
Abstract
Oligodendroglioma, IDH-mutant and 1p/19q-codeleted has highly variable outcomes that are strongly influenced by patient age. The distribution of oligodendroglioma age is non-Gaussian and reportedly bimodal, which motivated our investigation of age-associated molecular alterations that may drive poorer outcomes. We found that elevated HOXD12 expression was associated with both older patient age and shorter survival in the TCGA (FDR < 0.01, FDR = 1e−5) and the CGGA (p = 0.03, p < 1e−3). HOXD12 gene body hypermethylation was associated with older age, higher WHO grade, and shorter survival in the TCGA (p < 1e−6, p < 0.001, p < 1e−3) and with older age and higher WHO grade in Capper et al. (p < 0.002, p = 0.014). In the TCGA, HOXD12 gene body hypermethylation and elevated expression were independently prognostic of NOTCH1 and PIK3CA mutations, loss of 15q, MYC activation, and standard histopathological features. Single-nucleus RNA and ATAC sequencing data showed that HOXD12 activity was elevated in neoplastic tissue, particularly within cycling and OPC-like cells, and was associated with a stem-like phenotype. A pan-HOX DNA methylation analysis revealed an age and survival-associated HOX-high signature that was tightly associated with HOXD12 gene body methylation. Overall, HOXD12 expression and gene body hypermethylation were associated with an older, atypically aggressive subtype of oligodendroglioma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Oligodendroglioma, IDH-mutant and 1p/19q-codeleted is a type of adult-type diffuse glioma molecularly distinct from IDH-mutant astrocytoma and IDH-wildtype glioblastoma [43]. Patients diagnosed with oligodendroglioma generally have favorable outcomes compared to other adult-type diffuse gliomas, but individual patient outcomes are highly variable, with some patients living less than a year and others living decades [2, 11, 23, 30]. Chemoradiation, considered the standard of care treatment for oligodendrogliomas, is associated with short-term and long-term functional and cognitive toxicities [89]. Considering the toxicity of this non-curative treatment and the relatively favorable prognosis of oligodendroglioma, current guidelines offer varying definitions of high-risk tumors, which results in situations where it is unclear whether a patient should receive postoperative chemoradiation or whether IDH inhibitors, chemotherapy, or observation are more appropriate [49, 51, 83, 89]. For example, the American Society for Radiation Oncology (ASTRO) recommends all patients over age 40 should be treated with chemoradiation [31]; however, several other guidelines support delaying chemoradiation until disease progression based on patient and provider preference [51, 52, 83]. Patient-specific risk indicators are needed to identify patients with abnormally aggressive disease for whom the delay of chemoradiation would not be appropriate.
Oligodendroglioma age has been reported to be prognostic and bimodally distributed [17]. Based on previous studies of IDH-wildtype glioblastoma, we hypothesized that older oligodendroglioma patients may have molecular signatures indicative of poor survival [16, 58]. To date, only a few prognostic molecular biomarkers have been proposed (including PIK3CA mutations [74], NOTCH1 mutations [2, 30], CDKN2A homozygous deletion [3], loss of 15q [30], and MYC activation [38]) to complement known clinical factors associated with survival such as age, KPS, World Health Organization (WHO) grade, and extent of resection [17, 26]. In this study, we sought to discover and validate novel age-associated alterations that characterize aggressive oligodendroglioma. These age-related molecular features may reflect a more aggressive subtype of oligodendroglioma that occurs disproportionately in older patients. To interrogate this hypothesis, we investigated oligodendroglioma data across multiple cohorts, including those published by The Cancer Genome Atlas (TCGA), the Chinese Glioma Genome Atlas (CGGA), the Glioma Longitudinal AnalySiS consortium (GLASS), and Capper et al. along with single-nucleus sequencing data published by Blanco-Carmona et al. and Wang et al. to identify, validate, and characterize prognostically relevant genes [10, 13, 19, 82, 91].
Materials and methods
The cancer genome atlas (TCGA) molecular and clinical data
Precomputed DNA methylation beta values (Illumina 450 K) from 171 primary oligodendroglioma, IDH-mutant and 1p/19q-codeleted tumors from TCGA were downloaded from University of California Santa Cruz (UCSC) Xena (https://xena.ucsc.edu/) [28]. RNA-seq gene counts for TCGA oligodendroglioma were downloaded from the National Cancer Institute’s Genomic Data Commons (GDC) Data Portal (https://gdc.cancer.gov/) (N = 170), and TCGA transcript per million data (TPM) were computed from the recount2 dataset [18] (N = 169), as specified by Arora et al. [4] to be consistent with available Chinese Glioma Genome Atlas (CGGA) TPM data. The mutational status of NOTCH1 and PIK3CA and somatic copy number alteration (SCNA) status of CDKN2A and chromosome arm 15q were determined from TCGA somatic mutation calls and SCNA GISTIC calls as previously described [56, 57]. Chromosome 15q loss was called if more than half of the genes on 15q were lost (Supplemental Fig. 1). CDKN2A homozygous deletions were called if CDKN2A had a GISTIC score of -2 in at least two of three versions of TCGA SCNA calls [56]. MYC activation was called as described in Kamoun et al. [38]. Characteristics including age, central nervous system (CNS) WHO grade, and overall survival were also downloaded from UCSC Xena, and chemotherapy and radiation therapy treatment details (postoperative_rx_tx, radiation_therapy) were downloaded from the GDC Data Portal. Treatment was considered adjuvant if it was labeled “ADJUVANT” in the metadata field “therapy_regimen” or was administered within 8 months of diagnosis (“pharmaceutical_tx_started_days_to”) and was the first treatment administered (Supplemental Fig. 2). Drug names were ascertained from the metadata field “pharmaceutical_therapy_drug_name.” Patients who received any adjuvant combination of procarbazine, lomustine (CCNU), and vincristine were considered treated by PCV, with the exception of EORTC 26951 Phase III trial data, where PCV refers to treatment with procarbazine, CCNU, and vincristine. TCGA IDH-mutational status and 1p/19q codeletion data were ascertained as previously described [56].
TCGA histopathologic features
TCGA pathology reports for 156 oligodendroglioma patients were downloaded from the GDC Legacy Archive. Values for the Ki-67 proliferative index, presence of necrosis, presence of microvascular proliferation, and presence of mitotic figures were recorded. When the Ki-67 proliferative index was given as a range, we reported the upper bound. Ki-67 proliferative index values were considered high (≥ 10%) or low (< 10%) based on published reports [88]. We considered ≥ 6 mitoses per 10 high power fields (HPF) as high and < 6 mitoses per 10 HPF as low in line with WHO guidelines for tumor grading [44].
TCGA radiographic features
The Multimodal Brain Tumor Segmentation Challenge (BraTS) 2020 training and validation datasets provide pre-operative, multimodal MRI tumor scans for 27 oligodendroglioma patients in the TCGA [6, 50]. Ground-truth segmentations delineating the boundaries of tumor edema, enhancement, and necrotic/non-enhancing tissue were available for 13 oligodendrogliomas. We used a segmentation machine learning model trained on the BraTS training set to assign segmentations to the remaining 14 oligodendrogliomas followed by manual correction [54, 55]. These segmentation maps were used to calculate the volume of each tumor compartment, which was then binarized as high or low (Supplemental Fig. 3).
As described in Bakas et al. [6], all multimodal MRI volumes contained pre- and post-contrast T1-weighted sequences, T2-weighted sequences, and T2 Fluid-Attenuated Inversion Recovery (FLAIR) sequences that were co-registered to the same anatomic atlas using affine registration provided by the Brain Linear Image Registration Tool (FLIRT) from the Functional Magnetic Resonance Imaging of the Brain Software Library (FSL) developed by the Oxford Center for Functional MRI [6, 34,35,36, 70, 85]. All MRI volumes were subsequently resampled to 1 mm3 isotropic voxel resolution and skull stripped by the Brain Extraction Tool (BET) from FSL or by Multi-Atlas Skull-Stripping [21] as described in Bakas et al. [6]. Resampling MRI volumes to the same resolution made direct comparisons of tumor volumes valid.
Chinese glioma genome atlas (CGGA) dataset
Raw RNA gene counts for 48 CGGA [91] primary oligodendroglioma tumors were downloaded from recount2 [18] and used to compute TPM values [4]. Unsupervised dimension reduction [79] of CGGA and TCGA TPM data identified four CGGA oligodendroglioma outliers; these patients were excluded from our gene expression analysis (Supplemental Fig. 4). Clinical variables including age (N = 48), tumor CNS WHO grade (N = 48), and overall survival (N = 46) were deduced from published annotations [91].
Capper et al. dataset
DNA methylation IDAT files from the Illumina 450 K platform for 170 oligodendroglioma tumors (119 primary) made available by the authors of Capper et al. [13] were downloaded from the NCBI Gene Expression Omnibus (GEO) under accession number GSE109381. Beta values were determined using the R package minfi [5]. Patient age for tumors (N = 160 total; N = 111 primary) and WHO tumor grade (N = 80 total, N = 61 primary) were determined from published clinical variables. Outcome data were not available. Tumors were considered oligodendroglioma if the classification of the version 11 Capper et al. random forest classifier was oligodendroglioma (O_IDH or O IDH) or if their WHO 2016 classification was Anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted or Oligodendroglioma, IDH-mutant and 1p/19q-codeleted. For age and WHO grade analyses, we limited our analyses on this dataset to primary tumors classified as oligodendroglioma by methylation classification and by WHO 2021 diagnostic criteria (N = 113).
The glioma longitudinal analysis (GLASS) consortium dataset
The Synapse API was used to download gene expression TPM data (Primary N = 9; Total 19), DNA methylation beta values from the merged Illumina 450 K and EPIC platforms (beta.merged) (Primary N = 10; Total N = 24), as well as clinical variables age (N = 34), WHO grade (N = 34), and overall survival (N = 34) for oligodendroglioma tumors in the Glioma Longitudinal AnalySiS (GLASS) cohort (Data Release version 2021-11-16) [7, 19, 80]. GLASS tumors were determined to be oligodendrogliomas if the following conditions were met in the metadata for the patient’s primary tumor: "idh_status" was “IDHmut” and "codel_status" was "codel" or "idh_codel_subtype" was "IDHmut-codel." For our age and WHO grade analyses, we restricted our analyses to primary tumors whose "idh_codel_subtype" was "IDHmut-codel." All GLASS samples that appear in the TCGA were excluded.
Jonsson et al. dataset
Published clinical variables, including overall survival and age for 84 primary oligodendroglioma tumors were downloaded from cBioPortal [15, 25, 37].
Oligo nation
We downloaded metadata for 34 primary oligodendrogliomas with patient age annotations from the Oligo Nation cohort from PedcBioPortal (Data accessed 2024–02–06).
EORTC 26951 phase III clinical trial
DNA methylation data and metadata from 115 WHO grade 3 diffuse gliomas made available by the authors of van den Bent, et al. [78] were downloaded from the NCBI GEO under accession number GSE48462. Patients were filtered to include only those who (1) harbored 1p/19q codeletions (according to 2021 WHO CNS tumor classification guidelines), (2) were treated by radiation therapy and procarbazine, CCNU, and vincristine, and (3) were profiled by Illumina 450 k arrays, which contain all HOXD12 gene body probes we analyzed. After subsetting this cohort, only 10 tumors remained.
Bulk gene RNA expression analysis
Differential gene expression analysis for bulk RNA-seq data was conducted using the R package DESeq2 [45]. Genes shown in a volcano plot were limited to the genes that have raw gene counts data and TPM data available. When testing for gene expression prognostic implications during unbiased searches, we compared the outcomes of patients whose gene expression was above the median (elevated) to patients whose gene expression was equal or less than the median (low) based on the TPM values for the gene in question. Median HOXD12 expression was zero in the TCGA; thus, we considered HOXD12 expression elevated (HOXD12-postive) when it was non-zero and low otherwise (HOXD12-negative). Gene ontology analysis and gene set enrichment analysis were performed using the R package clusterProfiler [62] (R version 4.1.1).
HOXD12-specific methylation analysis
We used public annotations to determine DNA methylation probe genomic regions (5′UTR, TSS -1500, TSS -200, gene body, or 3′UTR) [9]. The mean value of the beta values of the three probes (cg23130254, cg03964958, cg03371669) in HOXD12’s gene body was used to determine HOXD12 hypermethylation. We determined a threshold by taking the mean value of this signature for patients with HOXD12-negative expression status (0.3034) and patients with HOXD12-positive expression status (0.4121) in the TCGA. Therefore, if a patient’s three gene body probe average exceeded this threshold (0.3577), they were considered HOXD12 hypermethylated; otherwise, we considered that patient HOXD12 hypomethylated.
Blanco–Carmona et al. single cell dataset
We downloaded paired single-nucleus RNA sequencing (snRNA-seq) and single-nucleus ATAC sequencing (snATAC-seq) data for 7817 nuclei from the NCBI Gene Expression Omnibus (GEO) under accession number GSE205771 [10]. These data were available pre-processed in the Seurat R object “GSM6261343_oligodendroglioma_atac_seurat_object.rds.gz” [32, 72]. We used the precomputed UMAP embedding, and Seurat clusters published for these data, but we repeated the quality control described in Blanco-Carmona et al., which excluded 88 cells (N = 7729). We also recomputed cell types and tumor cell states and found results that differed from the published annotations. Specifically, the somatic copy number alterations (SCNAs) called by the R package inferCNV version 1.18.1 suggested that many of the nuclei reported as neoplastic by Blanco-Carmona et al. lacked 1p/19q codeletions [75]. Therefore, we used our inferCNV results for chromosome arms 1p and 19q to determine neoplastic nuclei, as described below.
Wang et al. single cell dataset
Single-nucleus ATAC sequencing data (matrix, barcodes, peaks) for 10,732 nuclei were downloaded for two oligodendroglioma tumors—SF11612 (N = 5519) and SF11949 (N = 5213)—from the NCBI Gene Expression Omnibus (GEO) under accession number GSE138794 [82]. Quality control was performed separately on each tumor, yielding 10,281 nuclei (SF11612 N = 5323; SF11949 N = 4958). Single-nucleus RNA sequencing data was estimated using Signac’s function GeneActivity [73]. We then used the function merge to merge these datasets. Next, we normalized the merged, estimated snRNA-seq data using Seurat’s SCTransform. After this, we conducted dimension reduction using RunHarmony with 30 components. Following this, we used the functions RunUMAP, FindNeighbors, and FindClusters. These clusters were assigned non-neoplastic cell type and neoplastic cell state labels using SCNA calls from inferCNV and published gene markers as described below.
Computational analysis of single cell datasets
As a first pass, gene markers were used to assign cell types. RBFOX3 expression was used to identify neurons, MBP, MOG, and CNP expression were used to identify oligodendrocytes, SLC1A2, GFAP, AQP4, and ALDH1L1 expression were used to identify astrocytes, and PTPRC, CD163, and CD14 expression were used to identity microglia [76]. SOX6 and MAP2 expression were expected to be elevated in neoplastic nuclei, and FLT1 expression was expected to be elevated in non-neoplastic nuclei compared to neoplastic nuclei. To more definitively identify tumor cells, we iteratively computed inferred SCNAs using the R package inferCNV version 1.18.1 with meta-cells, first using suspected non-neoplastic clusters as controls, which we updated after identifying 1p/19q codeletions in the output of inferCNV. Nuclei in clusters that showed universal loss of chromosome 1p and 19q and intact 1q and 19p were considered neoplastic. Clusters showing no evidence of 1p/19q codeletions were labeled with the cell type previously assigned by cell markers. Nuclei in clusters where the presence or absence of 1p/19q codeletions was not universal were excluded. We recomputed neoplastic cells for Blanco-Carmona et al. because we found instances where the published neoplastic nuclei labels and our inferCNV results contradicted each other.
We next assigned neoplastic nuclei to one of four metaprograms (oligodendrocyte progenitor cell (OPC)-like, Astro-like, Cycling, and ribosomal-enriched (RE)) as described in Blanco-Carmona et al. using the permutation test the authors detailed in their study [10]. We refer to these metaprograms as cell states. In our permutation test, we performed 100,000 permutations, which yielded a minimum possible p-value of 1−e5. For each nucleus, we computed a score for each cell state and an associated adjusted p-value (FDR) from our permutation test. We assigned each nucleus the cell state with the lowest FDR if the lowest FDR was less than a threshold of 0.0125; otherwise, we assigned the nucleus the cell state “gradient.” The threshold 0.0125 represented 0.05 divided by the number of cell states we considered. We calculated stemness scores and lineage scores for AC and OC exactly as described in Tirosh et al. [76].
Pan-HOX methylation analysis
We pooled all TCGA (N = 171), Capper et al. (N = 170), and GLASS (N = 24) DNA methylation samples and subsetted the pooled dataset to gene body probes associated with genes in the HOXA, HOXB, HOXC, and HOXD loci. DNA methylation probe gene locations were deduced from Illumina 450 k metadata downloaded from USCS Xena and Illumina’s website. We found a total of 422 HOX gene body probes common between the TCGA, Capper et al., and GLASS cohorts. We batch-corrected samples in this pooled dataset using ComBat-seq to control for dataset source [90]. We subsequently trained 1000 UMAP models with different initializations and observed a clear, consistent separation of samples into two separated clusters. We excluded samples (N = 20) that switched clusters more than 10% of the time. Sample cluster membership was used to perform differential expression and differential DNA methylation. To compare the differential DNA methylation results of each HOX loci and individual HOX genes, we averaged the results of the probes associated with each HOX loci and each HOX gene.
Statistical analysis
The Shapiro–Wilk test was used to test distribution normality. Hartigans' dip test was used to test distributions for unimodality. The log-rank test was used to test for univariate survival-association. We used Fisher’s exact test (Fisher’s) to assess statistical differences in binary data across two groups. Unless otherwise noted, we used the two-tailed Student's t-test when comparing groups of continuous values, except for patient age and Ki-67 proliferation index, for which we used the Mann-Whitney U Test due to concerns over distribution normality. The Pearson correlation coefficient was used to assess correlation. Cox’s proportional hazard (CPH) regression was used to compute hazard ratios (HR) and for multivariate survival analysis. Covariates whose variance inflation factor (VIF) indicated collinearity (VIF > 10) were omitted from CPH regression. The R package rms was used to construct a nomogram. Statistical analyses were performed in Python version 3.8.6 or R version 4.0.5 unless otherwise specified.
Results
To study age and survival-associated molecular features of oligodendroglioma, we consulted 550 oligodendroglioma tumors from seven publicly available cohorts, including the TCGA, Chinese Glioma Genome Atlas (CGGA), Capper et al., Glioma Longitudinal AnalySiS Consortium (GLASS), Jonsson et al., Oligo Nation, and the EORTC 26951 Phase III clinical trial [7, 12, 13, 37, 41, 68, 77]. Patient age (N = 481), overall survival (N = 342), and bulk RNA-sequencing data (N = 232) from the TCGA, CGGA, and GLASS were used to nominate a molecular marker associated with an older, more aggressive subset of oligodendroglioma. The DNA methylation profile of this marker was subsequently analyzed in a group of 364 tumors from the TCGA, Capper et al., and GLASS cohorts and, along with RNA-sequencing data, subjected to association tests with features in 156 TCGA pathology reports and 27 pre-operative multimodal magnetic resonance imaging volumes. To isolate our analyses to neoplastic cells and interrogate proliferating phenotypes, we used single-nucleus RNA sequencing from 7817 nuclei and single-nucleus ATAC-sequencing data from 18,549 nuclei published by Blanco-Carmona et al. and Wang et al. [10, 82]. All data analyzed in this study are summarized in Supplemental Table 1.
HOXD12 expression is age-associated and prognostic in oligodendroglioma
Contrary to most cancer types, the age distribution of oligodendroglioma patients has been reported to appear bimodal, possibly indicating that certain age ranges are associated with different tumor biology [17, 64]. Although we were unable to statistically judge whether oligodendroglioma patient age distributions were multimodal in our data (Hartigans' dip test), we did find many cohorts exhibited a non-Gaussian distribution. Unlike IDH-wildtype glioblastoma and IDH-mutant astrocytoma adult-type diffuse gliomas in the TCGA, patient age in TCGA oligodendrogliomas was not normally distributed (p < 0.04, Shapiro–Wilk) (Supplemental Fig. 5). In addition to the TCGA, oligodendroglioma age was significantly or marginally significantly non-Gaussian distributed among primary tumors in cohorts published by Jonsson et al. (N = 84, p = 0.01, Shapiro–Wilk), Capper et al. (N = 111, p = 0.048), the CGGA (N = 48, p = 0.06), the GLASS Consortium (N = 31, p = 0.08), and Oligo Nation (N = 34, p = 0.09) (Fig. 1a) [7, 13, 19, 37, 80, 91]. In addition, all non-TCGA datasets were enriched for younger, longer surviving patients compared to the TCGA, suggesting that older patients may be underrepresented and obscure a clearer bimodal distribution in these datasets (Fig. 1b). Given that age is prognostic and non-Gaussian distributed in oligodendroglioma, we hypothesized that a distinctive, aggressive subtype of oligodendroglioma that occurs at older ages may exist.

Investigation of the non-Gaussian age distribution in oligodendroglioma identifies HOXD12 expression as age and survival associated. a The distribution of oligodendroglioma patient age was significantly and marginally significantly non-Gaussian in six independent oligodendroglioma cohorts. b Older age was associated with worse outcomes in cohorts with survival data. c Unbiased differential gene expression analysis between older and younger patients returned 16 significantly differentially expressed genes, including several genes in the HOX, GATA, and keratin gene families. d, e Of the four differentially expressed genes that also stratified TCGA oligodendroglioma survival, HOXD12 had the strongest association with poor outcomes. f In addition to the TCGA, older oligodendroglioma patients were also enriched for HOXD12-positive expression status in the CGGA. g HOXD12-positive expression status was prognostic in the CGGA, validating the results from the TCGA. h HOXD12-positive expression status was prognostic independent of patient age and CNS WHO grade in the TCGA and independent of CNS WHO grade in the CGGA. i Age distributions of TCGA and CGGA oligodendrogliomas when partitioned by HOXD12-positive and HOXD12-negative expression status
To test genes for age and survival association, we first performed an unbiased differential gene expression analysis comparing older (≥ 45 years) and younger TCGA oligodendrogliomas and identified 16 differentially expressed genes. These genes included developmental transcription factors in the HOX and GATA gene families, which were upregulated in older patients, and keratin genes, which were downregulated in older patients (Fig. 1c). Gene ontology analyses of the results from this differential gene expression analysis showed that the most significantly activated pathways in older TCGA oligodendrogliomas were linked to developmental transcription factors and included DNA-binding transcription activator activity (padj < 1e−6) and the development of the embryonic skeletal system (padj < 1e−7), appendages (padj < 1e−6), and limbs (padj < 1e−6) (Supplemental Fig. 6a,b). Suppressed pathways included cornification (padj < 1e−13) and keratinization (padj < 1e−10) and other pathways that involve structural proteins that control cell shape. Our results were supported by a gene set enrichment analysis, whose most significant results were the suppression of cornification (padj < 1e−13) and the activation of embryonic skeletal system development (padj < 1e−7) (Supplemental Fig. 6c).
We followed these age-related analyses with survival analyses focused on the 16 differentially expressed genes identified above. To compare high and low gene expression levels, we compared patients with gene expression above a gene’s median expression level (elevated) to patients whose gene expression is below or equal to that gene’s median expression level (low). Of these 16 genes, 4 genes conferred significantly worse outcomes when elevated, of which HOXD12 was the most significant and severe (HR = 9.3, FDR < 1e−5, log-rank) (Fig. 1d,e). Given that the median HOXD12 expression in the TCGA was zero, we refer to elevated HOXD12 expression as HOXD12-positive. HOXD12-positive expression status’ statistical associations with age and survival were validated in the CGGA (Fig. 1f,g). Older (≥ 45 years old) CGGA oligodendroglioma patients were significantly enriched for HOXD12-positive patients (p = 0.03, Fisher’s), and HOXD12-positive status was prognostic in the CGGA (p < 0.001, log-rank), incurring a similar hazard ratio (HR) as observed in the TCGA (TCGA HR = 9.3, CGGA HR = 8.4). In multivariate survival analyses, HOXD12 was the only one of the four genes identified as prognostic whose elevated expression was also prognostic independent of patient age and WHO grade in the TCGA (FDR = 0.02, CPH) (Fig. 1h, Supplemental Fig. 7). HOXD12-positive status was independently prognostic of WHO grade in the CGGA but lost its significance when patient age was accounted for, possibly because the sample size (N = 44) was relatively small for a multivariate analysis. For both TCGA and CGGA cohorts, when HOXD12-positive and HOXD12-negative patients were considered separately, their age distributions more closely resembled Gaussian distributions (Fig. 1i). Similar trends toward age and survival association were found in the GLASS dataset; however, low patient count limited these analyses (N = 9) (Supplemental Fig. 8).
HOXD12 gene body hypermethylation is age-associated and predictive of poor survival independent of HOXD12 expression
Given that DNA methylation and gene transcription are related, we sought to evaluate the relationship between HOXD12 methylation levels and patient age and survival. We found that every Illumina 450 k array DNA methylation probe associated with HOXD12 (N = 14) was positively correlated with HOXD12 expression in the TCGA, suggesting that HOXD12 hypermethylation was associated with higher transcriptional activity (Fig. 2a). Accordingly, we hypothesized that HOXD12 hypermethylation would be more prevalent in older oligodendroglioma patients and associated with poor survival. To test this hypothesis, we defined a HOXD12 methylation signature and set a threshold above which we deemed patients HOXD12 hypermethylated. This signature was developed by testing each probe for age and survival association in both univariate and multivariate analysis (Methods) (Fig. 2b, Supplemental Fig. 9). Interestingly, the only probes that reached significance (p < 0.05) in all tests were HOXD12’s three gene body probes, which is consistent with the notion that gene body methylation is often positively correlated with gene expression and a report that HOXD12 hypermethylation is linked to higher HOXD12 expression levels [69, 87]. To formally establish a HOXD12 gene body hypermethylation threshold, we considered the mean of the beta values for the three HOXD12 gene body methylation probes for patients with HOXD12-positive expression status and for patients with HOXD12-negative expression status. We chose the mean of these two values (0.3577) as the HOXD12 gene body hypermethylation threshold.

HOXD12 Hypermethylation is Associated with Age and Predictive of Poor Survival Independent of HOXD12 Expression. a All HOXD12-associated methylation probes (N = 14) were significantly positively correlated with HOXD12 expression in the TCGA. b HOXD12-associated probes were tested for age, univariate survival, and multivariate survival associations in the TCGA using specified thresholds. Only the three probes located within HOXD12’s gene body passed all such tests. c HOXD12 hypermethylation was associated with age in the TCGA and the Capper et al. cohort. d HOXD12 hypermethylated oligodendrogliomas from Capper et al. and the TCGA were enriched for higher CNS WHO grade. e HOXD12 hypermethylation was prognostic in the TCGA. f Nomogram for predicting 5- and 10-year overall survival using patient age, HOXD12 methylation level, and CNS WHO grade. g Multivariate survival analyses showed HOXD12 hypermethylation was prognostic independent of HOXD12-positive expression status, age, and CNS WHO grade. the TCGA. h Oligodendroglioma TCGA patients harboring HOXD12-positive expression status and HOXD12 gene body hypermethylation had the worst outcomes (overall survival = 2.6 years)
Using this gene body methylation signature and threshold, we found that HOXD12 gene body hypermethylation was associated with age in the TCGA (p < 1e-6, Mann–Whitney U) and Capper et al. cohorts (p = 0.002, Mann–Whitney U) (Fig. 2c, Supplemental Fig. 10). While this age association was not as strong in the Capper et al. cohort, this is likely because the Capper et al. cohort was disproportionally enriched for younger, WHO grade 3 tumors compared to all other datasets we analyzed (Supplemental Fig. 11). Furthermore, HOXD12 gene body hypermethylation was prognostic in the TCGA in a univariate analysis (HR = 4.2, p < 0.001, log-rank) and two multivariate analyses, the first of which accounted for HOXD12 expression status (p < 0.003, CPH) and the second accounted for HOXD12 expression status, patient age, and WHO grade (p < 0.007, CPH) (Fig. 2d,e). Patients harboring HOXD12-positive expression status and HOXD12 gene body hypermethylation in the TCGA formed the class of oligodendrogliomas with the worst outcomes, with a median overall survival of only 2.6 years (Fig. 2f). Although outcome data was not available for the Capper et al. cohort, oligodendrogliomas from this cohort were substantially enriched for central nervous system (CNS) WHO grade 3 tumors (p = 0.01, Fisher’s), as were their TCGA counterparts (p < 0.001, Fisher’s), indicating that HOXD12 gene body hypermethylation was likely prognostic in the Capper et al. cohort (Fig. 2g). HOXD12 gene body hypermethylation in the GLASS dataset showed similar patterns but suffered from a low sample count (N = 10) (Supplemental Fig. 12). To explore the clinical applicability of HOXD12 gene body methylation, we developed a nomogram predicting 5- and 10-year overall survival using patient age, HOXD12 gene body methylation level, and CNS WHO grade using TCGA data (Fig. 2h). Unfortunately, immunohistochemistry for HOXD12 was not a sufficiently sensitive surrogate marker to detect a difference between HOXD12 gene body methylation levels in a small set of oligodendrogliomas (N = 10) (Supplemental Fig. 13).
HOXD12-positive expression status and gene body hypermethylation are prognostic independent of key histopathologic, genomic, and radiographic features
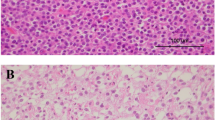
Having shown that HOXD12-positive expression status and HOXD12 gene body hypermethylation were markers for poor survival in oligodendroglioma, we interrogated their relationship with clinically relevant histopathologic, genomic, and radiographic features in the TCGA (Fig. 3a). Among histopathologic features, HOXD12 gene body hypermethylation was linked to a high Ki-67 proliferative index (≥ 10%) (p < 0.0001, Fisher’s) and the presence of mitotic figures (p = 0.015, Fisher’s) but not to the presence of necrosis or microvascular proliferation (MVP) (Fig. 3b). Among proposed prognostic genomic alterations, we found that HOXD12-positive expression status and HOXD12 gene body hypermethylation were associated with the loss of chromosome arm 15q (p < 0.001, p = 0.049, Fisher’s) and MYC activation (p = 0.04, p < 0.03, Fisher’s) but not with NOTCH1 mutations or PIK3CA mutations. CDKN2A homozygous deletions were not tested because fewer than 1% (1/171) of TCGA oligodendrogliomas harbored these alterations. Radiographically, HOXD12 gene body hypermethylation was associated with the presence of T1-post contrast enhancement in a set of 27 multimodal pre-operative MRIs from the TCGA (p = 0.03, Fisher’s), while HOXD12-positive expression status was not significantly associated with any tested radiographic feature. These associations suggest that tumors expressing HOXD12 and harboring HOXD12 gene body hypermethylation are not only prognostic, but they also appear more aggressive histopathologically, molecularly, and radiographically. To highlight these features, we identified a representative tumor harboring both HOXD12-positive expression status and HOXD12 gene body hypermethylation that displays mitotic figures, necrosis, and MVP (Fig. 3c), as well as contrast enhancement and substantial peritumoral edema (Fig. 3d).

HOXD12-Positive Expression and Gene Body Hypermethylation are Independently Prognostic of Key Histopathologic, Genomic, and Radiographic Features. a List of key tested histopathologic, genomic, and radiographic features. b HOXD12 gene body hypermethylation was linked to high Ki-67 proliferative index and the presence of mitotic figures. HOXD12-positive expression status and hypermethylation were associated with the loss of chromosome arm 15q and MYC activation. HOXD12 hypermethylation was associated with the presence of T1-post-contrast enhancement. c Representative H&E-stained sections of a HOXD12-positive and gene body hypermethylated TCGA oligodendroglioma (TCGA-DU-7018) showed increased mitotic activity, necrosis, and MVP. d MRI of the same patient showed strong contrast enhancement (blue) and extensive peritumoral edema (red) on T1-post contrast and FLAIR MRI sequences, respectively. e, f HOXD12-positive expression status and hypermethylation were independently prognostic of all tested histopathologic and genomic variables. g HOXD12-positive expression status was independently prognostic of all tested radiographic features
Remarkably, HOXD12-positive expression status and HOXD12 gene body hypermethylation were independently prognostic of all tested histopathologic and genomic variables (Fig. 3e, f). These multivariate analyses indicated that the statuses of HOXD12 expression and gene body methylation may be better indicators of outcome than histopathologic features used to determine CNS WHO grade (mitotic features, necrosis, MVP) and genomic markers nominated by other studies [2, 3, 30, 38, 74]. Similarly, HOXD12-positive expression status was independently prognostic of all testable radiographic features (total tumor volume omitted due to collinearity). However, HOXD12 gene body hypermethylation was not (p = 0.12), possibly because the MRI dataset was small (Fig. 3g, Supplemental Fig. 14). Regrettably, it was not possible to validate our findings in the CGGA or Capper et al. cohorts because of insufficient imaging or histopathology data.
Importantly, the TCGA survival associations observed here were likely not due to treatment differences because the use of adjuvant chemotherapy (N = 152), predominately temozolomide, and/or adjuvant radiation therapy (RT) (N = 151) were not statistically associated with HOXD12 gene body hypermethylation or HOXD12-positive expression status (Supplemental Fig. 15a, b, Supplemental Table 2). Of note, TCGA WHO grade 2 oligodendrogliomas were treated with postoperative chemoradiation dramatically less frequently than TCGA WHO grade 3 oligodendrogliomas (9% vs. 55%, p < 0.0001, Fisher’s) (Supplemental Fig. 15c). It is conceivable that if HOXD12 gene body methylation status had been incorporated as a molecular risk factor, patients with HOXD12 gene body hypermethylated tumors may have been treated more commonly with chemoradiation, as the case with WHO grade 3 tumors, and the survival differences we observed may have been slimmer.
To further control for confounding treatment differences and to test HOXD12 gene body hypermethylation in a prospective cohort, we also analyzed DNA methylation data from the prospective EORTC 26951 Phase III trial] [41, 77]. EORTC 26951 Phase III compared RT to RT and six cycles of chemotherapy agents procarbazine, CCNU, and vincristine (PCV) in WHO grade 3 diffuse gliomas. For our analysis, we included EORTC patients who met the following three criteria: (1) their tumor met the current WHO CNS classification system definition of oligodendroglioma (including 1p/19q-codeletion), (2) they were treated with the trial’s uniform chemoradiation regimen (RT + PCV), and (3) their tumor had methylation data sufficient to determine HOXD12 gene body methylation status. Unfortunately, most of the 115 tumors with profiled DNA methylation data were excluded because, unlike Illumina 450 k arrays, they were profiled with Illumina 27 k arrays, which lack two of the three HOXD12 gene body probes in our HOXD12 gene body methylation signature. Only 10 tumors met these three selection criteria, of which four were HOXD12 gene body hypomethylated and six were HOXD12 gene body hypermethylated. Our analysis of these data, although severely underpowered, did show a weak trend toward worse survival among HOXD12 gene body hypermethylated patients in both overall survival (HR = 2.8, p = 0.36) and progression-free survival (HR = 2.8, p = 0.36) (Supplemental Fig. 16). Overall, more extensive prospective follow-up data and more feasible clinical testing are necessary to definitively establish any prognostic utility of HOXD12-positive expression status or HOXD12 gene body methylation status.
Increased HOXD12 activity in neoplastic cells is associated with a proliferative phenotype
To determine HOXD12 activity in specific cell compartments and associated cell states, we analyzed high-quality paired oligodendroglioma single-nucleus ATAC sequencing (snATAC-seq) and single-nucleus RNA sequencing (snRNA-seq) data published by Blanco-Carmona et al. (N = 7729) and snATAC-seq data published by Wang et al. (N = 10,281), from which we estimated snRNA-seq reads using Signac’s GeneActivity function [10, 73, 82]. To identify neoplastic nuclei, we used inferCNV to compute the somatic copy number alteration status of chromosome arms 1p and 19q to determine whether they were codeleted, a unique feature of oligodendroglioma cells; we used published gene markers to assign cell types to non-neoplastic nuclei (Fig. 4a, Supplemental Figs. 17, 18) [48, 53, 75, 81, 84]. Neoplastic cell states were assigned using lineage scores for oligodendrocyte progenitor cell (OPC)-like, Astro-like, Cycling, and ribosomal-enriched (RE) cell states for each nucleus, which we computed using published gene sets [10, 81]. Using a previously described permutation test, we calculated and corrected p-values which we used for final cell state assignment [10]. All RE cells were excluded due to a low sample count. Stemness scores and lineage scores for OC-like and AC-like cell states were also computed as previously described [76].

HOXD12 activity is associated with neoplastic nuclei, proliferating cells, and an increased stem-like phenotype. a UMAP embeddings of single-nuclei RNA-seq and single-nuclei ATAC-derived single-nuclei RNA-seq from the Blanco-Carmona et al. and Wang et al. cohorts, respectively, labeled by neoplastic cell state or non-neoplastic cell type. Neoplastic nuclei are labeled as Gradient, Astro-like, OPC-like, or Cycling. b In both the Blanco-Carmona et al. and Wang et al. cohorts, single-nucleus HOXD12 RNA reads and single-nucleus ATAC HOXD12 reads were more prevalent in neoplastic tissue compared to non-neoplastic tissue. c, d Among neoplastic nuclei, HOXD12 RNA and ATAC reads were most common in OPC-like and cycling cells, which are associated with stem-like and proliferative phenotypes. e Lineage and stemness scores for oligodendroglioma tumors nuclei in the Blanco-Carmona et al. and Wang et al. cohorts. f Nucleus stemness scores are higher in tumor nuclei that have HOXD12 RNA or ATAC reads than those without in both the Blanco-Carmona et al. and Wang et al.
We found that HOXD12 expression was significantly more common in cycling/proliferative neoplastic cells than in non-proliferative neoplastic cells, which, in turn, had a higher rate of HOXD12 expression than non-neoplastic brain cells. In general, HOXD12 expression and chromatin accessibility were significantly more prevalent in neoplastic nuclei compared to non-neoplastic nuclei in the Blanco-Carmona et al. and Wang et al. datasets (Fig. 4b). After cycling/proliferative neoplastic cells, HOXD12 snRNA-seq expression was most prevalent in OPC-like neoplastic cells in both datasets, followed by gradient and astro-like neoplastic cells (Fig. 4c). Likewise, snATAC-seq confirmed that neoplastic cells classified as cycling or OPC-like had the highest prevalence of HOXD12 chromatin accessibility followed by cells classified as gradient or astro-like (Fig. 4d). Given that cycling cells are in the process of dividing and that OPC-like cells are known for their proliferative potential and stem-like properties, we computed stemness scores for all neoplastic cells. For both snRNA-seq and snATAC-seq data, and in both the Blanco-Carmona et al. and Wang et al. datasets, nuclei with HOXD12 activity had significantly higher stemness scores (Student’s t-test) (Fig. 4e,f). Altogether, these single-nuclei data suggested that HOXD12 activity is associated with a proliferative phenotype in neoplastic cells and indicated that our previous analyses of bulk sequencing data reflect biological processes within the neoplastic cell compartment, rather than the non-neoplastic cells of the tumor microenvironment.
A Pan-HOX methylation signature, driven by HOXD12, is associated with increased proliferation and poor outcomes
To place our HOXD12-centric findings in the context of all HOX genes, we analyzed the HOX gene body DNA methylation probes present in all 365 oligodendroglioma tumors from the TCGA (N = 171), Capper et al. (N = 170), and GLASS (N = 24) datasets. Two distinct, consistent clusters, not related to dataset membership, appeared across 1000 UMAP projections of these data generated using different initializations (Fig. 5a, Supplemental Fig. 19). We restricted our analysis to samples that remained in the same cluster in at least 90% of the UMAP projections (N = 345) (Supplemental Fig. 20). These two clusters were characterized by global HOX gene body DNA methylation levels: nearly all HOX-associated gene body probes had higher beta values in one cluster (HOX-high) compared to the other (HOX-low) (Fig. 5b, c). Patients in the HOX-high group were significantly older in the TCGA (p = 3e−15, Mann–Whitney U) and Capper et al. (p = 1e−8, Mann–Whitney U) datasets, and they disproportionally harbored higher CNS WHO grade (p = 2e−5, p = 0.0004, Fisher’s) (Fig. 5d, Supplemental Fig. 21). TCGA patients in the HOX-high group harbored more aggressive tumors, measured both by clinical outcomes and histological features. HOX-high TCGA patients suffered worse outcomes than those in the HOX-low group (HR = 7.6, p = 0.01). HOX-high TCGA tumors also more commonly had increased mitotic figures (p = 0.01, Fisher’s) and higher Ki-67 proliferative index (p < 0.01, Mann–Whitney U) (Fig. 5e, f). The presence of palisading necrosis and microvascular proliferation (MVP) were also elevated in HOX-high TCGA patients; however, their difference was less significant. Although the sample size of stable samples in the GLASS dataset was small (N = 9), we observed more movement from the HOX-low cluster to the HOX-high cluster as tumors evolved than vice versa (Supplemental Fig. 22a–c). The scarcity of these data made conclusions impossible to draw with confidence; however, GLASS samples in the HOX-high cluster tended to be recurrent, higher WHO grade, and shorter-lived (Supplemental Fig. 22d–f). As was the case with HOXD12 gene body hypermethylation and HOXD12-positive expression status, HOX-high survival associations were not linked to lower rates of adjuvant therapy in the TCGA (Supplemental Fig. 23). In fact, the only significant associations we found were higher rates of adjuvant chemotherapy (p = 0.01, Fisher’s) and chemoradiation (p < 0.05) in HOX-high patients.

A Pan-HOX signature led by HOXD12 is associated with poor outcomes and proliferation. a A UMAP embedding of all HOX gene body associated DNA methylation probes for oligodendroglioma patients in the TCGA, Capper et al., and GLASS datasets (N = 365) formed two clear clusters termed HOX-high and HOX-low. b The mean value of HOX gene body probes is significantly higher in the HOX-high cluster than in the HOX-low cluster. c The mean value of 98.4% of HOX gene body probes is higher in the HOX-high cluster than in the HOX-low cluster. d The HOX-high cluster is associated with patient age and tumor WHO grade in the cohorts of the TCGA and Capper et al. Differences in TCGA and Capper et al. age distributions are likely attributable to the abundance of young, WHO grade 3 oligodendrogliomas in the Capper et al. cohort. e The TCGA patients in the HOX-high cluster harbor significantly worse outcomes than those in the HOX-low cluster. f TCGA tumors in the HOX-high cluster more commonly had mitotic figures and high Ki67 index; the presence of palisading necrosis and MVP were also elevated. g In a differential gene expression analysis between the two methylation classes, HOXD12 had the highest RNA expression log fold change. h TCGA patients in the HOX-high cluster were associated with the mitotic cell cycle pathway in a gene ontology analysis. i In a differential methylation analysis, gene body probes on the HOXD locus had higher log fold change on average compared to gene body probes from other HOX loci. j In this differential methylation analysis, gene body probes associated with HOXD12 had the highest average log fold change compared to other HOX genes
Next, we performed unbiased differential gene expression and differential DNA methylation analyses by comparing patients in the HOX-high group to patients in the HOX-low group. Remarkably, HOXD12 was the gene with the highest RNA expression log-fold change between the HOX-high and HOX-low TCGA patients (4.8, padj < 7e−5) (Fig. 5g). Gene ontology analyses of the results from our differential gene expression analysis on TCGA oligodendroglioma tumors showed that the most significantly activated pathways in HOX-high patients were related to mitotic activity and cell cycle (padj < 1e−6) (Fig. 5h). In our differential DNA methylation analysis, gene body probes from the HOXD locus had more significant, higher log-fold change on average compared to gene body probes from other HOX loci (Fig. 5i). Among HOX genes, gene body probes associated with HOXD12 had the highest average log-fold change compared to gene body probes associated with other HOX genes (Fig. 5j, Supplemental Fig. 24). In general, gene body DNA methylation of HOX genes partitioned oligodendroglioma into two groups with lower and higher HOX gene body DNA methylation levels. The group of oligodendroglioma tumors with high HOX gene body DNA methylation levels was associated with poor survival, older patients, and more aggressive histology and was best differentiated from the HOX-low group by genes in the HOXD locus, especially HOXD12.
Discussion
Although patients diagnosed with oligodendroglioma typically have better prognoses than their IDH-mutant astrocytoma and IDH-wildtype glioblastoma counterparts, oligodendroglioma patients have more variable outcomes, which underscores the need to make therapy intensities proportionate to individual patient risk. However, assessing a patient’s risk is difficult as only a handful of oligodendroglioma risk factors have been proposed to date, notably including patient age [2, 3, 30, 38, 74]. Unlike other adult-type diffuse gliomas, the age distribution of oligodendroglioma patients is non-Gaussian and reportedly bimodal, suggesting that investigations into age-associated biological differences in oligodendroglioma tumors are merited [17]. In this study, we sought to discover molecular markers associated with age and patient survival to gain insight into the relationship between patient age and oligodendroglioma tumor biology. We propose that HOXD12 expression and gene body hypermethylation are molecular features associated with poor prognoses that may be linked to two age-associated subtypes of oligodendroglioma.
HOXD12 expression was identified in an unbiased manner by filtering for genes whose expression was associated with age and survival in the TCGA. In addition to HOXD12 expression, we also found that HOXD12 gene body hypermethylation was correlated with HOXD12 expression levels. Remarkably, HOXD12 gene body hypermethylation was prognostic independent of HOXD12 expression, as well as patient age and tumor WHO grade. Our results were validated in oligodendroglioma cohorts published by the CGGA, Capper et al., and GLASS. We also showed HOXD12 gene body hypermethylation and elevated expression were independently prognostic of published biomarkers and standard histopathological features, and we used single-nucleus RNA and ATAC sequencing to show that HOXD12 activity was localized to neoplastic cells rather than cells of the tumor microenvironment and was particularly strong within cycling and OPC-like cells. A pan-HOX DNA methylation analysis revealed an age and survival-associated HOX-high signature that was tightly associated with HOXD12 gene body methylation. Overall, elevated HOXD12 expression and gene body hypermethylation were associated with an older, atypically aggressive subtype of oligodendroglioma. We propose that future biomarker-driven clinical trials of oligodendroglioma collect HOXD12 gene body methylation data, possibly through whole-genome DNA methylation profiling. If HOXD12 gene body methylation’s prognostic value is confirmed in trials such as these, HOXD12 gene body methylation testing may be considered for risk-stratification in clinical, surgical neuropathology, as whole-genome DNA methylation array profiling has made its way into routine clinical practice across several academic medical centers [14, 24, 33, 39, 60, 61, 63, 65,66,67, 86].
HOXD12 is a gene in the HOX gene family located on the HOXD locus on chromosome 2—one of four HOX loci (HOXA, HOXB, HOXC, and HOXD) found on chromosomes 7, 17, 12, and 2, respectively. In healthy individuals, HOX genes are involved in embryonic brain development and are usually absent in the adult brain; however, abnormal HOX gene expression and regulation have been extensively documented in cancer [1, 8, 29, 59]. Numerous genes in the HOX gene family have been reported to be involved in tumors of the CNS (as reviewed by Gonçalves et al. [29]), particularly in glioblastoma [16, 20, 22, 27, 29, 40, 42], primarily as oncogenes. More recently, HOX genes in the HOXA and HOXD loci have been implicated in IDH-mutant adult-type diffuse glioma progression and aggression [46, 47]. A study by Mamatjan et al. analyzed HOX gene overexpression and hypermethylation and proposed a seven HOX gene signature (HOXA4, HOXA7, HOXA10, HOXA13, HOXD3, HOXD9, and HOXD10) that was shown to be prognostic in IDH mutant astrocytoma and oligodendrogliomas [47]. The gene HOXD13 has also been implicated in glioma progression in a ChIP-seq analyses of 14 IDH-mutant gliomas, but this phenotype was not validated in oligodendroglioma [46].
Our findings support and extend the existing literature regarding oligodendroglioma heterogeneity. Kamoun et al. identified three subtypes of oligodendroglioma from four microarray datasets, of which the largest subset was negatively prognostic in the TCGA, weakly associated with age, and characterized by MYC activation [38]. Interestingly, we showed that TCGA patients that express HOXD12 or harbor HOXD12 gene body hypermethylation were associated with age and MYC activation, but HOXD12 expression and gene body hypermethylation were prognostic independent of both. This suggests that HOXD12 expression and gene body hypermethylation may explain the reported prognostic value of MYC activation. A single-cell RNA-seq study by Tirosh et al. also interrogated oligodendroglioma molecular heterogeneity, albeit in an intra-tumoral setting, and linked neurodevelopmental transcription factor expression to a potentially proliferative stem/progenitor program in a small subpopulation of oligodendroglioma cells [76]. Our results corroborate these findings: HOXD12 is a neurodevelopmental transcription factor, HOXD12 expression was associated with a stemlike phenotype in single-nucleus sequencing data, and HOXD12 was expressed in a minority of neoplastic nuclei. The results of the study by Mamatjan et al. also align with our findings. Our unbiased pan-HOX analysis showed DNA methylation probes associated with HOX genes from the HOXA and HOXD loci were most differentially methylated between the prognostic HOX-high and HOX-low clusters. The unbiased nature of our analyses also boosts this study, as does our deep interaction of HOXD12 in multiple datasets and the supportive single-nucleus data we presented.
We do acknowledge several limitations in our study. Our analyses were conducted on retrospective data, which can suffer from sparse annotations, masked biases, and outdated treatment. To address these limitations, we confirmed our major results on at least two datasets, and we verified that there were no significant differences in treatment that would explain survival differences between compared groups in the TCGA. Another obstacle was our inability to detect HOXD12 by immunohistochemistry on a small set of patients; this must be further investigated before HOXD12 activity can be considered clinically relevant. Overall, more prospective human data must be collected and subjected to long-term follow-up to confirm any findings and subsequent use in risk stratification and clinical decision-making. Additionally, given the clinical benefit of IDH inhibitors in patients who have delayed chemoradiation, future study of HOXD12 in the context of mutant IDH inhibition is merited and may assist in guiding clinical decisions [49, 52, 71].
References
Abdel-Fattah R, Xiao A, Bomgardner D, Pease CS, Lopes MB, Hussaini IM (2006) Differential expression of HOX genes in neoplastic and non-neoplastic human astrocytes. J Pathol 209:15–24. https://doi.org/10.1002/path.1939
Aoki K, Nakamura H, Suzuki H, Matsuo K, Kataoka K, Shimamura T et al (2018) Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol 20:66–77. https://doi.org/10.1093/neuonc/nox132
Appay R, Dehais C, Maurage CA, Alentorn A, Carpentier C, Colin C et al (2019) CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol 21:1519–1528. https://doi.org/10.1093/neuonc/noz124
Arora S, Pattwell SS, Holland EC, Bolouri H (2020) Variability in estimated gene expression among commonly used RNA-seq pipelines. Sci Rep 10:2734. https://doi.org/10.1038/s41598-020-59516-z
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD et al (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363–1369. https://doi.org/10.1093/bioinformatics/btu049
Bakas S, Akbari H, Sotiras A, Bilello M, Rozycki M, Kirby JS et al (2017) Advancing the cancer genome atlas glioma MRI collections with expert segmentation labels and radiomic features. Sci Data 4:170117. https://doi.org/10.1038/sdata.2017.117
Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E et al (2019) Longitudinal molecular trajectories of diffuse glioma in adults. Nature 576:112–120. https://doi.org/10.1038/s41586-019-1775-1
Bhatlekar S, Fields JZ, Boman BM (2014) HOX genes and their role in the development of human cancers. J Mol Med (Berl) 92:811–823. https://doi.org/10.1007/s00109-014-1181-y
Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM et al (2011) High density DNA methylation array with single CpG site resolution. Genomics 98:288–295. https://doi.org/10.1016/j.ygeno.2011.07.007
Blanco-Carmona E, Narayanan A, Hernandez I, Nieto JC, Elosua-Bayes M, Sun X et al (2023) Tumor heterogeneity and tumor-microglia interactions in primary and recurrent IDH1-mutant gliomas. Cell Rep Med 4:101249. https://doi.org/10.1016/j.xcrm.2023.101249
Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA et al (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372:2481–2498. https://doi.org/10.1056/NEJMoa1402121
Cancer Genome Atlas Research N, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR et al (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372:2481–2498. https://doi.org/10.1056/NEJMoa1402121
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Capper D, Stichel D, Sahm F, Jones DTW, Schrimpf D, Sill M et al (2018) Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol 136:181–210. https://doi.org/10.1007/s00401-018-1879-y
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404. https://doi.org/10.1158/2159-8290.CD-12-0095
Cimino PJ, Kim Y, Wu HJ, Alexander J, Wirsching HG, Szulzewsky F et al (2018) Increased HOXA5 expression provides a selective advantage for gain of whole chromosome 7 in IDH wild-type glioblastoma. Genes Dev 32:512–523. https://doi.org/10.1101/gad.312157.118
Cimino PJ, Zager M, McFerrin L, Wirsching HG, Bolouri H, Hentschel B et al (2017) Multidimensional scaling of diffuse gliomas: application to the 2016 World Health Organization classification system with prognostically relevant molecular subtype discovery. Acta Neuropathol Commun 5:39. https://doi.org/10.1186/s40478-017-0443-7
Collado-Torres L, Nellore A, Kammers K, Ellis SE, Taub MA, Hansen KD et al (2017) Reproducible RNA-seq analysis using recount2. Nat Biotechnol 35:319–321. https://doi.org/10.1038/nbt.3838
Consortium G (2018) Glioma through the looking GLASS: molecular evolution of diffuse gliomas and the Glioma Longitudinal Analysis Consortium. Neuro Oncol 20:873–884. https://doi.org/10.1093/neuonc/noy020
Costa BM, Smith JS, Chen Y, Chen J, Phillips HS, Aldape KD et al (2010) Reversing HOXA9 oncogene activation by PI3K inhibition: epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res 70:453–462. https://doi.org/10.1158/0008-5472.CAN-09-2189
Doshi J, Erus G, Ou Y, Gaonkar B, Davatzikos C (2013) Multi-atlas skull-stripping. Acad Radiol 20:1566–1576. https://doi.org/10.1016/j.acra.2013.09.010
Duan R, Han L, Wang Q, Wei J, Chen L, Zhang J et al (2015) HOXA13 is a potential GBM diagnostic marker and promotes glioma invasion by activating the Wnt and TGF-beta pathways. Oncotarget 6:27778–27793. https://doi.org/10.18632/oncotarget.4813
Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H et al (2015) Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 372:2499–2508. https://doi.org/10.1056/NEJMoa1407279
Galbraith K, Vasudevaraja V, Serrano J, Shen G, Tran I, Abdallat N et al (2023) Clinical utility of whole-genome DNA methylation profiling as a primary molecular diagnostic assay for central nervous system tumors-A prospective study and guidelines for clinical testing. Neurooncol Adv 5:vdad076. https://doi.org/10.1093/noajnl/vdad076
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1. https://doi.org/10.1126/scisignal.2004088
Garton ALA, Kinslow CJ, Rae AI, Mehta A, Pannullo SC, Magge RS et al (2020) Extent of resection, molecular signature, and survival in 1p19q-codeleted gliomas. J Neurosurg 134:1357–1367. https://doi.org/10.3171/2020.2.JNS192767
Gaspar N, Marshall L, Perryman L, Bax DA, Little SE, Viana-Pereira M et al (2010) MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res 70:9243–9252. https://doi.org/10.1158/0008-5472.CAN-10-1250
Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A et al (2020) Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 38:675–678. https://doi.org/10.1038/s41587-020-0546-8
Goncalves CS, Le Boiteux E, Arnaud P, Costa BM (2020) HOX gene cluster (de)regulation in brain: from neurodevelopment to malignant glial tumours. Cell Mol Life Sci 77:3797–3821. https://doi.org/10.1007/s00018-020-03508-9
Halani SH, Yousefi S, Velazquez Vega J, Rossi MR, Zhao Z, Amrollahi F et al (2018) Multi-faceted computational assessment of risk and progression in oligodendroglioma implicates NOTCH and PI3K pathways. NPJ Precis Oncol 2:24. https://doi.org/10.1038/s41698-018-0067-9
Halasz LM, Attia A, Bradfield L, Brat DJ, Kirkpatrick JP, Laack NN et al (2022) Radiation therapy for IDH-mutant grade 2 and grade 3 diffuse glioma: an ASTRO clinical practice guideline. Pract Radiat Oncol 12:370–386. https://doi.org/10.1016/j.prro.2022.05.004
Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A et al (2021) Integrated analysis of multimodal single-cell data. Cell 184(3573–3587):e3529. https://doi.org/10.1016/j.cell.2021.04.048
Jaunmuktane Z, Capper D, Jones DTW, Schrimpf D, Sill M, Dutt M et al (2019) Methylation array profiling of adult brain tumours: diagnostic outcomes in a large, single centre. Acta Neuropathol Commun 7:24. https://doi.org/10.1186/s40478-019-0668-8
Jenkinson M, Bannister P, Brady M, Smith S (2002) Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage 17:825–841. https://doi.org/10.1016/s1053-8119(02)91132-8
Jenkinson M, Beckmann CF, Behrens TE, Woolrich MW, Smith SM (2012) Fsl. Neuroimage 62:782–790. https://doi.org/10.1016/j.neuroimage.2011.09.015
Jenkinson M, Smith S (2001) A global optimisation method for robust affine registration of brain images. Med Image Anal 5:143–156. https://doi.org/10.1016/s1361-8415(01)00036-6
Jonsson P, Lin AL, Young RJ, DiStefano NM, Hyman DM, Li BT et al (2019) Genomic correlates of disease progression and treatment response in prospectively characterized gliomas. Clin Cancer Res 25:5537–5547. https://doi.org/10.1158/1078-0432.CCR-19-0032
Kamoun A, Idbaih A, Dehais C, Elarouci N, Carpentier C, Letouzé E et al (2016) Integrated multi-omics analysis of oligodendroglial tumours identifies three subgroups of 1p/19q co-deleted gliomas. Nat Commun 7:11263. https://doi.org/10.1038/ncomms11263
Karimi S, Zuccato JA, Mamatjan Y, Mansouri S, Suppiah S, Nassiri F et al (2019) The central nervous system tumor methylation classifier changes neuro-oncology practice for challenging brain tumor diagnoses and directly impacts patient care. Clin Epigenet 11:185. https://doi.org/10.1186/s13148-019-0766-2
Kurscheid S, Bady P, Sciuscio D, Samarzija I, Shay T, Vassallo I et al (2015) Chromosome 7 gain and DNA hypermethylation at the HOXA10 locus are associated with expression of a stem cell related HOX-signature in glioblastoma. Genome Biol 16:16. https://doi.org/10.1186/s13059-015-0583-7
Lassman AB, Hoang-Xuan K, Polley MC, Brandes AA, Cairncross JG, Kros JM et al (2022) Joint final report of EORTC 26951 and RTOG 9402: phase III trials with procarbazine, lomustine, and vincristine chemotherapy for anaplastic oligodendroglial tumors. J Clin Oncol 40:2539–2545. https://doi.org/10.1200/JCO.21.02543
Le Boiteux E, Court F, Guichet PO, Vaurs-Barriere C, Vaillant I, Chautard E et al (2021) Widespread overexpression from the four DNA hypermethylated HOX clusters in aggressive (IDHwt) glioma is associated with H3K27me3 depletion and alternative promoter usage. Mol Oncol 15:1995–2010. https://doi.org/10.1002/1878-0261.12944
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D et al (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol: https://doi.org/10.1093/neuonc/noab106
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D et al (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 23:1231–1251. https://doi.org/10.1093/neuonc/noab106
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Malta TM, Sabedot TS, Morosini NS, Datta I, Garofano L, Vallentgoed W et al (2024) The epigenetic evolution of glioma is determined by the IDH1 mutation status and treatment regimen. Cancer Res 84:741–756. https://doi.org/10.1158/0008-5472.CAN-23-2093
Mamatjan Y, Voisin MR, Nassiri F, Moraes FY, Bunda S, So J et al (2023) Integrated molecular analysis reveals hypermethylation and overexpression of HOX genes to be poor prognosticators in isocitrate dehydrogenase mutant glioma. Neuro Oncol 25:2028–2041. https://doi.org/10.1093/neuonc/noad126
McKenzie AT, Wang M, Hauberg ME, Fullard JF, Kozlenkov A, Keenan A et al (2018) Brain cell type specific gene expression and co-expression network architectures. Sci Rep 8:8868. https://doi.org/10.1038/s41598-018-27293-5
Mellinghoff IK, van den Bent MJ, Blumenthal DT, Touat M, Peters KB, Clarke J et al (2023) Vorasidenib in IDH1- or IDH2-mutant low-grade glioma. N Engl J Med 389:589–601. https://doi.org/10.1056/NEJMoa2304194
Menze BH, Jakab A, Bauer S, Kalpathy-Cramer J, Farahani K, Kirby J et al (2015) The multimodal brain tumor image segmentation benchmark (BRATS). IEEE Trans Med Imaging 34:1993–2024. https://doi.org/10.1109/TMI.2014.2377694
Mohile NA, Messersmith H, Gatson NT, Hottinger AF, Lassman A, Morton J et al (2022) Therapy for diffuse astrocytic and oligodendroglial tumors in adults: ASCO-SNO guideline. J Clin Oncol 40:403–426. https://doi.org/10.1200/JCO.21.02036
National Comprehensive Cancer Network (2024) Central Nervous System Cancers (Version 1.2024) https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf. Accessed July 2, 2024
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ et al (2019) An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178(835–849):e821. https://doi.org/10.1016/j.cell.2019.06.024
Nuechterlein N, Li B, Feroze A, Holland EC, Shapiro L, Haynor D et al (2021) Radiogenomic modeling predicts survival-associated prognostic groups in glioblastoma. Neurooncol Adv 3:vdab004. https://doi.org/10.1093/noajnl/vdab004
Nuechterlein N, Mehta S (2018) 3D-ESPNet with pyramidal refinement for volumetric brain tumor image segmentation. Springer, Cham, City
Nuechterlein N, Shapiro LG, Holland EC, Cimino PJ (2021) Machine learning modeling of genome-wide copy number alteration signatures reliably predicts IDH mutational status in adult diffuse glioma. Acta Neuropathol Commun 9:191. https://doi.org/10.1186/s40478-021-01295-3
Nuechterlein N, Shelbourn A, Szulzewsky F, Arora S, Casad M, Pattwell S et al (2024) Haploinsufficiency of phosphodiesterase 10A activates PI3K/AKT signaling independent of PTEN to induce an aggressive glioma phenotype. Genes Dev 38:273–288. https://doi.org/10.1101/gad.351350.123
Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K et al (2014) Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 26:288–300. https://doi.org/10.1016/j.ccr.2014.06.005
Paco A, Garcia SAB, Leitao Castro J, Costa-Pinto AR, Freitas R (2020) Roles of the HOX proteins in cancer invasion and metastasis. Cancers. https://doi.org/10.3390/cancers13010010
Pages M, Uro-Coste E, Colin C, Meyronet D, Gauchotte G, Maurage CA et al (2021) The implementation of DNA methylation profiling into a multistep diagnostic process in pediatric neuropathology: a 2-year real-world experience by the french neuropathology network. Cancers. https://doi.org/10.3390/cancers13061377
Pickles JC, Fairchild AR, Stone TJ, Brownlee L, Merve A, Yasin SA et al (2020) DNA methylation-based profiling for paediatric CNS tumour diagnosis and treatment: a population-based study. Lancet Child Adolesc Health 4:121–130. https://doi.org/10.1016/S2352-4642(19)30342-6
Pojo M, Gonçalves CS, Xavier-Magalhães A, Oliveira AI, Gonçalves T, Correia S et al (2015) A transcriptomic signature mediated by HOXA9 promotes human glioblastoma initiation, aggressiveness and resistance to temozolomide. Oncotarget 6:7657–7674. https://doi.org/10.18632/oncotarget.3150
Priesterbach-Ackley LP, Boldt HB, Petersen JK, Bervoets N, Scheie D, Ulhoi BP et al (2020) Brain tumour diagnostics using a DNA methylation-based classifier as a diagnostic support tool. Neuropathol Appl Neurobiol 46:478–492. https://doi.org/10.1111/nan.12610
Qiu S, An Z, Tan R, He PA, Jing J, Li H et al (2021) Understanding the unimodal distributions of cancer occurrence rates: it takes two factors for a cancer to occur. Brief Bioinform. https://doi.org/10.1093/bib/bbaa349
Rajappa P, Eng KW, Bareja R, Bander ED, Yuan M, Dua A et al (2022) Utility of multimodality molecular profiling for pediatric patients with central nervous system tumors. Neurooncol Adv 4:vdac031. https://doi.org/10.1093/noajnl/vdac031
Santana-Santos L, Kam KL, Dittmann D, De Vito S, McCord M, Jamshidi P et al (2022) Validation of whole genome methylation profiling classifier for central nervous system tumors. J Mol Diagn 24:924–934. https://doi.org/10.1016/j.jmoldx.2022.04.009
Schepke E, Lofgren M, Pietsch T, Olsson Bontell T, Kling T, Wenger A et al (2022) DNA methylation profiling improves routine diagnosis of paediatric central nervous system tumours: a prospective population-based study. Neuropathol Appl Neurobiol 48:e12838. https://doi.org/10.1111/nan.12838
Shapiro JA, Gaonkar KS, Spielman SJ, Savonen CL, Bethell CJ, Jin R et al (2023) OpenPBTA: the open pediatric brain tumor atlas. Cell Genom 3:100340. https://doi.org/10.1016/j.xgen.2023.100340
Smith J, Sen S, Weeks RJ, Eccles MR, Chatterjee A (2020) Promoter DNA hypermethylation and paradoxical gene activation. Trends Cancer 6:392–406. https://doi.org/10.1016/j.trecan.2020.02.007
Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen-Berg H et al (2004) Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23(Suppl 1):S208-219. https://doi.org/10.1016/j.neuroimage.2004.07.051
Spitzer A, Gritsch S, Nomura M, Jucht A, Fortin J, Raviram R et al (2024) Mutant IDH inhibitors induce lineage differentiation in IDH-mutant oligodendroglioma. Cancer Cell. https://doi.org/10.1016/j.ccell.2024.03.008
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd et al (2019) Comprehensive Integration of single-cell data. Cell 177(1888–1902):e1821. https://doi.org/10.1016/j.cell.2019.05.031
Stuart T, Srivastava A, Madad S, Lareau CA, Satija R (2021) Single-cell chromatin state analysis with Signac. Nat Methods 18:1333–1341. https://doi.org/10.1038/s41592-021-01282-5
Tateishi K, Nakamura T, Juratli TA, Williams EA, Matsushita Y, Miyake S et al (2019) PI3K/AKT/mTOR pathway alterations promote malignant progression and xenograft formation in oligodendroglial tumors. Clin Cancer Res 25:4375–4387. https://doi.org/10.1158/1078-0432.CCR-18-4144
Tickle T TI, Georgescu C, Brown M, Haas B (2019) inferCNV of the Trinity CTAT Project. Klarman Cell Observatory, Broad Institute of MIT and Harvard
Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K et al (2016) Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539:309–313. https://doi.org/10.1038/nature20123
van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY et al (2013) Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 31:344–350. https://doi.org/10.1200/JCO.2012.43.2229
van den Bent MJ, Erdem-Eraslan L, Idbaih A, de Rooi J, Eilers PH, Spliet WG et al (2013) MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic Oligodendrogliomas and Oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res 19:5513–5522. https://doi.org/10.1158/1078-0432.CCR-13-1157
Van der Maaten L, Hinton G (2008) Visualizing data using t-SNE. City
Varn FS, Johnson KC, Martinek J, Huse JT, Nasrallah MP, Wesseling P et al (2022) Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 185(2184–2199):e2116. https://doi.org/10.1016/j.cell.2022.04.038
Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG et al (2017) Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. https://doi.org/10.1126/science.aai8478
Wang L, Babikir H, Muller S, Yagnik G, Shamardani K, Catalan F et al (2019) The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov 9:1708–1719. https://doi.org/10.1158/2159-8290.CD-19-0329
Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G et al (2021) EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol 18:170–186. https://doi.org/10.1038/s41571-020-00447-z
Weng Q, Wang J, Wang J, He D, Cheng Z, Zhang F et al (2019) Single-cell transcriptomics uncovers glial progenitor diversity and cell fate determinants during development and gliomagenesis. Cell Stem Cell 24(707–723):e708. https://doi.org/10.1016/j.stem.2019.03.006
Woolrich MW, Jbabdi S, Patenaude B, Chappell M, Makni S, Behrens T et al (2009) Bayesian analysis of neuroimaging data in FSL. Neuroimage 45:S173-186. https://doi.org/10.1016/j.neuroimage.2008.10.055
Wu Z, Abdullaev Z, Pratt D, Chung HJ, Skarshaug S, Zgonc V et al (2022) Impact of the methylation classifier and ancillary methods on CNS tumor diagnostics. Neuro Oncol 24:571–581. https://doi.org/10.1093/neuonc/noab227
Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G (2014) Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26:577–590. https://doi.org/10.1016/j.ccr.2014.07.028
Yuan Y, Xiang W, Yanhui L, Ruofei L, Shuang L, Yingjun F et al (2013) Ki-67 overexpression in WHO grade II gliomas is associated with poor postoperative seizure control. Seizure 22:877–881. https://doi.org/10.1016/j.seizure.2013.08.004
Zadeh G, Khan OH, Vogelbaum M, Schiff D (2015) Much debated controversies of diffuse low-grade gliomas. Neuro Oncol 17:323–326. https://doi.org/10.1093/neuonc/nou368
Zhang Y, Parmigiani G, Johnson WE (2020) ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom Bioinform 2:lqaa078. https://doi.org/10.1093/nargab/lqaa078
Zhao Z, Zhang KN, Wang Q, Li G, Zeng F, Zhang Y et al (2021) Chinese glioma genome atlas (CGGA): a comprehensive resource with functional genomic data from Chinese glioma patients. Genom Proteom Bioinform 19:1–12. https://doi.org/10.1016/j.gpb.2020.10.005
Acknowledgements
This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Funding
Open access funding provided by the National Institutes of Health. This work was supported by the National Science Foundation Graduate Research Fellowship Program DGE-1762114 (NN) and the Intramural Research Program of the National Institute of Neurological Disorders and Stroke at the National Institutes of Health (PJC).
Author information
Authors and Affiliations
Contributions
The study was conceived and designed by NN and PJC. Acquisition of data was performed by NN. Data was analyzed and interpreted by NN, SC, VH, SA, SR, AS, and PJC. Drafting of manuscript was completed by NN and PJC. Critical review of manuscript was additionally performed by LS, TM, MRG and ECH. All authors have approved this version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The use of human subject material was performed in accordance with the World Medical Association Declaration of Helsinki and with the approval of the National Institutes of Health Review Board.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nuechterlein, N., Cimino, S., Shelbourn, A. et al. HOXD12 defines an age-related aggressive subtype of oligodendroglioma. Acta Neuropathol 148, 41 (2024). https://doi.org/10.1007/s00401-024-02802-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00401-024-02802-1