
Abstract
Respiratory virus infections leads to coughing, sneezing, and increases in reflex parasympathetic bronchoconstriction and secretions. These responses to viral infection are exclusively or largely secondary to changes in the function of the nervous system. For many with underlying airway pathologies such as asthma and COPD, this neuroplasticity can lead to disease exacerbations and hospitalization. Relatively little is understood about the cellular and molecular mechanisms that underlie the changes in neuronal control of the respiratory tract during viral infection, but the evidence supports the idea that changes occur in the physiology of both the sensory and autonomic innervation. Virus infection can lead to acute increases in the activity of sensory nerves as well as to genetic changes causing alterations in sensory nerve phenotype. In addition, respiratory viral infections are associated with changes in the control of neurotransmitter release from cholinergic nerve endings terminating at the level of the airway smooth muscle.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The immune system is effective in clearing most respiratory viral infections within a period of a few weeks. In order to avoid the immune system, viruses have evolved mechanisms to escape the respiratory tract of the host where it can then go on to infect another person. This has been accomplished by causing profound alterations in airway neuronal control, leading to coughing, sneezing, and increases in reflex parasympathetic mucus secretions. The increase in parasympathetic drive can also lead to reflex bronchoconstriction. In most healthy subjects, these symptoms can be trying but are self-limiting and often little more than an inconvenience. In some cases, the symptoms such as urge to cough can persist into a chronic or sub acute state [1, 2]. For those suffering with underlying airway pathologies such as asthma or COPD, the changes in neural control may contribute to acute exacerbations of the disease [1, 3, 4].
Experimental viral infection in laboratory animals [5–11] and in human volunteers [12–15] leads to increases in cough and also increases in bronchial responsiveness to stimuli such as methacholine and histamine. The increase in cough associated with virus infection is consistent with alterations in the sensory nerve function in the airways. The increase in bronchial responsiveness involves multiple mechanisms but changes in both afferent (sensory) and efferent (parasympathetic) nerve function are likely involved.
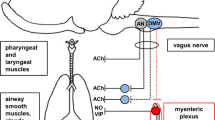
The hyperresponsiveness associated with airway inflammation in laboratory animals typically requires an intact vagal nervous system. The classical allergen-induced airway hyperresponsiveness to methacholine in mice is entirely prevented in animals in which either the vagus nerves are cut or if only the C-fiber neurons are depleted from the vagal sensory ganglia [16, 17]. Likewise, virus-induced airway hyperresponsiveness of guinea pigs to histamine challenge also requires intact vagus nerves [6, 18]. This indicates that the hyperresponsiveness to histamine and methacholine in these studies is actually a bronchial hyper-neuronal responsiveness (Fig. 1).

A schematic highlighting the potential effects of viral infection on vagus nerve activity. (I) Viral infection of epithelial cells can lead to release of mediators that stimulate action potential discharge in afferent nerves, thereby alerting the central nervous system. Viral infection can also lead to the production and release of neurotrophic factors that can (II) influence gene expression in the vagal jugular and nodose ganglia in a manner that can lead to relevant phenotypic changes in the airway sensory nerves. (III) Action potentials arriving in the brainstem are integrated, ultimately leading to sensations and to (IV) increases in preganglionic parasympathetic drive. (V) This will increase neurotransmission at the neuro-effector junctions, causing bronchoconstriction and mucus secretion; effects further amplified by inhibition of inhibitory muscarinic autoreceptors. See text for references
In human volunteers, experimental viral infections consistently lead to an increase in the bronchial responsiveness to histamine [12, 14]. Histamine effectively sensitizes vagal afferent C-fibers in a manner that would likely lead to increased reflex parasympathetic drive [19]. Indeed, the increase in response to histamine following respiratory viral infection was abolished by cholinergic muscarinic receptor antagonism [12]. Virus infection of rats and guinea pigs can also augment the cholinergic nerve evoked bronchoconstriction, even when the sensory input to the brainstem is bypassed [6, 7, 9]. Considered together, the evidence for increases in urge to cough and bronchial vagal responsiveness associated with viral infections likely involves neural plasticity within both the sensory and autonomic systems. The sensory and autonomic innervation of the respiratory tract has recently been extensively reviewed [20, 21].
Sensory Nerves
Sensory nerves innervating the airways are responsible for initiating respiratory sensations and reflexes, including coughing and sneezing. In addition by regulating parasympathetic reflex tone, sensory nerves play a major role in controlling mucus secretion and airway caliber. The majority of sensory nerve fibers that project to airways are derived from cell bodies located in the vagal jugular or nodose ganglia. The vagal C-fibers are relatively insensitive to the physiological milieu but respond vigorously to noxious stimuli and to mediators associated with inflammation [22, 23]. As Sherrington said of nociceptors in the somatosensory system they are equipped to provide the organ, “with a so to say sense of its own potential injury” [24]. In addition to nociceptive C-fibers, low-threshold mechanosensory sensory nerves innervate the lower respiratory tract that can be activated in a rapidly or slowly adapting fashion by the mechanical forces of respiration. These nerves are generally less susceptible to inflammatory-associated stimuli [22, 23].
Respiratory viruses commonly infect and replicate in airway epithelial cells leading to local inflammatory reactions. The inflammation is associated with the production of a host of inflammatory mediators, many of which can lead to the acute activation and sensitization of nociceptive terminals that reside within the epithelial layer [20]. This acute activation (action potential discharge) of sensory nerves can potentially lead to coughing, sneezing and parasympathetic driven bronchial smooth muscle contraction and secretions. These responses to viral infection would be expected to terminate as the infection and inflammation subside. Longer lasting effects of a viral infection may involve phenotypic changes to the nerves.
Airway inflammation not only activates airway C-fibers but also can phenotypically switch the low-threshold mechanosensitive A-fibers such that they take on characteristics of nociceptive C-fibers [25–29]. Induction of genes that are normally not expressed by a cell has been defined as “phenotypic switching.” In the somatosensory system, inflammation is associated with a phenotypic switch of low-threshold mechanosensitive Aβ-fibers that respond to light touch such that they begin to express the tachykinin substance P. Light touch of the inflamed skin can then lead to tachykinergic transmission in the spinal cord, altering the communication between the skin and central nervous system [30]. Large A-fiber neurons in the trachea and lungs typically do not express the genes for the synthesis of neuropeptides like substance P and calcitonin gene-related peptide (CGRP). In the presence of allergic inflammation of guinea pig airways, these neurons begin to express both substance P and CGRP [26, 28]. Likewise, four days after a respiratory viral infection substance P is synthesized by large Aδ cough receptors that terminate in the guinea pig trachea [31]. In addition to changing the neurotransmitters that A-fiber neurons produce, allergic inflammation also induces the synthesis of key ion channels involved in nerve activation. For example, during allergic airway inflammation transient receptor potential cation channel subfamily V member 1 (TRPV1) is turned on in rapidly and slowly adapting mechanosensitive A-fibers in rat lungs such that they become responsive to capsaicin [29]. Allergic inflammation also leads to de novo production of functional TRPV1 channels in tracheal Aδ cough receptors in the guinea pig trachea [32]. This is relevant because TRPV1 is not only the receptor for capsaicin but also a pivotal channel in the activation mechanisms of many disparate stimuli including heat, acid, and certain inflammatory mediators [33, 34]. We have now found that viral infection with intranasal parainfluenza 3 (PIV3) leads to TRPV1 induction in trachea Aδ cough receptors in guinea pig trachea. In vehicle treated animals, ~16 % of the tracheal-specific nodose neurons expressed TRPV1, whereas 4 days after PIV3 infection, ~50 % of the neurons expressed TRPV1. The most likely mechanism by which a local infection in the airway epithelium leads to changes in gene expression in the cell bodies involves the production of neurotrophic factors.
Neurotrophic factors can be produced during inflammatory reactions, including those associated with viral infections [35]. The neurotrophin family of neurotrophic factors comprise nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT 3), and neurotrophin 4 (NT 4) [36]. Unlike other inflammatory mediators, these factors can interact with high affinity receptors on nerve terminals in a manner that leads to changes in gene transcription in cell bodies situated within the distant sensory ganglia [37, 38]. Which neurotrophin is most likely to interact with airway sensory nerves depends on the nature of the neurotrophin receptors the nerve expresses. Although there is some overlap, NGF interacts selectively with the receptor TRKA, BDNF and neurotrophin-4 (NT4) interact with the receptor TRKB, and NT3 interacts with TRKC [36]. The neurons undergoing the phenotypic changes (increase in TRPV1 and neuropeptide gene expression) after a viral infection were the tracheal nodose Aδ cough receptors. We have previously reported that these nerves nearly uniformly express TRKB, whereas only a minority of the nodose tracheal Aδ neurons expressed TRKA or TRKC [32]. Moreover, applying exogenous BDNF to the trachea mimics the effect to virus infection with respect to induction of TRPV1 expression in A-fiber neurons [27]. We perfused guinea pig isolated trachea with PIV3 and found that already within 8 h, there was nearly tenfold increase in BDNF mRNA in the tissue (P < 0.05). It is therefore possible that the phenotypic changes in nodose A-fiber neurons may be secondary to TRKB activation by BDNF or NT4.
Evaluation of cough sensitivity is a reasonable monitor of sensory nerve function in the whole animal. Ye et al. noted nearly a fourfold increase in the cough responses to capsaicin in guinea pigs that had previously been inoculated intranasally with PIV3 [39]. We too have found that PIV3 infection of the guinea pig airway epithelium leads to two to three times more coughs than those treated with the viral growth medium in response to capsaicin, BK, and citric acid. Capsaicin stimulation of sensory nerves depends entirely on TRPV1, whereas bradykinin and acid stimulation depend in part on TRPV1 [40–42]. These results support the hypothesis that the virus-induced TRPV1 and neuropeptide gene expression by the vagal sensory neurons may be physiologically relevant.
Sensory nerve stimulation will lead to increases in neurotransmission in the brainstem where the integrated signals can lead to sensations (urge to cough and dyspnea) and to subconscious increases autonomic parasympathetic drive. In addition, activation of vagal C-fibers can lead to the local release of neuropeptides in the tissue in a manner that can augment the local inflammatory response. Respiratory viral infections have been associated with an augmentation of the sensory neurogenic inflammatory system [43, 44].
Parasympathetic Nerves
Experimental viral infection can augment the vagal nerve mediated bronchoconstriction independently of the sensory nerves. This can be shown by evaluating the parasympathetic nerve response after severing the vagus nerve and stimulating the peripheral cut end. In this design, the vagal sensory nerves cannot communicate with the CNS and lead to reflex parasympathetic drive. When the peripheral end of the cut nerve is stimulated, action potentials are evoked, which synapse with nicotinic receptors in the local airway ganglia. This leads to activation of the postganglionic nerve and stimulation of acetylcholine release onto muscarinic receptors that causes rapid smooth muscle contraction and airway narrowing [21]. Four or 5 days following a respiratory viral infection in guinea pigs and rats, the bronchoconstriction following vagus nerve stimulation is substantially enhanced [6, 7, 9]. This may involve increases in synaptic transmission within the ganglia, increases in release of acetylcholine from the postganglionic terminals, and/or increases in the response of the muscle to the released acetylcholine. With respect to the latter possibility, there is little evidence that viral infection increases the response to exogenously applied acetylcholine at the level of the smooth muscle [7]. However, there is good evidence that viral infections can increase the amount of acetylcholine released from the postganglionic nerve terminals. A key mechanism that contributes to the effect is a reduction in the activity of prejunctional muscarinic M2 receptors [7, 9, 11, 45]. Muscarinic M2 receptors are expressed in the postganglionic nerve terminal membrane where they serve as feedback inhibitory autoreceptors. When the acetylcholine release is increased, it acts on these terminal M2 receptors leading to a reduction in the release of acetylcholine per impulse [46]. Following viral infections, this inhibitory feedback mechanism is inhibited in guinea pig and rat airways [9, 10]. The virally mediated decrease in the cholinergic inhibitory feedback loop is associated with cytokine mediated decreases in M2 receptor gene expression in neurons within the airway parasympathetic ganglia [45].
Conclusions
In summary, it is likely that respiratory viral infections can lead to airway inflammation and the production of inflammatory mediators that would be expected to stimulate nociceptive C-fibers. In addition, viral infection may lead to changes in gene expression within the sensory cell bodies causing phenotypic changes in a manner that is relevant to their neurotransmission (neuropeptide production) and activation profile (e.g., TRP channel expression). The phenotypic changes in sensory nerves following viral infection is likely due to increases in the production of neurotrophic factors by the epithelial cells and may persist beyond the period of active infection. Viral infection may also alter the function of parasympathetic nerves to further increase the reflex bronchoconstriction and secretions. The net effect of this sensory and autonomic neuroplasticity is an increase in the classical neuronal symptoms such as sneezing, coughing, reflex secretions, and bronchoconstriction. For those with underlying airway disorders, the viral-associated neuromodulation likely contributes to disease exacerbations. A better understanding of the molecular mechanisms underlying this neuroplasticity may lead to novel therapeutic approaches aimed at decrease viral-associated airways disorders.
References
Footitt J, Johnston SL (2009) Cough and viruses in airways disease: mechanisms. Pulm Pharmacol Ther 22(2):108–113
McGarvey LP, Nishino T (2004) Acute and chronic cough. Pulm Pharmacol Ther 17(6):351–354. doi:10.1016/j.pupt.2004.09.017
Dulek DE, Peebles RS Jr (1810) Viruses and asthma. Biochim Biophys Acta 11:1080–1090. doi:10.1016/j.bbagen.2011.01.012
Sethi S (2011) Molecular diagnosis of respiratory tract infection in acute exacerbations of chronic obstructive pulmonary disease. Clin Infect Dis 52(Suppl 4):S290–295. doi:10.1093/cid/cir044
Broadley KJ, Blair AE, Kidd EJ, Bugert JJ, Ford WR (2010) Bradykinin-induced lung inflammation and bronchoconstriction: role in parainfluenze-3 virus-induced inflammation and airway hyperreactivity. J Pharmacol Exp Ther 335(3):681–692. doi:10.1124/jpet.110.171876
Buckner CK, Songsiridej V, Dick EC, Busse WW (1985) In vivo and in vitro studies on the use of the guinea pig as a model for virus-provoked airway hyperreactivity. Am Rev Respir Dis 132(2):305–310
Fryer AD, Jacoby DB (1991) Parainfluenza virus infection damages inhibitory M2 muscarinic receptors on pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol 102(1):267–271
Elwood W, Lotvall JO, Barnes PJ, Chung KF (1993) Airway hyperresponsiveness to acetylcholine and to tachykinins after respiratory virus infection in the guinea pig. Ann Allergy 70(3):231–236
Sorkness R, Clough JJ, Castleman WL, Lemanske RF Jr (1994) Virus-induced airway obstruction and parasympathetic hyperresponsiveness in adult rats. Am J Respir Crit Care Med 150(1):28–34. doi:10.1164/ajrccm.150.1.8025764
Lee AM, Fryer AD, van Rooijen N, Jacoby DB (2004) Role of macrophages in virus-induced airway hyperresponsiveness and neuronal M2 muscarinic receptor dysfunction. Am J Physiol Lung Cell Mol Physiol 286(6):L1255–1259. doi:10.1152/ajplung.00451.2003
Nie Z, Scott GD, Weis PD, Itakura A, Fryer AD, Jacoby DB (2011) Role of TNF-alpha in virus-induced airway hyperresponsiveness and neuronal M(2) muscarinic receptor dysfunction. Br J Pharmacol 164(2b):444–452. doi:10.1111/j.1476-5381.2011.01393.x
Laitinen LA, Elkin RB, Empey DW, Jacobs L, Mills J, Nadel JA (1991) Bronchial hyperresponsiveness in normal subjects during attenuated influenza virus infection. Am Rev Respir Dis 143(2):358–361
Grunberg K, Kuijpers EA, de Klerk EP, de Gouw HW, Kroes AC, Dick EC, Sterk PJ (1997) Effects of experimental rhinovirus 16 infection on airway hyperresponsiveness to bradykinin in asthmatic subjects in vivo. Am J Respir Crit Care Med 155(3):833–838. doi:10.1164/ajrccm.155.3.9117013
Grunberg K, Timmers MC, Smits HH, de Klerk EP, Dick EC, Spaan WJ, Hiemstra PS, Sterk PJ (1997) Effect of experimental rhinovirus 16 colds on airway hyperresponsiveness to histamine and interleukin-8 in nasal lavage in asthmatic subjects in vivo. Clin Exp Allergy 27(1):36–45
Curley FJ, Irwin RS, Pratter MR, Stivers DH, Doern GV, Vernaglia PA, Larkin AB, Baker SP (1988) Cough and the common cold. Am Rev Respir Dis 138(2):305–311. doi:10.1164/ajrccm/138.2.305
McAlexander MA, Gavett SH, Kollarik M, Undem BJ (2015) Vagotomy reverses established allergen-induced airway hyperreactivity to methacholine in the mouse. Respir Physiol Neurobiol 212–214:20–24. doi:10.1016/j.resp.2015.03.007
Trankner D, Hahne N, Sugino K, Hoon MA, Zuker C (2014) Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proc Natl Acad Sci USA 111(31):11515–11520. doi:10.1073/pnas.1411032111
Costello RW, Evans CM, Yost BL, Belmonte KE, Gleich GJ, Jacoby DB, Fryer AD (1999) Antigen-induced hyperreactivity to histamine: role of the vagus nerves and eosinophils. Am J Physiol 276(5 Pt 1):L709–714
Lee LYMR (1993) Histamine enhances vagal pulmonary C-fiber responses to capsaicin and lung inflation. Respir Physiol 93(1):83–96
Lee LY, Yu J (2014) Sensory nerves in lung and airways. Compr Physiol 4(1):287–324. doi:10.1002/cphy.c130020
Undem BJ, Potenzieri C (2012) Autonomic neural control of intrathoracic airways. Compr Physiol 2(2):1241–1267. doi:10.1002/cphy.c110032
Lee L-Y, Undem BJ (2005) Bronchopulmonary vagal afferent nerves. Advances in vagal afferent neurobiology. CRC Taylor and Francis, Boca Raton
Coleridge HMCJ (1977) Impulse activity in afferent vagal C-fibres with endings in the intrapulmonary airways of dogs. Respir Physiol 29:125–142
Sherrington C (1906) The integrated action of the nervous system, vol 1. Yale University Press, New Haven
Fischer AMG, Saria A, Philippin B, Kummer W (1996) Induction of tachykinin gene and peptide expression in guinea pig nodose primary afferent neurons by allergic airway inflammation. J Clin Invest 98:2284–2291
Chuaychoo BHD, Myers AC, Kollarik M, Undem BJ (2005) Allergen-induced substance P synthesis in large-diameter sensory neurons innervating the lungs. J Allergy Clin Immunol 116(2):325–331
Lieu TUB (2011) Allergen and BDNF-induced TRP channel expression in nodose cough-fiber neurons. FASEB J 25(864):814
Myers ACKR, Undem BJ (2002) Allergic inflammation-induced neuropeptide production in rapidly adapting afferent nerves in guinea pig airways. Am J Physiol Lung Cell Mol Physiol 282:L775–L781
Zhang GLR, Wiggers M, Snow DM, Lee LY (2008) Altered expression of TRPV1 and sensitivity to capsaicin in pulmonary myelinated afferents following chronic airway inflammation in the rat. J Physiol 586:5771–5786
Neumann S, Doubell TP, Leslie T, Woolf CJ (1996) Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature 384(6607):360–364. doi:10.1038/384360a0
Carr MJ, Hunter DD, Jacoby DB, Undem BJ (2002) Expression of tachykinins in nonnociceptive vagal afferent neurons during respiratory viral infection in guinea pigs. Am J Respir Crit Care Med 165(8):1071–1075
Lieu TKM, Myers AC, Undem BJ (2011) Neurotrophin and GDNF family ligand receptor expression in vagal sensory nerve subtypes innervating the adult guinea pig respiratory tract. Am J Physiol Lung Cell Mol Physiol 300(5):L790–L798
Caterina MJJD (2001) The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci 24:487–517
Veldhuis NA, Poole DP, Grace M, McIntyre P, Bunnett NW (2015) The G protein-coupled receptor-transient receptor potential channel axis: molecular insights for targeting disorders of sensation and inflammation. Pharmacol Rev 67(1):36–73. doi:10.1124/pr.114.009555
Tortorolo L, Langer A, Polidori G, Vento G, Stampachiacchere B, Aloe L, Piedimonte G (2005) Neurotrophin overexpression in lower airways of infants with respiratory syncytial virus infection. Am J Respir Crit Care Med 172(2):233–237. doi:10.1164/rccm.200412-1693OC
Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24:677–736. doi:10.1146/annurev.neuro.24.1.677
Chowdary PD, Che DL, Cui B (2012) Neurotrophin signaling via long-distance axonal transport. Annu Rev Phys Chem 63:571–594. doi:10.1146/annurev-physchem-032511-143704
Zweifel LSKR, Ginty DD (2005) Functions and mechanisms of retrograde neurotrophin signalling. Nat Rev Neurosci 22(3):146–152
Ye XM, Zhong NS, Liu CL, Chen RC (2011) Cough reflex sensitivity is increased in guinea pigs with parainfluenza virus infection. Exp Lung Res 37(3):186–194. doi:10.3109/01902148.2010.540768
Carr MJKM, Meeker SN, Undem BJ (2003) A role for TRPV1 in bradykinin-induced excitation of vagal airway afferent nerve terminals. J Pharmacol Exp Ther 304:1275–1279
Kollarik MUB (2002) Mechanisms of acid-induced activation of airway afferent nerve fibres in guinea-pig. J Physiol 543:591
Grace M, Birrell MA, Dubuis E, Maher SA, Belvisi MG (2012) Transient receptor potential channels mediate the tussive response to prostaglandin E2 and bradykinin. Thorax 67(10):891–900. doi:10.1136/thoraxjnl-2011-201443
Tan Y, Yang T, Liu S, Liu H, Xiang Y, Qu F, Li H, Qin X (2008) Infection with respiratory syncytial virus alters peptidergic innervation in the lower airways of guinea-pigs. Exp Physiol 93(12):1284–1291. doi:10.1113/expphysiol.2008.043521
Auais A, Adkins B, Napchan G, Piedimonte G (2003) Immunomodulatory effects of sensory nerves during respiratory syncytial virus infection in rats. Am J Physiol Lung Cell Mol Physiol 285(1):L105–113. doi:10.1152/ajplung.00004.2003
Rynko AE, Fryer AD, Jacoby DB (2014) Interleukin-1beta mediates virus-induced m2 muscarinic receptor dysfunction and airway hyperreactivity. Am J Respir Cell Mol Biol 51(4):494–501. doi:10.1165/rcmb.2014-0009OC
Fryer AD, Maclagan J (1984) Muscarinic inhibitory receptors in pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol 83(4):973–978
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
None.
Rights and permissions
About this article

Cite this article
Zaccone, E.J., Undem, B.J. Airway Vagal Neuroplasticity Associated with Respiratory Viral Infections. Lung 194, 25–29 (2016). https://doi.org/10.1007/s00408-015-9832-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-015-9832-5