardiac TnT. Note that cTnT is not restricted to the heart muscle but is also present in skeletal muscle [44]. The present analysis demonstrates that degradation of both proteins occurs rather late and involves the appearance of two split products. Thus, the degradation of SERCA1 and cTnT may be of interest particularly for the late postmortem phase.
Calpains, a family of Ca2+-dependent proteolytic enzymes, have been shown to be involved in the degradation of cytoskeletal muscle and intermediate filament proteins, such as titin, nebulin, and desmin [19, 20, 30, 43]. The present data show that calpain 1 and calpain 2 are initially active in a native form but within the first 24 hpm also in an autolyzed form. The native and the autolyzed forms have been shown to be active and bind tightly to intracellular structures such as myofibrils, where they proteolyze titin, nebulin, and desmin [31, 36]. Native calpain 1 activity decreased faster than calpain 2 activity which is in agreement with the results in pig and lamb [32, 45]. This is supported by Western blot analysis in bovine and ovine muscle, which demonstrated that the 80-kDa subunit of calpain 1 almost entirely degraded into a 76-kDa subunit within the first 7 days postmortem, while the 80-kDa subunit of calpain 2 remained undegraded in this period [42, 46]. Within the first 24 hpm, a 76-kDa subunit of calpain 1 has also been shown in pigs [27]. Thus, the activities of these enzymes also characterize specific postmortem phases which makes them promising candidates for PMI delimitation.
In summary, the present study has identified a series of changes in protein presence and enzymatic activities that are each specific for certain points of time after death (Fig. 5). Analysis of the proteins identified allows a so far unknown accuracy in the characterization of PMI in the first 10 days pm in the porcine system. As a similar pattern of protein degradation has been described for several other vertebrate species, it is likely that the degradation pattern in humans will follow the same scheme. This offers a unique chance to establish a new method of long-term PMI determination for use in forensic casework which provides two important advantages: (i) The method is very simple to apply in practice and provides reliable results within 24 h, and (ii) once implemented in analysis of human muscle, the method will steadily upgrade itself by allowing to generate and expand a database compiling information on the times of presence (or absence) of a diverse array of human muscle proteins, and on their 95 % confidence intervals of band change under distinct physical/environmental conditions postmortem. This is presently tested in a follow-up study in our lab and is thought to offer a major improvement in actual forensic case analysis in the future.

Chronological arrangement of 95 % confidence intervals (CI) of band changes. Throughout the investigated 10-day period, changes in protein expression and enzymatic activities are found. These changes are characteristic for certain periods of time after death. Vertical lines and error bars within the gray bars mark the mean and SE. The survival analysis was performed using the Kaplan-Meier estimator
References
Henssge C, Madea B (2007) Estimation of the time since death. Forensic Sci Int 165:182–184
Madea B (1994) Importance of supravitality in forensic medicine. Forensic Sci Int 69:221–241
Henssge C, Althaus L, Bolt J, Freislederer A, Haffner HT, Henssge CA, Hoppe B, Schneider V (2000) Experiences with a compound method for estimating the time since death. I. Rectal temperature nomogram for time since death. Int J Legal Med 113:303–319
Henssge C, Althaus L, Bolt J, Freislederer A, Haffner HT, Henssge CA, Hoppe B, Schneider V (2000) Experiences with a compound method for estimating the time since death. II. Integration of non-temperature-based methods. Int J Legal Med 113:320–331
Muñoz Barús JI, Suárez-Peñaranda J, Otero XL, Rodríguez-Calvo MS, Costas E, Miguéns X, Concheiro L (2002) Improved estimation of postmortem interval based on differential behaviour of vitreous potassium and hypoxantine in death by hanging. Forensic Sci Int 125:67–74
Jackson ARW, Jackson JM (2011) Forensic science.In: Pearson Education Limited, 3rd edn. pp 376–381
Kimura A, Ishida Y, Hayashi T, Nosaka M, Kondo T (2010) Estimating time of death based on the biological clock. Int J Legal Med 125:385–391
Amendt J, Richards CS, Campobasso CP, Zehner R, Hall MJR (2011) Forensic entomology: applications and limitations. Forensic Sci Med Pathol 7:379–392
Metcalf JL, Wegener Parfrey L, Gonzalez A, Lauber CL, Knights D, Ackermann G, Humphrey GC, Gebert MJ, Van Treuren W, Berg-Lyons D, Keepers K, Guo Y, Bullard J, Fierer N, Carter DO, Knight R (2013) A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system. elife 2(e01104):1–19
Young ST, Wells JD, Hobbs GR, Bishop CP et al (2013) Estimating postmortem interval using RNA degradation and morphological changes in tooth pulp. Forensic Sci Int 229:163.e1–163.e6
Itani M, Yamamoto Y, Doi Y, Miyaishi S (2011) Quantitative analysis of DNA degradation in the dead body. Acta Med Okayama 65:299–306
Alibegović A (2014) Cartilage: a new parameter for the determination of the postmortem interval? J Forensic Legal Med 27:39–45
Tomita Y, Nihira M, Ohno Y, Sato S (2004) Ultrastructural changes during in situ early postmortem autolysis in kidney, pancreas, liver, heart and skeletal muscle of rats. Legal Med 6:25–31
Collan Y, Salmenperä M (1976) Electron microscopy of postmortem autolysis of rat muscle tissue. Acta Neuropathol 35:219–233
Tokunaga I, Takeichi S, Yamamoto A, Gotoda M, Maeiwa M (1993) Comparison of postmortem autolysis in cardiac and skeletal muscle. J Forensic Sci 38:1187–1193
Kang S, Kassam N, Gauthier ML, O'Day DH (2003) Post-mortem changes in calmodulin binding proteins in muscle and lung. Forensic Sci Int 131:140–147
Poloz YO, O'Day DH (2009) Determining time of death: temperature-dependent postmortem changes in calcineurin A, MARCKS, CaMKII, and protein phosphatase 2A in mouse. Int J Legal Med 123:305–314
Sanoudou D, Kang PB, Haslett JN, Han M, Kunkel LM, Beggs AH (2004) Transcriptional profile of postmortem skeletal muscle. Physiol Genomics 16:222–228
Taylor RG, Geesink GH, Thompson VF, Koohmaraie M, Goll DE (1995) Is Z-disk degradation responsible for postmortem tenderization? J Anim Sci 73:1351–1367
Huff-Lonergan E, Mitsuhashi T, Beekman DD, Parrish FC Jr, Olson DG, Robson RM (1996) Proteolysis of specific muscle structural proteins by mu-calpain at low pH and temperature is similar to degradation in postmortem bovine muscle. J Anim Sci 74:993–1008
Huff-Lonergan E, Mitsuhashi T, Parrish FC Jr, Robson RM (1996) Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blotting comparisons of purified myofibrils and whole muscle preparations for evaluating titin and nebulin in postmortem bovine muscle. J Anim Sci 74:779–785
Tomaszewska-Gras J, Kijowski J, Schreurs FJ (2002) Quantitative determination of titin and nebulin in poultry meat by SDS-PAGE with an internal standard. Meat Sci 62:61–66
Szalata M, Pospiech E, Greaser ML, Lyczynski A, Grzes B, Mikolajczak B (2005) Titin and troponin T changes in relation to tenderness of meat from pigs of various meatiness. Pol J Food Nutr Sci 14:139–144
Wu G, Clerens S, Farouk MM (2014) LC MS/MS identification of large structural proteins from bull muscle and their degradation products during post mortem storage. Food Chem 150:137–144
Wu G, Farouk MM, Clerens S, Rosenvold K (2014) Effect of beef ultimate pH and large structural protein changes with aging on meat tenderness. Meat Sci 98:637–645
Tomaszewska-Gras J, Schreurs FJ, Kijowski J (2011) Post mortem development of meat quality as related to changes in cytoskeletal proteins of chicken muscles. Br Poult Sci 52:189–201
Melody JL, Lonergan SM, Rowe LJ, Huiatt TW, Mayes MS, Huff-Lonergan E (2004) Early postmortem biochemical factors influence tenderness and water-holding capacity of three porcine muscles. J Anim Sci 82:1195–1205
Matsuura T, Kimura S, Ohtsuka S, Maruyama K (1991) Isolation and characterization of 1,200 kDa peptide of alpha-connectin. J Biochem 110:474–478
Warren CM, Krzesinski PR, Greaser ML (2003) Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis 24:1695–1702
Koohmaraie M, Geesink GH (2006) Contribution of postmortem muscle biochemistry to the delivery of consistent meat quality with particular focus on the calpain system. Meat Sci 74:34–43
Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83:731–801
Pomponio L, Ertbjerg P (2012) The effect of temperature on the activity of μ- and m-calpain and calpastatin during post-mortem storage of porcine longissimus muscle. Meat Sci 91:50–55
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Raser KJ, Posner A, Wang KK (1995) Casein zymography: a method to study mu-calpain, m-calpain, and their inhibitory agents. Arch Biochem Biophys 319:211–216
Meyer LC, Wright NT (2013) Structure of giant muscle proteins. Physiol 4 Article 368:1–12
Huff Lonergan E, Zhang W, Lonergan SM (2010) Biochemistry of postmortem muscle - lessons on mechanisms of meat tenderization. Meat Sci 86:184–195
Ho CY, Stromer MH, Rouse G, Robson RM (1997) Effects of electrical stimulation and postmortem storage on changes in titin, nebulin, desmin, troponin-T, and muscle ultrastructure in Bos indicus crossbred cattle. J Anim Sci 75:366–376
Geesink GH, Bekhit AD, Bickerstaffe R (2000) Rigor temperature and meat quality characteristics of lamb longissimus muscle. J Anim Sci 78:2842–2848
Koohmaraie M, Shackelford SD, Wheeler TL, Lonergan SM, Doumit ME (1995) A muscle hypertrophy condition in lamb (callipyge): characterization of effects on muscle growth and meat quality traits. J Anim Sci 73:3596–3607
Rowe LJ, Maddock KR, Lonergan SM, Huff-Lonergan E (2004) Oxidative environments decrease tenderization of beef steaks through inactivation of μ-calpain. J Anim Sci 82:3254–3266
Zhang WG, Lonergan SM, Gardner MA, Huff-Lonergan E (2006) Contribution of postmortem changes of integrin, desmin and μ-calpain to variation in water holding capacity of pork. Meat Sci 74:578–585
Geesink GH, Koohmaraie M (1999) Postmortem proteolysis and calpain/calpastatin activity in callipyge and normal lamb biceps femoris during extended postmortem storage. J Anim Sci 77:1490–1501
Baron CP, Jacobsen S, Purslow PP (2004) Cleavage of desmin by cysteine proteases: Calpains and cathepsin B. Meat Sci 68:447–456
Bodor GS, Survant L, Voss EM, Smith S, Porterfield D, Apple FS (1997) Cardiac troponin T composition in normal and regenerating human skeletal muscle. Clin Chem 43:476–484
Veiseth E, Shackelford SD, Wheeler TL, Koohmaraie M (2001) Effect of postmortem storage on μ-calpain and m-calpain in ovine skeletal muscle. J Anim Sci 79:1502–1508
Boehm ML, Kendall TL, Thompson VF, Goll DE (1998) Changes in the calpains and calpastatin during postmortem storage of bovine muscle. J Anim Sci 76:2415–2434
Acknowledgments
The authors are grateful to Fiona Bergmann, Magdalena Brandauer, and Christian Platzl for providing excellent technical assistance, as well as Roman Fuchs and Arne Bathke for advices and assistance in statistics. Our gratitude also goes to Elena Esra Foditsch and Edith Tutsch-Bauer for exchange of ideas and discussion throughout the study.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pittner, S., Monticelli, F.C., Pfisterer, A. et al. Postmortem degradation of skeletal muscle proteins: a novel approach to determine the time since death. Int J Legal Med 130, 421–431 (2016). https://doi.org/10.1007/s00414-015-1210-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-015-1210-6