
Abstract
Larnite (β-Ca2SiO4) has previously been reported as an inclusion in sub-lithospheric diamonds and is generally interpreted as a retrograde reaction product of calcium silicate perovskite. In this study, we review the controls on the stability of the Ca2SiO4 polymorphs and show that phosphorus is likely essential for the preservation of β-Ca2SiO4. We also report a detailed study of the solubility of water and its incorporation mechanisms in γ-Ca2SiO4 and phosphorus-doped β-Ca2SiO4 using FTIR spectroscopy on high-pressure experiments quenched from 4–9.5 GPa and 1000–1200 °C combined with ab initio calculations. The experimentally determined water solubilities are in the range of 107–178 ppm. Our FTIR spectra and ab initio calculations indicate that for phosphorus-free γ-Ca2SiO4 the incorporation mechanism involves protonated Si and Ca1 vacancies. For phosphorus-bearing β-Ca2SiO4, our preferred incorporation mechanism involves one Si4+ ion replaced by one P5+ ion with a single protonated Ca2 vacancy. The low water solubility observed here for larnite implies that if primary calcium silicate perovskite inclusions trap high water concentrations during diamond growth from a volatile-rich fluid, measurements of the concentration of water in larnite will not provide a useful record of the initial volatile concentration. Instead, water would be hosted in other retrograde reaction products, possibly including exsolved fluids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Natural occurrences of calcium silicate minerals in the crust are rare and most often represent reactions between CaCO3 and silicates where either might be a melt phase (Tilley 1929; Mason 1957; Sokol et al. 2014). However, in the Earth’s lower mantle, calcium perovskite (CaSiO3) is a major phase, and other calcium silicate minerals could be stabilised at shallower depths in the mantle in Ca-enriched regions, perhaps by reactions between Ca-rich carbonate melts and silicate rocks. Evidence of the role of calcium silicate minerals in the mantle is provided by the presence of various inclusions in diamonds of sub-lithospheric origin (Anzolini et al. 2016, 2018; Brenker et al. 2005, 2007, 2021; Bulanova et al. 2010; Burnham et al. 2016; Daver et al. 2022; Genzel et al. 2023; Joswig et al. 1999; Nasdala et al. 2003; Regier et al. 2023; Smith et al. 2016, 2018; Stachel et al. 2000; Timmerman et al. 2023; Zedgenizov et al. 2016; Zhang et al. 2024). These inclusions include larnite (β-Ca2SiO4), titanite-structured CaSi2O5, breyite (CaSiO3) and wollastonite (CaSiO3). One explanation for the origin of these inclusions is that they represent retrograde transformation products of CaSiO3-perovskite originally entrapped in the lower part of the transition zone or the lower mantle (Ko et al. 2022; Trønnes 2010). This interpretation is supported by experimental phase equilibria, which show that calcium perovskite is the only pure calcium silicate phase stable in peridotitic or eclogitic lithologies. Alternatively, some of these inclusions could have been trapped directly in the upper mantle in anomalously Ca-rich regions even though these phases are not stable in bulk peridotitic or eclogitic compositions. In either case, it is likely that calcium silicate inclusions were formed by processes such as reactions between a slab-derived carbonatite liquid and the peridotitic mantle (Thomson et al. 2014, 2016; Walter et al. 2008) or the reduction of CaCO3-CaSiO3 melts generated from carbonaceous metasediments (Fedoraeva et al. 2019; Woodland et al. 2020; Brenker et al. 2021; Daver et al. 2022).
One motivation for the present study is specific to the model of larnite and other Ca silicate phases in diamond representing retrograde reaction products of deep mantle calcium perovskite and the potential for the use of these phases to reconstruct the original dissolved water concentration of perovskite. This is an important step in determining if CaSiO3-perovskite might contribute significantly to the water storage capacity of the deep mantle, as current solubility estimates are contradictory, ranging from 1 wt.% to near zero (Ross et al. 2003; Chen et al. 2020). Moreover, there is limited water solubility data for the retrograde transformation products of CaSiO3-perovskite. This type of data is essential to determine if the measured water content in naturally occurring larnite (β-Ca2SiO4), titanite-structured CaSi2O5 and breyite (CaSiO3) inclusions in diamonds represent the maximum solubility limit imposed by crystal structure during retrogression or the original water content of the CaSiO3-perovskite. In the case of larnite (β-Ca2SiO4), which is metastable under ambient conditions, this strategy is complicated by the special circumstances required for its recovery in both experiments and as inclusions in diamond. In this paper, we focus on the factors that control this metastability, as well as the solubility and incorporation mechanisms of water in the larnite structure (β-Ca2SiO4) as well as the other phases with Ca2SiO4 stoichiometry.
Review of the literature on Ca2SiO4
Thermodynamic stability of Ca2SiO4 polymorphs
There are five polymorphs of pure dicalcium silicate (Ca2SiO4): γ, β, αʹL, αʹH and α (Bredig 1950; Figs. 1 and 2). The orthorhombic olivine-type γ phase is stable under ambient conditions (Udagawa et al. 1980). The αʹL, αʹH, and α phases become stable sequentially on heating (Remy et al. 1997). At ambient pressure, the monoclinic β phase (also known as larnite in the mineralogical literature or belite in the cement literature) appears only as a metastable phase during cooling but reverts to the γ phase before reaching room temperature. Nevertheless, it is clearly possible to recover the β phase to ambient conditions because all known diamond-hosted inclusions with the Ca2SiO4 stoichiometry have this structure where it has been determined, even when the inclusions are exposed to ambient conditions. Metastability in this system is known to depend on grain size, cooling rate, synthesis conditions, and the presence of impurities (Pritts and Daugherty 1976; Chan et al. 1992; Lai et al. 1992; Remy et al. 1995; Saidani et al. 2020).
The β to γ transition involves a symmetry change from monoclinic to orthorhombic. This transformation leads to a reduction in the coordination number of calcium atoms and rotations of the SiO4 tetrahedra (Smith et al. 1961). Although Si-O bond lengths change only slightly during recovery to ambient conditions, the Ca-O bond lengths change significantly, leading to the collapse of the CaO polyhedra. Neighbouring SiO4 tetrahedra adjust their position in response, reducing the Si-Ca bond distance sufficiently (Jost et al. 1977; Kim et al. 1992) to generate electrostatic repulsive forces strong enough to break the Ca-O bonds and overcome the substantial activation energy associated with this transition. These structural changes result in a 12 % increase in lattice volume, causing the phase to crack and self-pulverise during the transition. An example of this process is shown in the video in the supplementary material 1.
At ambient pressure, pure β-Ca2SiO4 might be synthesised by rapidly quenching from the αʹL, αʹH or α phase fields. However, this method is difficult and often produces a two-phase mixture of β-Ca2SiO4 and γ-Ca2SiO4 depending on the cooling rate, grain size, and synthesis temperature, which in turn controls the degree of lattice strain and charge repulsion in β-Ca2SiO4 (Remy et al. 1997). A grain size below 10 μm prevents transformation to γ-Ca2SiO4 on cooling (Chan et al. 1992) by reducing the strain gradient (Saidani et al. 2018). Rapid cooling from the αʹL, αʹH or α phase fields produces a large spontaneous strain that triggers a ferroelastic transformation to β-Ca2SiO4 at ambient pressure, the structure of which is better suited to accommodate lattice strain than the γ structure (Kim et al. 1992). β-Ca2SiO4 crystals produced in this way are expected to be highly stressed due to strong electrostatic charge repulsion between Si and Ca and as a result often exhibit twinning. Two twinning modes have been observed in β-Ca2SiO4: along both the {100} and {001} planes or along the {100} planes alone, which is energetically favoured and therefore more common. Crystals recovered from high pressure at room temperature display the same twinning modes as those recovered from high temperatures at ambient pressure (Remy et al. 1995); twinning in β-Ca2SiO4 is not characteristic of a particular P-T path or the presence or absence of impurities (Ghose et al. 1983). Kim et al. (1992) report the development of a lattice superstructure in β-Ca2SiO4 when quenched from the αʹ field, the observation of which would suggest a high-temperature origin.
The β to γ transition can be inhibited by trace amounts of stabilising impurities (Saalfeld 1975; Dehghan-Manshadi et al. 2014; Saidani et al. 2020). Stabiliser atoms in β-Ca2SiO4 decrease the free energy of the lattice and increase the energy barrier associated with the transformation (Sakurada et al. 2015) by producing vacancies that accommodate the lattice strain generated by charge repulsion. Many stabilising impurities are known, including P2O5, SrO, K2O, Na2O, MgO, TiO2 and Cr2O3 (Lai et al. 1992; Guo et al. 2016). In contrast, other impurities such as FeO and Al2O3 can counteract this stabilising effect (Zhang et al. 2012; Guo et al. 2016). P2O5 appears to be the most efficient enhancer of β-Ca2SiO4 larnite stability; a concentration of as little as 0.3 wt.% can prevent any β to γ transformation (Zhang et al. 2012). A maximum of 3.3 % of silicon sites can be replaced by phosphorus in β-Ca2SiO4 (Nastac et al. 2007); at higher P concentrations the αʹ-Ca2SiO4 modification is stabilised.
Synthesis and stabilisation of β-Ca2SiO4 in previous experiments at mantle conditions
β-Ca2SiO4 has been recovered with complete or partial success from high-pressure and high-temperature experiments (Kanzaki et al. 1991, 2013; Gasparik et al. 1994; Remy et al. 1995; Reynard et al. 1997; Xiong et al. 2016; Németh et al. 2017), but these studies fail to fully evaluate the factors that control metastability such as impurities or grain size. Xiong et al. (2016) sythesised β-Ca2SiO4 at 6 GPa and 1473 K without any added impurities but then produced γ-Ca2SiO4 in a repeat experiment. Successful synthesis is likely to have been facilitated by variable B2O3 contamination from BN capsules surrounding the sample (Brooker et al. 1998; Matjuschkin et al. 2015) as B2O3 concentrations of < 0.2 wt.% are known to stabilise β-Ca2SiO4 (Remy et al. 1997). Reynard et al. (1997) recovered larnite from one diamond anvil cell (DAC) experiment at 24 GPa and room temperature, but another sample quenched from 2.5 GPa partially reverted to γ-Ca2SiO4. This particular lack of repeatability is probably the result of grain size, which ranged from 5 to 20 μm, straddling the 10 μm threshold for stabilising β-Ca2SiO4 at 1 atm (Chan et al. 1992; Saidani et al. 2018). Remy et al. (1995) also performed DAC experiments that recovered β-Ca2SiO4 but again the grains were in the 10s of micron range allowing stabilisation. Finally, Németh et al. (2017), sythesised β-Ca2SiO4 in a multi-anvil press at 17 GPa and 1773 K with grain sizes exceeding 10 μm. However, microprobe data indicate the presence of up to 0.2 wt.% of other oxides (not detailed by the authors) and have low totals, such that stabilisation by contaminants cannot be ruled out.
Evidence for water dissolution in β-Ca2SiO4
β-Ca2SiO4 has also been recovered from high-pressure high-temperature experiments under hydrous conditions. Németh et al. (2017) reported structural hydroxyl on the basis of attenuated total reflection infrared spectroscopy (ATR-IR) analyses of β-Ca2SiO4 crystals (50–100 μm) recovered from 17 GPa and 1773 K using a hydrous starting material. The OH defect bands at ~ 3500 cm−1 are poorly defined with a broad molecular water component as well as the clear presence of Ca(OH)2. ATR-IR spectra were not quantified, but a water content of 4.06 wt.% was estimated by electron-probe microanalysis (EPMA) of the same crystals. However, ~ 2 wt.% H2O was reported from experiments performed under nominally dry conditions at the same pressure and temperature in the same study. Furthermore, Németh et al. (2017) interpreted superlattice reflections in electron-diffraction patterns as evidence of crystallographic ordering of hydroxyl group defects in hydrous β-Ca2SiO4. However, these experiments were quenched to the β-Ca2SiO4 field from the αʹ field which is known to produce an equivalent superlattice structure (Kim et al. 1992) without the presence of structural water. There is therefore some doubt as to the reliability of the Németh et al. (2017) result. In this paper, we present a comprehensive study of Ca2SiO4 polymorphs using both high-pressure/temperature experiments and ab initio calculations. We determine the solubility and incorporation mechanism of water in β-Ca2SiO4 as well as the likely mechanism of its stabilisation.
Experimental and computational methods
Strategy
Here, we address both the stability of β-Ca2SiO4 under hydrous conditions and the solubility of water in β-Ca2SiO4 at high pressure. Our method involved heating samples of high purity Ca2SiO4 or Ca2SiO4 doped with P2O5 in the stability field of the αʹL phase at high pressure under water-saturated conditions followed by a rapid quenching to room temperature and recovery at ambient pressure. The conditions were chosen to simulate a normal mantle adiabat along which sub-lithospheric diamonds are likely re-equilibrated during exhumation from depth (Fig. 2). Recovered samples were analysed using Raman and Fourier transform infrared (FTIR) spectroscopy.
To complement our experimental results, ab initio calculations were performed to provide information on the thermodynamic stability of P and OH bearing defects in β-Ca2SiO4 as well as the vibrational spectrocopic properties of selected OH defect models in β-Ca2SiO4 for comparison with our analysis of the FTIR spectra.
Synthesis experiments
Two starting materials were used: ND1, a mixture of 80 wt.% γ-Ca2SiO4 and 20 wt.% Ca(OH)2 and ND2, a mixture of 80 wt.% P-doped β-Ca2SiO4 and 20 wt.% Ca(OH)2. The γ-Ca2SiO4 was prepared by grinding SiO2 and CaCO3 in a molar ratio of 1:2 under ethanol, pressing this mixture into a pellet and decarbonating at 1 atm and 1273 K for 5 days in a Pt crucible. The γ-Ca2SiO4 structure was confirmed by Raman spectroscopy and the pellet was then crushed and stored at 393 K in a vacuum oven. P-doped β-Ca2SiO4 was prepared by mixing 99.5 wt.% γ-Ca2SiO4 and 0.5 wt.% P2O5 and heating at 1473 K and 1 bar for 24 h in a Pt crucible. For these samples, the β-Ca2SiO4 structure was also confirmed by Raman spectroscopy. Only 0.16 wt.% P2O5 was measured in the final high-pressure products, so some of the P2O5 may have been lost via devolatilization during this heating stage. For water saturated conditions to be present during the high-pressure experiments, the Ca(OH)2 is required to break down to CaO and H2O which is not a clearly defined reaction at the experimental conditions used (see results).
Starting materials were sealed into 3 mm diameter capsules made of either Au or Pt. High pressure synthesis experiments were performed at the University of Bristol at 4–8 GPa and 1273–1473 K for 24 h using a Walker-style 6–8 multianvil press (Walker et al. 1990), equipped with WC cubes with corners truncated at 11 mm and a Cr-doped MgO octahedral cell assembly with 18 mm edge length. The target pressure was approached at ≤ 50 tonnes/hour. Experiments were heated using stepped graphite furnaces and temperature monitored to a precision of ± 1 K using 0.12 mm W5Re95–W26Re74 (type C) thermocouple wires. A summary of run conditions is given in Table 1. All experiments were performed in the αʹL stability field and then rapidly quenched in temperature by turning off the power to the heater, and recovered to ambient pressure, in an attempt to preserve the β-Ca2SiO4 structure.
Analytical methods
Run products were examined using a confocal Thermo Scientific DXRxi Raman microscope with a 532 nm laser source for phase identification. Major element analyses were obtained by electron probe microanalysis (EPMA) using a Cameca SX100 microprobe instrument equipped with a field emission gun (FEG). Measurements were made at 20 kV using a beam current of 5 nA and a beam size of 5 μm. Fourier-transform infrared (FTIR) spectroscopy measurements were made using a Thermo-Nicolet iN10MX IR microscope at room temperature. Unoriented single crystals of at least 100–200 μm dimensions were recovered from the capsule, cleaned, and placed on BaF2 plates. Unpolarised absorbance spectra were produced by combining up to 2048 scans in the wavenumber range of 450–4000 cm−1 with a spectral resolution of 4 cm−1 using a 200 × 100 μm aperture. Care was taken to target areas with limited fractures and no visible inclusions. Background spectra were collected for adjacent areas of the BaF2 plate prior to each analysis. To improve the spectra for β-Ca2SiO4, selected crystals were mounted on glass slides with Crystalbond™, double polished, and then removed by soaking in acetone in an ultrasonic bath for 2 h. Unpolarised and polarised spectra were then collected from Individual double polished crystals. The γ-Ca2SiO4 polycrystals are brittle and therefore not suitable for double polishing. Each spectrum for both β-Ca2SiO4 and γ-Ca2SiO4 requires a complex baseline correction involving the subtraction from our raw spectra of four separate components that define a broad non-linear feature and absorption peaks that do not appear to be related to the presence of structural OH in the β-Ca2SiO4 or γ-Ca2SiO4 lattice. These absorption components arise from molecular water, organic contaminants, portlandite, and an unknown component (Figs. 3 and 4). After each component was individually subtracted, we were able to isolate the OH defect bands in each spectrum with confidence and then characterise each of the overlapping peaks by deconvolution using Lorentzian or Gaussian lineshapes. Using the corrected spectra, the concentration of OH groups in the Ca2SiO4 phases studied were determined by integrating the IR spectrum from 3300 to 3700 cm−1 where absorption by OH bonds is observed. Quantification is difficult because there are currently no calibrated mineral-specific absorption coefficients (Ɛ ) for OH peaks in Ca2SiO4. This is required for quantification using the Beer-Lambert law,
where 18.02 is the relative formula mass of H2O, A is the integrated peak area of the OH absorption band, L is the sample thickness (cm) (equivalent to the path length of light through the sample), ρ is the density of the mineral (synthetic γ-Ca2SiO4 = 2940 g L−1; natural β-Ca2SiO4 = 3320 g L−1) and Ɛ is the molar absorption coefficient (L mol− 1 cm−2). To overcome this lack of a mineral-specific absorption coefficient, two methods were used to calculate water contents. First, we used the Withers et al. (2012) calibration for olivine with an absorption coefficient of 45,200 L mol−1 cm−2 and summed integrated absorption, with the rationale being that both polymorphs of Ca2SiO4 are orthosilicates like olivine. The second method is a non-mineral specific IR calibration (2), where the integrated molar absorption coefficient is calculated assuming it is frequency dependent using the expression
where ν is the mean wavenumber (cm−1) of the OH stretching band (Libowitzky and Rossman 1997). Crystal thicknesses were measured using a confocal microscope with a computer-controlled calibrated stage by determining the difference in stage height between the top and bottom of the crystal.
Computational methods
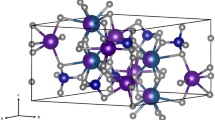
Ab initio calculations were performed for defects in β-Ca2SiO4 and γ-Ca2SiO4 using density functional theory (Kohn and Sham 1965) as implemented in version 6.3 of the Quantum-ESPRESSO (QE) package (Giannozzi et al. 2009). Calculations were performed using the Perdew-Burke-Ernzerhof (PBE) functional within the Generalised Gradient Approximation (GGA) using the exchange-correlation functional (Perdew et al. 1996). A convergence criterion of 10−12 Ry on the total energy in the self-consistent field calculations was used while the ionic positions were relaxed until the residual forces on the ions were less than 10−5 Ry/a.u. A planewave basis set was employed with a cutoff of 520 Ry for primitive cells and 120 Ry for supercells. Optimised norm-conserving Vanderbilt (ONVC) pseudopotentials (Ca_ONCV_PBE-1.2.upf, Si_ONCV_PBE-1.2.upf, O_ONCV_PBE-1.2.upf, H_ONCV_PBE-1.2.upf, P_ONCV_PBE-1.2.upf) were used (Hamann 2013; Schlipf and Gygi 2015). Simulations were performed on γ-Ca2SiO4 using a primitive cell (28 atoms) and a 2 × 2 × 2 supercell (227 atoms) containing up to four additional hydrogen atoms to model OH defects using the PWscf code with an initial structural model from Gobechiya et al. (2008). Simulations were performed on β-Ca2SiO4 using a primitive cell (30 atoms) and a 2 × 2 × 2 supercell (227 atoms), again using the PWscf code, this time with an initial structural model from Yamnova et al. (2011), with the supercell containing up to four hydrogens. To model combined OH and P defects in β-Ca2SiO4, a larger 3 × 2 × 2 supercell was employed containing up to three hydrogen and two phosphorus atoms (up to 336 atoms). For primitive cells, the Brillouin zone was sampled using a 3 × 3 × 3 or 4 × 4 × 4 Monkhorst-Pack grid for β-Ca2SiO4 and γ-Ca2SiO4 respectively, whereas supercells were only sampled at the Γ point (Monkhorst and Pack 1976). The size of the supercells provides sufficient separation between the defects to minimise any artificial interactions between them. The relaxed cell parameters of anhydrous β-Ca2SiO4 are a = 5.62, (experimental: 5.51 Å, theoretical: 5.57 Å ), b = 6.84 Å (6.76 Å, 6.81 Å), and c = 9.40 Å (9.31 Å, 9.39 Å) (P21/n space group) and agree well with experimental (Yamnova et al. 2011) and theoretical values (Xiong et al. 2016) with our calculated volumes being 4.50 % and 1.65 % larger, respectively. The relaxed cell parameters of anhydrous γ-Ca2SiO4 are a = 5.15 Å (5.08 Å, 5.12 Å), b = 11.42 Å (11.34 Å, 11.22 Å) and c = 6.83 Å (6.78 Å, 6.77 Å) (Pbnm space group) and are also in good agreement with experimental (Udagawa et al. 1980) and theoretical values (Rejmak et al. 2019) with our calculated volumes being larger by 3.87 % and 2.06 %, respectively. The GGA approximation commonly produces significant distortions in the relaxed structure due to the systematic overestimation of Si-O bond lengths, which leads to incorrect bond angles and a larger unit cell volume.
To model hydrogen incorporation in both phases, a single Ca2+ or Si4+ ion was removed from the relaxed supercell and the vacancy decorated with two or four hydrogen ions. For the β-Ca2SiO4 phase, additional defect models were tested in which one or more Si4+ ions were replaced with P5+ ions and charge balanced by hydrogen atoms located within Ca or Si vacancies. Supercell models have a maximum concentration of 0.66 wt.% H2O or 1.49 wt.% P2O5. All atoms in the supercell were allowed to fully relax including cell parameters and were checked to ensure the crystal symmetry had been preserved. This method has been previously applied to forsterite, enstatite, and wadsleyite (Balan et al. 2011, 2013; Blanchard et al. 2013). The effect of P defects on unit cell volume is negligible, with a unit cell difference of 0.009 % between the P-doped and pure system (Lai et al. 1992). Additional calculations were performed on CaSiO3 (breyite) using a 4 × 3 × 2 k-point grid, monoclinic titanite-structured CaSi2O5 using a 3 × 2 × 3 k-point grid, Ca(OH)2 (portlandite) using a 6 × 6 × 4 k-point grid, CaO using a 2 × 4 × 2 k-point grid and P2O5 using a 3 × 2 × 1 k-point grid. The simulated cell energies are reported in Table S2 of the supplementary material.
Vibrational frequencies were calculated from the centre of the Brillouin zone (Γ point) using linear response theory (Baroni et al. 2001) as implemented in the PHonon package (Giannozzi et al. 2009). High-frequency OH stretching modes can be accurately calculated by considering only the displacement of the oxygen and hydrogen atoms involved in the H-bearing defect as they are decoupled from the other vibrational modes occurring at lower frequencies. A Lorentzian damping parameter of 4 cm−1 was added to the expression for the low-frequency dielectric tensor of the system to account for the width of the absorption bands for calculated OH frequencies (Balan et al. 2001, 2008; Balan and Kloprogge 2017). Total molar absorption coefficients were also calculated for OH defect models using PDielec (Kendrick and Burnett 2016). Unfortunately, using theoretical absorption coefficients to estimate water contents from experimental FTIR spectra is not recommended because of systematic approximations in the calculations; nevertheless, the theoretical values can be used as a basis for comparison with experimental data.
Results
Experimental and analytical results
The phases obtained in our experiments are reported in Table 1. All recovered samples synthesised using the starting material ND1 have the γ-Ca2SiO4 structure, as indicated by strong Raman bands at 815, 840, 887 and 925 cm−1 (Remy et al. 1997). The samples consisted of a fine-grained powder with a few larger, polycrystalline but brittle aggregates around ~ 150 μm across with a similar morphology to that found by Udagawa et al. (1980). The samples recovered from the experiments using the ND2 starting material consisted of euhedral single crystals with common {100}-type twinning. They have the β-Ca2SiO4 (larnite) structure, as indicated by Raman bands at 842, 855 and 975 cm−1 (Remy et al. 1997; Brenker et al. 2005) and have an average composition of 33.79(16) wt.% SiO2, 64.39(22) wt.% CaO and 0.157(5) wt.% P2O5 as determined by EPMA. Typical Raman spectra are shown in Fig. 5 from crystals taken from experiments LA5 and LA16. Micrographs of these crystals are shown in Fig. 6. It was not possible to observe any excess water escaping during the opening of recovered capsules, which would have confirmed the breakdown of Ca(OH)2 into CaO and H2O. However, in any runs where the capsule was clearly breached, no large crystals had grown and the sample remained as a powdered aggregate. In addition, the IR spectra of single crystals show evidence of molecular water that is not in the crystal structure, but presumably was trapped as inclusions during the experiment. Thus, there is good evidence that excess water was present during all runs that did produce crystals large enough for FTIR measurements. It is possible that excess water would have recombined with CaO during the quench to recreate portlandite rather than remaining as a fluid phase.
The use of FTIR to constrain dissolved water concentrations in nominally anhydrous minerals ideally requires single crystals with dimensions of around 100–150 μm (depending on the water concentration). In the case of γ-Ca2SiO4, the only sample to yield suitable polycrystalline grains was LA5. FTIR measurements performed on a 150 μm polycrystalline aggregate from this sample (Fig. 7) show an extensive set of absorption peaks at 3374, 3523, 3532, 3557, 3576, 3599, 3614 and 3641 cm−1. The peak at 3641 cm−1 is associated with Ca(OH)2 (portlandite) (Chukanov 2014), while the absorption peaks from 3374 to 3614 cm−1 are attributed to structural OH groups. None of the absorption peaks can be attributed to calcium silicate hydrides (C-S-H) on the crystal surface such as afwillite (Ca3(HSiO4)2.2H2O), jennite (Ca9Si6O18(OH)6.8H2O), tobermorite (Ca5Si6O16(OH)2·4H2O), hillebrandite (Ca2SiO3(OH)2), chemgemite (Ca5(SiO4)2(OH)2) (Speakman et al. 1967; Galuskin et al. 2009), gyrolite (Ca8Si12O30(OH)4.7H2O) (Baltakys et al. 2008) or C-S-H gels (Yu et al. 1999; Chukanov 2014). The peak positions are significant as they are very close to the positions of the four strongest peaks for protonated Si vacancies in forsterite (3613, 3580, 3566 and 3555 cm−1) (Lemaire et al. 2004). Therefore, it is likely that most of the peaks above 3500 cm−1 in γ-Ca2SiO4 are also related to protonated Si vacancies. The large peak at 3374 cm−1 does not have an analogue in pure forsterite but is close to the region for protonated 3+ cations incorporated in forsterite.
All experiments using the starting material ND2 produced single transparent crystals of β-Ca2SiO4, and suitable crystals were recovered from all experiments for FTIR analysis. Initially, crystals were picked and analysed without polishing, but these spectra contained a large component related to portlandite (Ca(OH)2) and a broad component that we attribute to molecular water in fluid inclusions, grain boundaries or other extended defects. Therefore, the data presented here are from selected crystals that were doubly polished and show several absorption peaks at 3472, 3514, 3643 and 3691 cm−1 (Figs. 4 and 8). The IR absorption peak at 3643 cm−1 is again associated with Ca(OH)2 (portlandite). The peaks at 3514 cm−1 and 3691 cm−1 are likely associated with an unknown contaminant as the peaks are not consistently observed in our study. There are no peaks associated with calcium silicate hydrides, hydrated phosphates, or hydroxylapatite (Ca5(PO4)3(OH) (Tortet et al. 1997; Ishikawa et al. 2000; Koutsopoulos 2002). The broad absorption peak at 3476 cm−1 is attributed to structural OH groups in β-Ca2SiO4. Polarised FTIR measurements show that the main OH absorption peak at 3476 cm−1 has a strong vibration parallel to the crystallographic y-axis ({001}) (Fig. 9).
Quantifying water content
Dissolved water contents were determined for one phosphorus free γ-Ca2SiO4 sample, and three samples of phosphorus doped β-Ca2SiO4 each synthesised at different P-T conditions. Each calculated water content was determined by integrating the FTIR spectrum of each fitted Lorentzian or Gaussian lineshape from the main OH stretching region as shown in Figs. 7 and 8 using both the Withers et al. (2012) and the Libowitzky and Rossman (1997) calibrations. Gaussian lineshapes were preferred for β-Ca2SiO4 due to internal crystal strain indicated by twinning. Reported water contents are an average of measurements from multiple crystals from each experiment and are reported in Table 1. There is no clear trend in the dissolved water concentration as a function of pressure in β-Ca2SiO4 but the three samples were not all synthesised at the same temperature. The water concentration in the single sample of γ-Ca2SiO4 is not significantly different from those in the β-Ca2SiO4 samples.
Hydrogen incorporation mechanisms in γ-Ca2SiO4
The orthorhombic olivine-like γ-Ca2SiO4 unit cell contains two non-equivalent Ca atoms (Ca1 and Ca2), one Si atom, and four O atoms, two of which are equivalent (O1, O2, O3). Ca1 and Ca2 are both 6-fold coordinated, and the Si atom is 4-fold coordinated. Calculated relative energies, OH bond lengths and corresponding theoretical IR wavenumbers of the lowest energy geometry of both (2H)Ca and (4H)Si defects in γ-Ca2SiO4 at 0 GPa are presented in Table 2 and Fig. 10. Energetics of all 15 possible geometries of OH groups in both 6-fold coordinated, doubly protonated Ca1 and Ca2 vacancies were calculated. The lowest energy geometry involves the decoration of the (2H)Ca1 defect and is favoured energetically over the lowest energy geometry for the (2H)Ca2 defect by 45 kJ mol−1. The lowest-energy geometry for the (2H)Ca1 defect has two hydrogen atoms each bonded to oxygen atoms O3 and O1 with no apparent hydrogen bonds. These two bonds have comparable bond lengths and produce similar OH-stretching modes at 3360 and 3363 cm−1, while differing OH bond lengths for the (2H)Ca2 defect produce stretching modes at 3348 and 3598 cm−1. The (4H)Si defect has four OH bonds and produces OH stretching modes at 3493, 3501, 3567, and 3624 cm−1 with four apparent hydrogen bonds at O1-H∙∙∙O3 (2.41 Å), O2-H∙∙∙O1 (2.17 Å), O3-H∙∙∙O1 (2.20 Å) and O3-H∙∙∙O1 (2.34 Å).
Hydrogen incorporation mechanisms in β-Ca2SiO4
The β-Ca2SiO4 unit cell is also composed of two non-equivalent Ca atoms (Ca1 and Ca2), one Si atom, and four non-equivalent O atoms (O1, O2, O3 and O4). The Ca1 and Ca2 atoms are 8-fold and 7-fold coordinated, respectively, and the Si atom is 4-fold coordinated. The SiO4 tetrahedra are separated from each other but share corners or edges with neighbouring Ca-O7/8 polyhedra. The Ca-O polyhedra connect with each other by sharing faces or edges.
Calculated relative energies, OH bond lengths and corresponding theoretical IR wavenumbers of the lowest energy geometry of both (2H)Ca and (4H)Si defects in β-Ca2SiO4 at 0 GPa are presented in Table 3 and Fig. 11. The formation enthalpy of the lowest energy geometry of the (2H)Ca2 defect is 50 kJ mol−1 lower than that of the lowest energy geometry of the (2H)Ca1 defect and therefore only (2H)Ca2 will be considered further. The lowest-energy geometry for the (2H)Ca2 defect has two hydrogen atoms each bonded to O4 and O1. The two OH bonds (O4-H and O1-H) point towards the centre of the vacancy with dissimilar bond lengths (Table 3), creating two OH-stretching modes at 2966 cm−1 and 3560 cm−1.
All silicon sites in β-Ca2SiO4 are equivalent. The (4H)Si defect has four hydrogen atoms bonded to O1, O2, O3, and O4 in a silicon site vacancy (Fig. 9). Though there is only one permutation for four hydrogens within a 4-fold coordinated Si vacancy, we have assessed four initial geometries to test the sensitivity of the ionic relaxation to initial conditions. Each calculated geometry varied in energy by no more than 5 kJ mol−1. The minimum energy geometry has H atoms bonded to O1, O2, O3 and O4, pointing away from the centre of the vacancy and is the only one considered further. The OH bonds produce four stretching modes at 3242, 3533, 3584 and 3625 cm−1 with the lowest wavenumber corresponding to the longest bond, O2-H. The three highest frequencies are related to the shorter OH bonds. All OH groups display hydrogen bonding in the (4H)Si defect, with O4-H∙∙∙O1 (2.0 Å), O1-H∙∙∙O2 (2.1 Å) and O2-H∙∙∙O3 (2.2 Å) having similar lengths, while O3-H∙∙∙O1 (1.8 Å) is relatively shorter.
Following a similar methodology to Walker et al. (2007), hydrogen incorporation in β-Ca2SiO4 may be represented by reactions (3) and (4) using Ca(OH)2 as the hydrogen source.
The energies of reaction (3) and (4) are calculated using the total energy of the supercell with either a (2H)Ca2 or (4H)Si defect respectively at 0 GPa and thus provide a way to determine the formation energy of an isolated protonated defect. This yields a formation energy of 101.5 kJ mol−1 per (2H)Ca2 defect (reaction 3) and 61.0 kJ mol−1 per (4H)Si defect (reaction 4).
Incorporation of phosphorus in β-Ca2SiO4
The charge-balanced incorporation of phosphorus in the anhydrous β-Ca2SiO4 structure may be achieved by the creation of one Ca vacancy and the substitution of two Si atoms for two P atoms (Diouri et al. 1997; Fukuda and Taguchi 1999; Benarchid et al. 2004).
where open square brackets [ ] indicate a calcium vacancy. Energies were calculated at 0 GPa for the replacement of two Si atoms with two P atoms coupled with the creation of either a Ca1 or Ca2 vacancy. From reaction (5), the formation energy of a Ca2 vacancy (−335.7 kJ mol−1) is significantly lower than that of a Ca1 vacancy (−277.6 kJ mol−1). Both formation energies are negative. This indicates that the formation of P doped β-Ca2SiO4 is energetically favoured relative to β-Ca2SiO4 + P2O5 and that the formation of a Ca2 vacancy with the substitution of two Si atoms for two P atoms is the preferred incorporation mechanism.
Incorporation of phosphorus coupled with hydrogen in β-Ca2SiO4
Four incorporation mechanisms involving both P and H are considered in this study: (i) replacement of one Si4+ ion by one P5+ ion with a single H+ ion occupying a Ca2+ vacancy (reaction 6); (ii) replacement of two Si4+ ions by two P5+ ions with two H+ ions decorating a Ca2+ vacancy with an additional undecorated Ca2+ vacancy (reaction 7); (iii) replacement of two Si4+ ions by two P5+ ions and two singly protonated Ca2+ vacancies (reaction 8); or (iv) the replacement of a Si4+ ion by a P5+ ion with three H+ ions occupying an additional Si4+ vacancy (reaction 9). The energetics of these mechanisms, as well as their resulting IR frequencies were evaluated using ab initio calculations.
Mechanism i (reaction 6)
For mechanism (i), we considered either a Ca1 or a Ca2 ion being replaced by a single H+ with an adjacent Si4+ replaced by a P5+ ion. We considered each of the seven possible geometries for the H+ ion occupying a Ca1 or a Ca2 vacancy site. The lowest energy H+ geometry in the Ca2 site (denoted (H)Ca2(P)Si) is 127 kJ mol−1 lower than the lowest energy geometry in the Ca1 site. This lowest energy geometry has the hydrogen bonded to an O1 atom pointing into the centre of the Ca2 vacancy (Fig. 12). A single OH-stretching IR mode is expected for a (H)Ca2(P)Si defect in β-Ca2SiO4 at 3478 cm−1. Reaction 6 yields a formation energy for the (H)Ca2(P)Si defect of −495.3 kJ mol−1, indicating that the reaction is preferred relative to the defect-free structure.
Mechanism ii (reaction 7)
Two separate configurations were considered for mechanism (ii) whereby either a Ca1 or Ca2 site is doubly protonated while in both configurations a Ca2 site remains vacant (denoted by [ ] in reaction 7). The configuration with the doubly protonated Ca2 vacancy, denoted (2H)Ca22(P)Si, is energetically favoured by 146 kJ mol−1 compared to a doubly protonated Ca1 vacancy. Of the 21 geometries for a doubly protonated Ca2 vacancy, the lowest-energy geometry has two hydrogen atoms each bonded to O1 and O4. The two OH bonds (O1-H and O4-H) point towards the centre of the vacancy with similar bond lengths and a hydrogen bond at O1-H∙∙∙O2 (2.04 Å), leading to two OH-stretching modes at 3533 cm−1 and 3614 cm−1 (Fig. 12). Reaction 7 yields a formation energy for the (2H)Ca22(P)Si defect of −438.9 kJ mol−1 indicating the reaction is favoured relative to the defect-free structure.
Mechanism iii (reaction 8)
Mechanism (iii) has the same stoichiometry as mechanism (ii) but differs in defect configuration in that two Ca2 vacancies are singly protonated, denoted as (H)Ca2(H)Ca22(P)Si (Table 3). Only 13 geometries were explored to limit the number of computations. This was done iteratively by first fixing a hydrogen atom to a randomly chosen O site (O2) within a Ca2 vacancy and simulating the seven hydrogen geometries in the adjacent Ca2 vacancy. The adjacent vacancy was then fixed to the geometry with the lowest energy, and finally, the hydrogen in the first vacancy was moved between the available oxygen sites until the lowest energy geometry was found. Using this method, we found that the lowest energy geometry for the (H)Ca2(H)Ca22(P)Si defect is where two hydrogen atoms are bonded to O1 and O4 sites in adjacent Ca2 vacancies (Fig. 12) each pointing towards the centre of the vacancy. The lowest energy geometry has two OH-stretching modes at 3172 cm−1 and 3530 cm−1 (Fig. 12). Relative to mechanism (ii) with the same stoichiometry, the (H)Ca2(H)Ca22(P)Si defect is preferred by 28.9 kJ mol−1. Reaction (8) yields an enthalpy of −467.9 kJ mol−1, indicating that the (H)Ca2(H)Ca22(P)Si defect is preferred relative to the defect-free structure.
Mechanism iv (reaction 9)
For this mechanism, denoted (3H)Si(P)Si, there are four unique geometries for three hydrogen atoms partially occupying the Si4+ vacancy. Of these, the lowest energy geometry has hydrogen atoms bonded with O1, O2 and O3. A hydrogen bond, O2-H∙∙∙O4 (2.1 Å), points toward the unbonded O4 vertex and is near parallel to the edges of the vacant tetrahedron (Fig. 12). The hydrogen bonds for O1-H∙∙∙O2 (2.1 Å) and O2-H∙∙∙O4 (2.1 Å) have similar lengths. The defect geometry leads to calculated OH-stretching modes at 3329 cm−1, 3470 cm−1 and 3568 cm−1 (Fig. 12). The short O2-H bond corresponds to the higher wavenumber at 3568 cm−1. The lower wavenumbers at 3329 cm−1 and 3470 cm−1 originate from the relatively longer O3-H and O1-H bonds (Table 3). Reaction (9) yields an enthalpy of −222.6 kJ mol−1, indicating that the reaction is preferred relative to the defect-free structure.
Discussion
Ca2SiO4 inclusions in diamond
Ca2SiO4 inclusions in diamond appear to exist exclusively as monoclinic β-Ca2SiO4, which is surprising as β-Ca2SiO4 is a highly unstable phase at ambient conditions and will readily revert to the γ-Ca2SiO4 structure (Anzolini et al. 2016; Brenker et al. 2005, 2007, 2008; Daver et al. 2022; Joswig et al. 1999; Nasdala et al. 2003; Regier et al. 2023; Smith et al. 2016, 2018; Stachel et al. 2000; Zedgenizov et al. 2016). Most inclusions are studied in-situ using Raman spectroscopy or X-ray diffraction under residual pressure that may stabilise the β-Ca2SiO4 structure. However, inclusions studied by Joswig et al. (1999), Smith et al. (2016) and Zedgenizov et al. (2016) confirm that β-Ca2SiO4 is metastable even on exposure at ambient pressure. The presence of β-Ca2SiO4 inclusions in diamond and the absence of the γ, αʹL, αʹH, and α phases is surprising given the well-established materials science literature on β-Ca2SiO4 stability. Metastability in this system is known to depend on grain size, cooling rate, synthesis conditions and the presence of impurities. Here, we discuss the evidence for each mechanism.
Grain size
Reducing the grain size below 10 μm prevents transformation to γ-Ca2SiO4 from β-Ca2SiO4 on cooling as strain gradients decrease (Chan et al. 1992; Saidani et al. 2018). This mechanism can be excluded for reported β-Ca2SiO4 inclusions in diamonds because they all exceed 10 μm. Smaller grains for which this mechanism is important may exist in nature but have not yet been reported.
Cooling path
β-Ca2SiO4 can be stabilised by rapid quenching from the stability field of the αʹL, αʹH, or α phases. For example, larnite inclusions in diamonds from the Kankan district of Guinea were trapped at P-T conditions (Fig. 2) at which the αʹ phase would have been stable (Joswig et al. 1999; Xiong et al. 2016), suggesting a cooling path mechanism for stabilising β-Ca2SiO4 in diamond. However, a normal mantle geotherm exceeds the maximum temperature of the αʹ stability field (Fig. 2). Kim et al. (1992) observed a superlattice structure in β-Ca2SiO4 on quench from the αʹ field with or without the presence of impurities. Observations of such a superlattice in natural β-Ca2SiO4 inclusions in diamonds would support entrapment at high temperature but has not yet been observed.
Residual pressure
Room temperature high-pressure experiments show that the phase transformation from γ to β occurs spontaneously at ~ 2 GPa (Remy et al. 1995). In-situ measurements on diamond inclusions commonly indicate a residual pressure over 2 GPa (Nasdala et al. 2003) which could explain the observation of the β-Ca2SiO4 structure in unexposed inclusions but does not explain its persistence upon exposure (Joswig et al. 1999; Zedgenizov et al. 2016).
Stabilising impurities
The β to γ transition can be prevented by trace amounts of stabilising impurities (Saalfeld 1975; Dehghan-Manshadi et al. 2014; Saidani et al. 2020). The only β-Ca2SiO4 inclusion to be analysed for its minor and trace components (Joswig et al. 1999) contains 0.54 wt.% P2O5, 0.32 wt.% SrO, 0.28 wt.% K2O, 0.06 wt.% Na2O, 0.02 wt.% MgO and ≤ 0.01 wt.% TiO2 and Cr2O3. P2O5 has the greatest stabilising effect relative to the other oxide components and exceeds the concentration of 0.3 wt.% required to prevent any β to γ transformation (Zhang et al. 2012). The P2O5 concentration is lower than the 4 wt.% that would prevent the αʹ to β transformation and explains why αʹ-Ca2SiO4 is not found as inclusions in diamonds.
CaSiO3-perovskite has a maximum P2O5 solubility of 0.03 wt.% (equivalent to 131 ppm P) (Steinberg 2005). The P content of calcium silicate inclusions interpreted to be former CaSiO3-perovskite ranges from 0.2 to 1 wt.% (Thomson et al. 2016). A low P2O5 solubility will be insufficient to stabilize β-Ca2SiO4 inverted from CaSiO3-perovskite. The source of P2O5 could be P-bearing fluids surrounding the inclusion. This is supported by the observation of the phosphorus bearing mineral florencite adjacent to a β-Ca2SiO4 inclusion (Brenker et al. 2008). Florencite is not stable in diamond forming regions of the mantle and therefore likely precipitated from a P-bearing fluid. The discovery of merrillite (Ca3(PO4)2) inclusions found in a sub-lithospheric diamond containing calcium silicate inclusions further indicates phosphorus is present in diamond forming regions in the mantle (Kaminsky and Zedgenizov 2022). In future all new examples of Ca2SiO4 inclusions found in diamonds should be analysed for P2O5 and the structural polymorph determined.
Given the evidence presented in this study and the existing literature, we propose that the presence of stabilising impurities in β-Ca2SiO4, specifically P2O5, is the most likely explanation for the occurrence of this polymorph in exposed diamond inclusions.
Observations on Ca2SiO4 polymorphs recovered from experiments and calculations
In all experiments with P2O5-free starting composition ND1 a fine powder of γ-Ca2SiO4 is observed that consists of white laths that likely represent former twins of β-Ca2SiO4. The fine powder is a result of the large volume increase across the β to γ transition that causes self-pulverisation during decompression to ambient conditions. The failure to recover metastable β-Ca2SiO4 in these experiments indicates that the grain sizes prior to decompression were large enough to generate strain gradients that exceeded the strength of the β-Ca2SiO4 crystals. We conclude that it is not possible to recover metastable β-Ca2SiO4 under hydrous conditions due to grain growth aided by hydrous fluid at run conditions.
In contrast, in all experiments using the P2O5-bearing starting composition ND2, stabilisation of β-Ca2SiO4 under hydrous conditions was achieved. The P2O5 concentration of 0.16 wt.% found in our P2O5-doped β-Ca2SiO4 crystals is lower than the concentration proposed by Zhang et al. (2012) and insufficient to stabilise the αʹ phase that is the stable polymorph at run conditions.
Our calculations indicate that P dissolution into β-Ca2SiO4 is energetically favoured, as reflected by the high solubility in synthetic samples produced at ambient pressure (up to 5 wt.% P2O5; Fix et al., 1969) and high-pressure (up to 4 wt.%; Steinberg 2005) and natural larnite samples (up to 1.92 wt.% P2O5; Midgley and Bennett 1971; Sokol et al. 2014, 2015). It is possible that the addition of P to β-Ca2SiO4 changes the phase diagram sufficiently to make β-Ca2SiO4 the thermodynamically stable phase at ambient conditions. Alternatively, P-doped β-Ca2SiO4 could remain thermodynamically unstable at ambient conditions but be preserved on quench from the higher temperature polymorph because of an increase in the energy barrier of the β to γ transition due to the reduction of lattice strain by the creation of Ca2 vacancies.
Comparison of spectral features between ab initio simulations and FTIR spectroscopy
Our ab initio calculations and FTIR spectroscopic measurements reveal several OH defect incorporation mechanisms are possible in γ-Ca2SiO4, β-Ca2SiO4, and P doped β-Ca2SiO4.
We established three different hydrogen incoporation mechanisms in γ-Ca2SiO4: (2H)Ca1, (2H)Ca2, and (4H)Si. Our ab initio calculations indicate a strong preference for two hydrogens that occupy the Ca1 vacancy compared to the Ca2 vacancy. Our calculated IR OH bond wavenumbers for the (2H)Ca1 defect (3360, 3363 cm−1) correlate well with the IR band at 3374 cm−1 determined by FTIR (Fig. 7) while the (2H)Ca2 OH bond wavenumbers do not correlate with any observed IR bands. The calculated IR OH stretching modes for the (4H)Si defect (3493, 3501, 3568 and 3624 cm−1) are not in agreement with the IR band at 3374 cm−1 measured by FTIR. These calculated OH frequencies are in agreement and are probably associated with four of the six observed peaks from 3500 to 3620 cm−1 in our γ-Ca2SiO4 FTIR spectrum. Only four peaks are calculated for the (4H)Si defect and could be assigned to the observed IR peaks at 3523, 3532, 3557 and 3614 cm−1. However, the two remaining, minor, IR bands at 3599 and 3615 cm−1 cannot be assigned by theoretical OH-defect modelling in this study. Overall, there is a strong suggestion that the incorporation of hydrogen in γ-Ca2SiO4 is through a combination of (2H)Ca1 and (4H)Si defects.
We investigated the same mechanisms for pure β-Ca2SiO4: (2H)Ca1, (2H)Ca2, and (4H)Si. Our calculations indicate a preference for (2H)Ca2 over (2H)Ca1, but none of these models produce IR OH bond wavenumbers (Table 3) that are a convincing match for those observed in our experimental FTIR measurements (3479 cm−1) (Fig. 11).
Our simulations indicate that coupled substitution of P and H is energetically favoured relative to phosphorus only defects in β-Ca2SiO4. Of the defects studied for β-Ca2SiO4 we can constrain the likely mechanism of hydrogen incorporation from our theoretical IR spectra compared to our FTIR measurements. Our theoretical IR OH bonds for P and H coupled substitution produce a range of frequencies (Table 3; Fig. 12). The main band seen in our FTIR spectra at 3479 cm−1 (Fig. 7) matches precisely with the calculated 3478 cm−1 band for (H)Ca2(P)Si. Lattice strain, indicated by twinning in our single crystals, and accommodated by the Ca2 vacancies where the hydrogen is incorporated, may contribute to the broadness of the main OH absorption band through proton disordering. Alternatively, the predicted bands from (2H)Ca2(P)Si and (3H)Si(P)Si, which are similar, may overlap, increasing the apparent width of the measured IR band. However, we would expect to observe additional OH peaks in our FTIR spectra from these types of defects. The calculated OH frequencies for the (H)Ca2(H)Ca2(P)Si defect were not considered because they do not match the observed FTIR spectra.
Relative energy differences between water incorporation mechanisms
The calculations mentioned previously are designed to establish the lowest-energy configuration for each type of defect investigated and to provide the structural model for the calculation of vibrational spectra. However, they are unable to predict the relative importance of dissimilar defects, e.g., protonated Ca vacancies vs. protonated Si vacancies. Therefore, we performed additional calculations to investigate the relative stabilities of the (4H)Si and (2H)Ca defect mechanisms in P-free β-Ca2SiO4 at 0 K. In reactions (10) and (11), the (4H)Si defect is on the left-hand side of the reaction and the (2H)Ca defect is on the right-hand side.
Each of reactions 10 and 11 can be used to calculate the difference in energy between the two competing defect mechanisms (under high or low SiO2 chemical environments, respectively). Reaction 10 has a defect formation energy of −55 kJ mol−1 per OH defect as compared to 141.9 kJ mol−1 per OH defect for reaction 11. This indicates that at 0 K the formation of hydrated silicon vacancies in a low SiO2 chemical environment is favoured. In contrast, in a chemical environment with high SiO2, the formation of hydrated calcium vacancies is preferred. Our calculations are consistent with experimental observations for forsterite, where protonated silicon vacancies are dominant at low SiO2 activity and protonated Mg vacancies are dominant at high SiO2 activity (Lemaire et al. 2004; Fei and Katsura 2020).
In the phosphorus and hydrogen bearing defect structures studied, we can assess the relative stability between different incorporation mechanisms.
Reaction 12 determines the free energy difference between the defects (H)Ca2(P)Si (mechanism i) and (3H)Si(P)Si (mechanism iv). Reaction 13 determines the difference in free energy between (H)Ca2(P)Si (mechanism i) and (2H)Ca22(P) (mechanism ii). Reaction 14 determines the free energy difference between (2H)Ca22(P) (mechanism ii) and (3H)Si(P)Si (mechanism iv) defects.
The difference in free energy between the defects for reactions 12 and 13 is 169.8 kJ mol−1 and 56.4 kJ mol−1, respectively. The positive reaction energy in both reactions strongly favours the (H)Ca2(P)Si substitution mechanism in β-Ca2SiO4 represented by the left-hand side of reactions 13 and 14. The calculated energy change for reaction 15 has a positive formation energy of 283.2 kJ mol−1 that favours the creation of (2H)Ca22(P). The preference for the formation of (H)Ca2(P)Si over defects (2H)Ca22(P) and (3H)Si(P)Si further supports the interpretation that the calculated 3478 cm−1 band of (H)Ca2(P)Si that strongly coincides with the band at 3479 cm−1 measured by FTIR is the result of this defect (Fig. 7).
Water solubility in β-Ca2SiO4 and implications for inclusions in diamonds
We have shown here that under mantle conditions, the solubility of water in P-doped β-Ca2SiO4 is only 68–178 ppm H2O. The range of values reflects the range of pressure and temperature conditions in our study, as well as uncertainties in the spectral fits, crystal thicknesses, and FTIR calibrations (Table 1). Our measured solubility is completely different from the value of 4.06 wt.% H2O reported by Németh et al. (2017) at 17 GPa and 1500 °C. Although the pressure and temperature of the two studies do not overlap, there are differences in the interpretation of the FTIR spectra. The broad peak in the spectrum is interpreted by Németh et al. (2017) as water dissolved in larnite, whereas we consider it to be non-structural water. Furthermore, the water quantification in Németh et al. (2017) was performed using EPMA shortfall, a notoriously unreliable method. Finally, Németh et al. (2017) suggested that the superstructure reflections observed in their X-ray diffraction data are indicative of the ordering of OH groups in β-Ca2SiO4. However, Kim et al. (1992) have shown that these reflections can occur when samples are quenched from the αʹ field.
The water content of γ-Ca2SiO4 inverted from β-Ca2SiO4 during decompression was found here to be either 107 or 71 ppm, a similar range of values to those of P-doped β-Ca2SiO4. If larnite inclusions are trapped directly, the solubilities measured here would provide an upper limit to any expected water concentrations in larnite inclusions that could be measured in the future. Lower values would indicate a lower water fugacity for the diamond-forming fluid than pure water. If larnite inclusions are a back-transformation product from perovskite, it is notable that the measured water solubility in β-Ca2SiO4 reported in the present study is significantly less than the 0.5–1 wt.% reported for CaSiO3-perovskite by Chen et al. (2020). If that report is accurate, then β-Ca2SiO4 inclusions are unlikely to provide much constraint on the initial water concentration of CaSiO3-perovskite prior to unmixing. Instead, most of the water dissolved in CaSiO3-perovskite would have to be hosted in other crystalline phases or exsolved fluids. The common observation of cracks around former CaSiO3-perovskite inclusions in diamonds would be consistent with the presence of fluids. A complete understanding of the fate of water trapped in deep mantle CaSiO3-perovskite inclusions requires studies of the solubility of water in all phases formed by the breakdown of CaSiO3-perovskite during exhumation of diamonds. This includes breyite, CaTiO3 perovskite, and titanite-structured CaSi2O5. The solubility data for these phases will be reported elsewhere (Gregson et al., in prep). More extensive studies of the water solubility in CaSiO3-perovskite are also required.
Summary
Our high-pressure, high-temperature experiments and ab initio computer simulations on Ca2SiO4 indicate that:
-
i.
the incorporation of phosphorus is a key stabilising mechanism for inclusions of β-Ca2SiO4 in sub-lithospheric diamonds and that 0.16 wt.% P2O5 is sufficient to stabilise β-Ca2SiO4. The mechanism of incorporation of phosphorus in β-Ca2SiO4 is predicted to involve the substitution of two Si4+ ions with two P5+ ions, charge balanced by a Ca2+ vacancy on the Ca2 site. Calculations imply that this is energetically favourable compared with the defect-free structure.
-
ii.
the solubility of water in all Ca2SiO4 polymorphs is low (71–178 ppm H2O) regardless of the presence of phosphorus and under a range of high pressure and temperature conditions. The preferred mechanism of hydrogen substitution in γ-Ca2SiO4 involves the protonation of Ca1 and Si vacancies. For phosphorus-bearing β-Ca2SiO4, the preferred mechanism involves one Si4+ ion being replaced by one P5+ ion with a single protonated Ca2 vacancy.
-
iii.
the low water solubility observed in larnite implies that even if primary CaSiO3-perovskite inclusions can incorporate high water concentrations during diamond growth from a volatile rich fluid, measurements of water concentration in larnite inclusions formed by retrograde reactions will not provide a constraint or reflect the initial water concentration of CaSiO3-perovskite.

(Modified from Xiong et al. 2016)
Ca2SiO4 phase transformation sequence at ambient conditions.


FTIR spectrum of an inclusion-bearing γ-Ca2SiO4 polycrystal and the procedure for removing contaminant components. (A) Unprocessed unpolarised FTIR spectrum of γ-Ca2SiO4 from experiment LA5 (B) Processed spectrum with OH defect bands between 3370 cm–1 and 3615 cm–1 removed. This was achieved by subtracting a spectrum where the broad component related to fluid inclusions is weak from one in which it is strong, scaled so that the γ-Ca2SiO4 OH defect peaks were the same size and hence completely removed. The resulting spectrum contains four features relating to contaminants that were deconvolved by fitting: molecular H2O (C), organics (D), portlandite Ca(OH)2 (E) and an unknown component (F). Different proportions of these four components were subtracted from each raw spectrum to leave only the γ-Ca2SiO4 peaks (G). Spectra shown are not on a common scale

FTIR spectrum of inclusion-bearing β-Ca2SiO4 and the procedure for removing contaminant components as described in the caption to Fig. 3 (A) Unprocessed unpolarised FTIR spectrum of β-Ca2SiO4 from experiment LA16. (B) Processed spectrum with OH defect bands between 3370 cm–1 and 3615 cm–1 removed to leave four features related to molecular H2O (C), organics (D), portlandite Ca(OH)2 (E) and OH vibrations from an unknown component (F). (G) Final processed IR spectrum showing the OH defect at 3479 cm–1 with contributions C-F removed. Spectra shown are not on a common scale

Raman spectra from γ-Ca2SiO4 and P2O5-doped β-Ca2SiO4 recovered from high-pressure experiments LA5 and LA16, respectively

A polycrystalline opaque grain of γ-Ca2SiO4 consisting of orientated laths (left) and a transparent single crystal of β-Ca2SiO4 with {100}-type twinning (right) from high-pressure experiments LA5 and LA16, respectively

Unpolarised FTIR spectra showing OH bands in γ-Ca2SiO4 from experiment LA5 (see Fig. 3). From top to bottom, the lines represent: (A) the processed FTIR spectrum with a baseline correction, (B) the sum of fitted Lorentzian lineshapes for bands associated with protonated defects in γ-Ca2SiO4, (C) the individual deconvolved Lorentzian lineshapes and (D) the residual from the fitting procedure

Unpolarised FTIR spectra showing OH bands in β-Ca2SiO4 (see Fig. 4). The band at 3645 cm−1 corresponds to portlandite (Ca(OH)2). From top to bottom, the lines represent: (A) the processed FTIR spectrum with a baseline correction, (B) the sum of fitted Gaussian lineshapes for bands associated with protonated defects in β-Ca2SiO4, (C) the individual deconvolved Gaussian lineshapes and (D) the residual from the fitting procedure

Polarised and background-corrected FTIR spectra showing OH defect bands and silicate overtones in β-Ca2SiO4 synthesized at 4 GPa and 1273 K. Only the broad molecular water component has been removed from each spectrum. The band at 3641 cm−1 is portlandite (Ca(OH)2) and the band at 3691 cm−1 corresponds to an unknown hydrous phase contaminant. In the spectrum marked as 0° the infrared light is polarised parallel to the crystallographic Z axis; in the spectrum marked as 90° it is polarised parallel to the X axis. Spectra are offset for clarity. The polarisation of the broad OH band at 3479 cm−1 appears to be weak

Local structural models and theoretical IR spectra for (2H)Ca2 and (4H)Si defects in γ-Ca2SiO4: A) Charge-balanced protonated Ca2 vacancy. B) Si vacancy charged-balanced by 4 protons. A dashed line in the structural model is a O-H∙∙∙O hydrogen bond. Red and white spheres are oxygen and hydrogen atoms. Blue shaded areas are the experimentally observed IR bands (Fig. 7). All models shown are the lowest energy configuration and the calculated spectra are not on a common scale

Local structural models and theoretical IR spectra for (2H)Ca2 and (4H)Si defects in β-Ca2SiO4: A) Charge-balanced protonated Ca2 vacancy. B) Si vacancy charged-balanced by 4 protons. Dashed lines in the structural model indicate O-H∙∙∙O hydrogen bonds. Models are generated from the simulation with the lowest energy. Blue shaded areas are the experimentally observed IR bands (Fig. 8). Calculated spectra are not on a common scale

Local structural models and theoretical IR spectra for defects in β-Ca2SiO4: A) Mechanism (i) (reaction 5): a single protonated Ca2 vacancy with a neighbouring P atom substituting for a Si atom; B) Mechanism (ii) (reaction 7): a protonated Ca2 vacancy and an unprotonated Ca2 vacancy with two neighbouring P atoms substituting for two Si atoms; C) Mechanism (iii) two adjacent singly protonated Ca2 vacancies with two neighbouring P atoms substituting for two Si atoms; (D) Mechanism (iv): a Si vacancy containing three hydrogen atoms with a neighbouring P atom substituting for a Si atom. Dashed lines indicate O-H∙∙∙O hydrogen bonds. All models shown are the lowest energy configuration. Blue shaded areas in theoretical IR spectra are the observed experimental IR bands (Fig. 8)
Data availability
The CIF files and the DynG files of the computer simulations described in this study are available in the University of Bristol data repository, data.bris, at https://doi.org/10.5523/bris.db057m97guck1zxjpstqld26u.
References
Anzolini C, Angel RJ, Merlini M, Derzsi M, Tokár K, Milani S, Krebs MY, Brenker FE, Nestola F, Harris JW (2016) Depth of formation of CaSiO3-walstromite included in super-deep diamonds. Lithos 265:138–147. https://doi.org/10.1016/j.lithos.2016.09.025
Anzolini C, Prencipe M, Alvaro M, Romano C, Vona A, Lorenzon S, Smith EM, Brenker FE, Nestola F (2018) Depth of formation of super-deep diamonds: Raman barometry of CaSiO3-walstromite inclusions. Am Mineral 103:69–74. https://doi.org/10.2138/am-2018-6184
Balan E, Kloprogge JT (2017) Theoretical aspects of Infrared and Raman Spectroscopies, 1st edn. Elsevier Ltd.
Balan E, Saitta AM, Mauri F, Calas G (2001) First-principles modeling of the infrared spectrum of kaolinite. Am Mineral 86:1321–1330. https://doi.org/10.2138/am-2001-11-1201
Balan E, Refson K, Blanchard M, Delattre S, Lazzeri M, Ingrin J, Mauri F, Wright K, Winkler B (2008) Theoretical infrared absorption coefficient of OH groups in minerals. Am Mineral 93:950–953. https://doi.org/10.2138/am.2008.2889
Balan E, Ingrin J, Delattre S, Kovács I, Blanchard M (2011) Theoretical infrared spectrum of OH-defects in forsterite. Eur J Mineral 23:285–292. https://doi.org/10.1127/0935-1221/2011/0023-2090
Balan E, Blanchard M, Yi H, Ingrin J (2013) Theoretical study of OH-defects in pure enstatite. Phys Chem Min 40:41–50. https://doi.org/10.1007/s00269-012-0544-6
Baltakys K, Siauciunas R, Kitrys S (2008) Surface microstructure and specific surface area of pure and Na-substituted gyrolites. Mater Sci Pol 26:633–645
Baroni S, Gironcoli S, De, Corso AD, Scuola S, Superiore I, Istituto I, Materia F, Trieste I-, Giannozzi P (2001) Phonons and related crystal properties from density-functional perturbation theory. Rev Mod Phys 73:515–562. https://doi.org/10.1103/RevModPhys.73.515
Benarchid MY, Diouri A, Boukhari A, Aride J, Rogez J, Castanet R (2004) Elaboration and thermal study of iron-phosphorus-substituted dicalcium silicate phase. Cem Concr Res 34:1873–1879. https://doi.org/10.1016/j.cemconres.2004.01.030
Blanchard M, Roberge M, Balan E, Fiquet G, Bureau H (2013) Infrared signatures of OH-defects in wadsleyite: a first-principles study. Am Mineral 98:2132–2143. https://doi.org/10.2138/am.2013.4468
Bredig MA (1950) Polymorphism of Calcium Orthosilicate. J Am Ceram Soc 33:188–192. https://doi.org/10.1111/j.1151-2916.1950.tb12789.x
Brenker FE, Vincze L, Vekemans B, Nasdala L, Stachel T, Vollmer C, Kersten M, Somogyi A, Adams F, Joswig W, Harris JW (2005) Detection of a Ca-rich lithology in the Earth’s deep (> 300 km) convecting mantle. Earth Planet Sci Lett 236:579–587. https://doi.org/10.1016/j.epsl.2005.05.021
Brenker FE, Vollmer C, Vincze L, Vekemans B, Szymanski A, Janssens K, Szaloki I, Nasdala L, Joswig W, Kaminsky F (2007) Carbonates from the lower part of transition zone or even the lower mantle. Earth Planet Sci Lett 260:1–9. https://doi.org/10.1016/j.epsl.2007.02.038
Brenker FE, Vincze L, Vekemans B, Szymanski A, Nolf W, De, Janssens K, Stachel T, Harris J (2008) Detection of a REE-rich, F- and P-bearing fluid component in superdeep diamonds from Kankan (Guinea). In: International Kimberlite Conference: Extended Abstracts
Brenker FE, Nestola F, Brenker L, Peruzzo L, Harris JW (2021) Origin, properties, and structure of breyite: the second most abundant mineral inclusion in super-deep diamonds. Am Mineral 106:38–43. https://doi.org/10.2138/am-2020-7513
Brooker R, Holloway JR, Hervig RL (1998) Reduction in piston-cylinder experiments: the detection of carbon infiltration into platinum capsules. Am Mineral 83:985–994. https://doi.org/10.2138/am-1998-9-1006
Bulanova GP, Walter MJ, Smith CB, Kohn SC, Armstrong LS, Blundy J, Gobbo L (2010) Mineral inclusions in sublithospheric diamonds from Collier 4 kimberlite pipe, Juina, Brazil: subducted protoliths, carbonated melts and primary kimberlite magmatism. Contrib Mineral Petrol 160:489–510. https://doi.org/10.1007/s00410-010-0490-6
Burnham AD, Bulanova GP, Smith CB, Whitehead SC, Kohn SC, Gobbo L, Walter MJ (2016) Diamonds from the Machado River alluvial deposit, Rondônia, Brazil, derived from both lithospheric and sublithospheric mantle. Lithos 265:199–213. https://doi.org/10.1016/j.lithos.2016.05.022
Chan CJ, Kriven WM, Young JF (1992) Physical stabilization of the β→γ Transformation in Dicalcium Silicate. J Am Ceram Soc 75:1621–1627. https://doi.org/10.1111/j.1151-2916.1992.tb04234.x
Chen H, Leinenweber K, Prakapenka V, Prescher C, Meng Y, Bechtel H, Kunz M, Shim SH (2020) Possible H2O storage in the crystal structure of CaSiO3 perovskite. Phys Earth Planet Inter 299:106412. https://doi.org/10.1016/j.pepi.2019.106412
Chukanov NV (2014) Infrared spectra of mineral species: extended library. Springer Netherlands
Daver L, Bureau H, Boulard É, Gaillou É, Cartigny P, Pinti DL, Belhadj O, Guignot N, Foy E, Estève I, Baptiste B (2022) From the lithosphere to the lower mantle: an aqueous-rich metal-bearing growth environment to form type IIb blue diamonds. Chem Geol 613:121163. https://doi.org/10.1016/j.chemgeo.2022.121163
Dehghan-Manshadi A, Manuel J, Hapugoda S, Ware N (2014) Sintering characteristics of titanium containing iron ores. ISIJ Int 54:2189–2195. https://doi.org/10.2355/isijinternational.54.2189
Diouri A, Boukhari A, Aride J, Puertas F, Vazquez T (1997) Stable Ca3SiO5 solid solution containing manganese and phosphorus. Cem Concr Res 27:1203–1212. https://doi.org/10.1016/S0008-8846(97)00110-5
Fedoraeva AS, Shatskiy A, Litasov KD (2019) The join CaCO3-CaSiO3 at 6 GPa with implication to Ca-rich lithologies trapped by kimberlitic diamonds. High Press Res 39:547–560. https://doi.org/10.1080/08957959.2019.1660325
Fei H, Katsura T (2020) Pressure Dependence of Proton Incorporation and Water Solubility in Olivine. J Geophys Res Solid Earth 125:1–13. https://doi.org/10.1029/2019JB018813
Fukuda K, Taguchi H (1999) Hydration of α′L- and β-dicalcium silicates with identical concentration of phosphorus oxide. Cem Concr Res 29:503–506. https://doi.org/10.1016/S0008-8846(98)00195-1
Galuskin EV, Gazeev VM, Lazic B, Armbruster T, Galuskina IO, Zadov AE, Pertsev NN, Wrzalik R, Dzierżanowski P, Gurbanov AG, Bzowska G (2009) Chegemite Ca7(SiO4)3(OH)2 a new humite-group calcium mineral from the Northern Caucasus, Kabardino-Balkaria, Russia. Eur J Mineral 21:1045–1059. https://doi.org/10.1127/0935-1221/2009/0021-1962
Gasparik T, Wolf K, Smith CM (1994) Experimental determination of phase relations in the CaSiO3 system from 8 to 15 GPa. Am Mineral 79:1219–1222
Genzel PT, Pamato MG, Novella D, Santello L, Lorenzon S, Shirey SB, Pearson DG, Nestola F, Brenker FE (2023) Geobarometric evidence for a LM/TZ origin of CaSiO3 in a sublithospheric diamond. Geochemical Perspect Lett 25:41–45. https://doi.org/10.7185/geochemlet.2313
Ghose A, Chopra S, Young JF (1983) Microstructural characterization of doped dicalcium silicate polymorphs. J Mater Sci 18:2905–2914. https://doi.org/10.1007/BF00700771
Giannozzi P, Baroni S, Bonini N, Calandra M, Car R, Cavazzoni C, Ceresoli D, Chiarotti GL, Cococcioni M, Dabo I, Dal Corso A, De Gironcoli S, Fabris S, Fratesi G, Gebauer R, Gerstmann U, Gougoussis C, Kokalj A, Lazzeri M, Martin-Samos L, Marzari N, Mauri F, Mazzarello R, Paolini S, Pasquarello A, Paulatto L, Sbraccia C, Scandolo S, Sclauzero G, Seitsonen AP, Smogunov A, Umari P, Wentzcovitch RM (2009) QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J Phys Condens Matter 21. https://doi.org/10.1088/0953-8984/21/39/395502
Gobechiya ER, Yamnova NA, Zadov AE, Gazeev VM (2008) Calcio-olivine γ-Ca2SiO4: I. Rietveld refinement of the crystal structure. Crystallogr Rep 53:404–408. https://doi.org/10.1134/S1063774508030073
Guo P, Wang B, Bauchy M, Sant G (2016) Misfit stresses caused by atomic size mismatch: the origin of Doping-Induced destabilization of Dicalcium Silicate. Cryst Growth Des 16:3124–3132. https://doi.org/10.1021/acs.cgd.5b01740
Hamann DR (2013) Optimized norm-conserving Vanderbilt pseudopotentials. Phys Rev B - Condens Matter Mater Phys 88:1–10. https://doi.org/10.1103/PhysRevB.88.085117
Ishikawa T, Teramachi A, Tanaka H, Yasukawa A, Kandori K (2000) Fourier Transform Infrared Spectroscopy Study of Deuteration of Calcium Hydroxyapatite Particles. Langmuir 16:10221–10226. https://doi.org/10.1021/la0004855
Jost KH, Ziemer B, Seydel R (1977) Redetermination of the structure of β-dicalcium silicate. Acta Crystallogr Sect B Struct Crystallogr Cryst Chem 33:1696–1700. https://doi.org/10.1107/s0567740877006918
Joswig W, Stachel T, Harris JW, Baur WH, Brey GP (1999) New Ca-silicate inclusions in diamonds - tracers from the lower mantle. Earth Planet Sci Lett 173:1–6. https://doi.org/10.1016/S0012-821X(99)00210-1
Kaminsky FV, Zedgenizov DA (2022) First find of merrillite, Ca3(PO4)2, in a terrestrial environment as an inclusion in lower-mantle diamond. Am Mineral 107:1652–1655. https://doi.org/10.2138/am-2022-8175
Kanzaki M, Stebbins JF, Xue X (1991) Characterization of quenched high pressure phases in CaSiO3 system by XRD and 29Si NMR. Geophys Res Lett 18:463–466. https://doi.org/10.1029/91GL00463
Kanzaki M, Stebbins JF, Xue X (2013) Characterization of Crystalline and Amorphous silicates quenched from high pressure by 29Si MAS NMR spectroscopy. In high-pressure research: application to Earth and Planetary sciences. (Syono, Y. and Manghnani M.H Eds.). Terra Scientific Publishing Company/AGU, Tokyo/Washington, D.C., 89–100. https://doi.org/10.1029/GM067p0089
Katsura T, Yoneda A, Yamazaki D, Yoshino T, Ito E, Suetsugu D, Bina C, Inoue T, Wiens D, Jellinek M (2010) Adiabatic temperature profile in the mantle. Phys Earth Planet Inter 183:212–218. https://doi.org/10.1016/j.pepi.2010.07.001
Kendrick J, Burnett AD (2016) PDielec: the calculation of infrared and terahertz absorption for powdered crystals. J Comput Chem 37:1491–1504. https://doi.org/10.1002/jcc.24344
Kim YJ, Nettleship I, Kriven WM (1992) Phase transformations in Dicalcium Silicate: II, TEM studies of Crystallography, microstructure, and mechanisms. J Am Ceram Soc 75:2407–2419. https://doi.org/10.1111/j.1151-2916.1992.tb05593.x
Ko B, Greenberg E, Prakapenka V, Alp EE, Bi W, Meng Y, Zhang D, Shim S-H (2022) Calcium dissolution in bridgmanite in the Earth’s deep mantle. Nature 611:88–92. https://doi.org/10.1038/s41586-022-05237-4
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140:A1133–A1138. https://doi.org/10.1103/PhysRev.140.A1133
Koutsopoulos S (2002) Synthesis and characterization of hydroxyapatite crystals: a review study on the analytical methods. J Biomed Mater Res 62:600–612. https://doi.org/10.1002/jbm.10280
Lai GC, Nojiri T, Nakano Kichi (1992) Studies of the stability of β-Ca2SiO4 doped by minor ions. Cem Concr Res 22:743–754. https://doi.org/10.1016/0008-8846(92)90097-F
Lemaire C, Kohn SC, Brooker RA (2004) The effect of silica activity on the incorporation mechanisms of water in synthetic forsterite: a polarised infrared spectroscopic study. Contrib Mineral Petrol 147:48–57. https://doi.org/10.1007/s00410-003-0539-x
Libowitzky E, Rossman GR (1997) An IR absorption calibration for water in minerals. Am Mineral 82:1111–1115. https://doi.org/10.2138/am-1997-11-1208
Mason B (1957) Larnite, Scawtite, and Hydrogrossular from Tokatoka, New Zealand. Am Mineral 42:379–392
Matjuschkin V, Brooker RA, Tattitch B, Blundy JD, Stamper CC (2015) Control and monitoring of oxygen fugacity in piston cylinder experiments. Contrib Mineral Petrol 169:1–16. https://doi.org/10.1007/s00410-015-1105-z
Midgley HG, Bennett M (1971) A microprobe analysis of larnite and bridigite from scawt hill, larne, Northern Ireland. Cem Concr Res 1:413–418. https://doi.org/10.1016/0008-8846(71)90034-2
Monkhorst HJ, Pack JD (1976) Special points for Brillouin-Zone integrations. Phys Rev B - Condens Matter Mater Phys 13:5188–5192. https://doi.org/10.1103/PhysRevB.16.1748
Nasdala L, Brenker FE, Glinnemann J, Hofmeister W, Gasparik T, Harris JW, Stachel T, Reese I (2003) Spectroscopic 2D-tomography: residual pressure and strain around mineral inclusions in diamonds. Eur J Mineral 15:931–935. https://doi.org/10.1127/0935-1221/2003/0015-0931
Nastac DC, Kääntee U, Liimatainen J, Hupa M, Muntean M (2007) Influence of P(V) on the characteristics of calcium silicates and the hydration of clinkers. Adv Cem Res 19:93–100. https://doi.org/10.1680/adcr.2007.19.3.93
Németh P, Leinenweber K, Ohfuji H, Groy T, Domanik KJ, Kovács IJ, Kovács JS, Buseck PR (2017) Water-bearing, high-pressure Ca-silicates. Earth Planet Sci Lett 469:148–155. https://doi.org/10.1016/j.epsl.2017.04.011
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868. https://doi.org/10.1103/PhysRevLett.77.3865
Pritts IM, Daugherty KE (1976) The effect of stabilizing agents on the hydration rate of β-C2S. Cem Concr Res 6:783–795. https://doi.org/10.1016/0008-8846(76)90008-9
Regier ME, Smit KV, Chalk TB, Stachel T, Stern RA, Smith EM, Foster GL, Bussweiler Y, DeBuhr C, Burnham AD, Harris JW, Pearson DG (2023) Boron isotopes in blue diamond record seawater-derived fluids in the lower mantle. Earth Planet Sci Lett 602:117923. https://doi.org/10.1016/j.epsl.2022.117923
Rejmak P, Dolado JS, Aranda MAG, Ayuela A (2019) First-principles calculations on polymorphs of Dicalcium Silicate - Belite, a Main Component of Portland Cement. J Phys Chem C 123:6768–6777. https://doi.org/10.1021/acs.jpcc.8b10045
Remy C, Guyot F, Madon M (1995) High pressure polymorphism of dicalcium silicate Ca2SiO4. A transmission electron microscopy study. Phys Chem Min 22:419–427. https://doi.org/10.1007/BF00200319
Remy C, Reynard B, Madon M (1997) Raman Spectroscopic Investigations of Dicalcium Silicate: polymorphs and high-temperature phase transformations. J Am Ceram Soc 80:413–423. https://doi.org/10.1111/j.1151-2916.1997.tb02846.x
Reynard B, Remy C, Takir F (1997) High-pressure Raman spectroscopic study of Mn2GeO4, Ca2GeO4, Ca2SiO4, and CaMgGeO4 olivines. Phys Chem Min 24:77–84. https://doi.org/10.1007/s002690050019
Ross NL, Gibbs GV, Rosso KM (2003) Potential docking sites and positions of hydrogen in high-pressure silicates. Am Mineral 88:1452–1459. https://doi.org/10.2138/am-2003-1009
Saalfeld H (1975) X-ray investigation of single crystals of β-Ca2SiO4 (larnite) at high temperature. Am Mineral 60:824–827
Saidani S, Smith A, El Hafiane Y, Ben Tahar L (2018) Re-examination of the β→γ transformation of Ca2SiO4. J Eur Ceram Soc 38:4756–4767. https://doi.org/10.1016/j.jeurceramsoc.2018.06.011
Saidani S, Smith A, El Hafiane Y, Ben Tahar L (2020) Role of dopants (B, P and S) on the stabilization of β-Ca2SiO4. J Eur Ceram Soc. https://doi.org/10.1016/j.jeurceramsoc.2020.07.037
Sakurada R, Kawazoe Y, Singh AK (2015) First-principles study on structural stability of belite. ACI Mater J 112:85–93. https://doi.org/10.14359/51687128
Schlipf M, Gygi F (2015) Optimization algorithm for the generation of ONCV pseudopotentials. Comput Phys Commun 196:36–44. https://doi.org/10.1016/j.cpc.2015.05.011
Smith D, Majumdar AJ, Ordway F (1961) Re-examination of the polymorphism of Dicalcium Silicate. J Am Ceram Soc 44:405–411. https://doi.org/10.1111/j.1151-2916.1961.tb15472.x
Smith EM, Shirey SB, Nestola F, Bullock ES, Wang J, Richardson SH, Wang W (2016) Large gem diamonds from metallic liquid in Earth’s deep mantle. Science 354:1403–1405. https://doi.org/10.1126/science.aal1303
Smith EM, Shirey SB, Richardson SH, Nestola F, Bullock ES, Wang J, Wang W (2018) Blue boron-bearing diamonds from Earth’s lower mantle. Nature 560:84–87. https://doi.org/10.1038/s41586-018-0334-5
Sokol EV, Kokh SN, Vapnik Y, Thiéry V, Korzhova SA (2014) Natural analogs of belite sulfoaluminate cement clinkers from Negev Desert, Israel. Am Mineral 99:1471–1487. https://doi.org/10.2138/am.2014.4704
Sokol EV, Seryotkin YV, Kokh SN, Vapnik Y, Nigmatulina EN, Goryainov SV, Belogub EV, Sharygin VV (2015) Flamite, (Ca,Na,K)2(Si,P)O4, a new mineral from ultrahightemperature combustion metamorphic rocks, Hatrurim Basin, Negev Desert, Israel. Mineral Mag 79:583–596. https://doi.org/10.1180/minmag.2015.079.3.05
Speakman K, Taylor HFW, Bennett JM, Gard JA (1967) Hydrothermal reactions of γ-dicalcium silicate. J Chem Soc A 1052–1060. https://doi.org/10.1039/J19670001052
Stachel T, Harris JW, Brey GP, Joswig W (2000) Kankan diamonds (Guinea) II: lower mantle inclusion parageneses. Contrib Mineral Petrol 140:16–27. https://doi.org/10.1007/s004100000174
Steinberg HK (2005) Hochdruckexperimente an Calciumsilikatphasen zur Rekonstruktion der Aufstiegsgeschichte von Diamanten. Frankfurt am Main
Thomson AR, Kohn SC, Bulanova GP, Smith CB, Araujo D, Walter EIMF MJ (2014) Origin of sub-lithospheric diamonds from the Juina-5 kimberlite (Brazil): constraints from carbon isotopes and inclusion compositions. Contrib Mineral Petrol 168. https://doi.org/10.1007/s00410-014-1081-8
Thomson AR, Kohn SC, Bulanova GP, Smith CB, Araujo D, Walter MJ (2016) Trace element composition of silicate inclusions in sub-lithospheric diamonds from the Juina-5 kimberlite: evidence for diamond growth from slab melts. Lithos 265:108–124. https://doi.org/10.1016/j.lithos.2016.08.035
Tilley CE (1929) On larnite (calcium orthosilicate, a new mineral) and its associated minerals from the limestone contact-zone of Scawt Hill. Co Antrim Mineral Mag J Mineral Soc 22:77–86. https://doi.org/10.1180/minmag.1929.022.125.01
Timmerman S, Stachel T, Koornneef JM, Smit KV, Harlou R, Nowell GM, Thomson AR, Kohn SC, Davies JHFL, Davies GR, Krebs MY, Zhang Q, Milne SEM, Harris JW, Kaminsky F, Zedgenizov D, Bulanova G, Smith CB, Cabral Neto I, Silveira FV, Burnham AD, Nestola F, Shirey SB, Walter MJ, Steele A, Pearson DG (2023) Sublithospheric diamond ages and the supercontinent cycle. Nature 623:752–756. https://doi.org/10.1038/s41586-023-06662-9
Tortet L, Gavarri JR, Nihoul G, Dianoux AJ (1997) Study of Protonic mobility in CaHPO4·2H2O (Brushite) and CaHPO4(Monetite) by Infrared Spectroscopy and Neutron Scattering. J Solid State Chem 132:6–16. https://doi.org/10.1006/jssc.1997.7383
Trønnes RG (2010) Structure, mineralogy and dynamics of the lowermost mantle. Mineral Petrol 99:243–261. https://doi.org/10.1007/s00710-009-0068-z
Udagawa S, Urabe K, Natsume M, Yano T (1980) Refinement of the crystal structure of γ-Ca2SiO4. Cem Concr Res 10:139–144. https://doi.org/10.1016/0008-8846(80)90070-8
Walker D, Carpenter MA, Hitch CM (1990) Some simplifications to multianvil devices for high pressure experiments. Am Mineral 75:1020–1028
Walker AM, Hermann J, Berry AJ, O’Neill HSC (2007) Three water sites in upper mantle olivine and the role of titanium in the water weakening mechanism. J Geophys Res Solid Earth 112:1–12. https://doi.org/10.1029/2006JB004620
Walter MJ, Bulanova GP, Armstrong LS, Keshav S, Blundy JD, Gudfinnsson G, Lord OT, Lennie AR, Clark SM, Smith CB, Gobbo L (2008) Primary carbonatite melt from deeply subducted oceanic crust. Nature 454:622–625. https://doi.org/10.1038/nature07132
Wang Y, Weidner DJ (1994) Thermoelasticity of CaSiO3 perovskite and implications for the lower mantle. Geophys Res Lett 21:895–898. https://doi.org/10.1029/94GL00976
Withers AC, Bureau H, Raepsaet C, Hirschmann MM (2012) Calibration of infrared spectroscopy by elastic recoil detection analysis of H in synthetic olivine. Chem Geol 334:92–98. https://doi.org/10.1016/j.chemgeo.2012.10.002
Woodland AB, Girnis AV, Bulatov VK, Brey GP, Höfer HE (2020) Breyite inclusions in diamond: experimental evidence for possible dual origin. Eur J Mineral 32:171–185. https://doi.org/10.5194/ejm-32-171-2020
Xiong Z, Liu X, Shieh SR, Wang S, Chang L, Tang J, Hong X, Zhang Z, Wang H (2016) Some thermodynamic properties of larnite (β-Ca2SiO4) constrained by high T/P experiment and/or theoretical simulation. Am Mineral 101:277–288. https://doi.org/10.2138/am-2016-5425
Yamnova NA, Zubkova NV, Eremin NN, Zadov AE, Gazeev VM (2011) Crystal structure of larnite β-Ca2SiO4 and specific features of polymorphic transitions in dicalcium orthosilicate. Crystallogr Rep 56:210–220. https://doi.org/10.1134/S1063774511020209
Yu P, Kirkpatrick RJ, Brent P, McMillan PF, Cong X (1999) Structure of calcium silicate hydrate (C-S-H): Near-, mid-, and far-infrared spectroscopy. J Am Ceram Soc 82:742–748. https://doi.org/10.1111/j.1151-2916.1999.tb01826.x
Zedgenizov DA, Ragozin AL, Kalinina VV, Kagi H (2016) The mineralogy of Ca-rich inclusions in sublithospheric diamonds. Geochem Int 54:890–900. https://doi.org/10.1134/S0016702916100116
Zhang X, Zhao M, Zhang Y (2012) Preparation and properties of self-pulverizing calcium sulfoaluminate cement. Constr Build Mater 34:107–113. https://doi.org/10.1016/j.conbuildmat.2012.02.006
Zhang Q, Timmerman S, Stachel T, Chinn I, Stern RA, Davies J, Nestola F, Luth R, Pearson DG (2024) Sublithospheric diamonds extend paleoproterozoic record of cold deep subduction into the lower mantle. Earth Planet Sci Lett 634:118675. https://doi.org/10.1016/j.epsl.2024.118675
Acknowledgements
CG was supported by a NERC GW4 + Doctoral Training Partnership studentship from the Natural Environment Research Council [NE/L002434/1]. CG is grateful for supervision and support from Mike Walter. We thank Stuart Kearns for helping with EPMA. OTL acknowledges support from the Royal Society in the form of a University Research Fellowship [UF150057]. This work was carried out using the computational facilities of the Advanced Computing Research Centre, University of Bristol - http://www.bris.ac.uk/acrc/. The authors are grateful for the helpful comments made by three anonymous reviewers and Hans Keppler.
Author information
Authors and Affiliations
Contributions
Chris Gregson: Conceptualisation, experimental design and construction, experimental and theoretical investigation, data acquisition, validation, visualization, writing – original draft, review & editing, funding acquisition. Richard Brooker, Simon Kohn, and Oliver Lord: Writing – original draft, IR data processing, data review & editing, supervision, funding acquisition.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Communicated by Dante Canil.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gregson, C., Brooker, R., Kohn, S. et al. Thermodynamic and kinetic controls on phase stability and incorporation of water in larnite (β-Ca2SiO4): implications for calcium silicate inclusions in diamonds. Contrib Mineral Petrol 179, 82 (2024). https://doi.org/10.1007/s00410-024-02153-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-024-02153-w