
Abstract
Progressive meningiomas that have failed surgery and radiation have a poor prognosis and no standard therapy. While meningiomas are more common in females overall, progressive meningiomas are enriched in males. We performed a comprehensive molecular characterization of 169 meningiomas from 53 patients with progressive/high-grade tumors, including matched primary and recurrent samples. Exome sequencing in an initial cohort (n = 24) detected frequent alterations in genes residing on the X chromosome, with somatic intragenic deletions of the dystrophin-encoding and muscular dystrophy-associated DMD gene as the most common alteration (n = 5, 20.8%), along with alterations of other known X-linked cancer-related genes KDM6A (n =2, 8.3%), DDX3X, RBM10 and STAG2 (n = 1, 4.1% each). DMD inactivation (by genomic deletion or loss of protein expression) was ultimately detected in 17/53 progressive meningioma patients (32%). Importantly, patients with tumors harboring DMD inactivation had a shorter overall survival (OS) than their wild-type counterparts [5.1 years (95% CI 1.3–9.0) vs. median not reached (95% CI 2.9–not reached, p = 0.006)]. Given the known poor prognostic association of TERT alterations in these tumors, we also assessed for these events, and found seven patients with TERT promoter mutations and three with TERT rearrangements in this cohort (n = 10, 18.8%), including a recurrent novel RETREG1–TERT rearrangement that was present in two patients. In a multivariate model, DMD inactivation (p = 0.033, HR = 2.6, 95% CI 1.0–6.6) and TERT alterations (p = 0.005, HR = 3.8, 95% CI 1.5–9.9) were mutually independent in predicting unfavorable outcomes. Thus, DMD alterations identify a subset of progressive/high-grade meningiomas with worse outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although most meningiomas are WHO (World Health Organization) grade I benign tumors, up to 20% are atypical (grade II) or malignant (III) [35]. WHO grades II and III meningiomas relapse in 50–90% of cases after surgical resection [10], indicative of an accelerated natural history. There is currently no effective systemic therapy to offer patients after surgery or radiation failure, due to the limited understanding of the genetic drivers underlying meningioma recurrence and progression. NF2 alterations appear to be enriched in progressive cases [21]. Genomic sequencing of non-NF2 meningiomas found mutations in SMO, AKT1, KLF4, and TRAF7 [7, 14], which are more common in the benign subset of the disease [7, 14, 43, 53].
In contrast, large-scale genomic studies in higher-grade meningiomas consistently reported a heterogeneous mutation spectrum with few recurrently mutated genes aside from NF2 [4, 22, 27]. In addition, in a novel DNA methylation profiling analysis of 497 meningiomas, Sahm et al. described six methylation classes with different patterns of cytogenetic aberrations, mutations, and histology [46]. Interestingly, the classification predicted more accurately the risk of disease progression in WHO I meningioma in comparison with the current WHO grading system [46], suggesting that tumors may harbor or acquire genomic alterations that predispose towards more aggressive biology.
Indeed, in focused-sequencing studies, Koelsche et al. were the first to identify mutations in the telomerase reverse transcriptase promoter (TERTp) region in meningiomas [28], and subsequent studies have found these mutations to be associated with worse outcome in a subset of higher-grade meningiomas [20, 24, 45]. As another example, we reported on frequent breast cancer (BRCA)1-associated protein-1 tumor (BAP1) suppressor gene inactivation in tumors with rhabdoid histology, associated with a worse prognosis [48].
Many progressive/higher-grade meningiomas lack known genetic alterations that account for their aggressive biology. As genomic alterations in meningiomas evolve over time during treatment, a longitudinal genomic analysis across the spectrum of initial and recurrent disease is likely required to discover new genetic alterations driving recurrence. Therefore, to identify additional drivers of progressive/higher-grade meningiomas, we performed comprehensive molecular analyses in 169 samples collected from 53 patients with progressive/higher-grade disease, including matched pairs of primary meningiomas and their recurrences.
Materials and methods
Cohort and samples’ description
We collected 169 samples from 53 patients with progressive/higher-grade disease (24 females and 29 males). The cohort spanned WHO grades II and III at original diagnosis (39 relapsed grade II, and 14 de novo grade III), as determined by two experienced neuropathologists (M.M-L. and E.A.W.) who independently reviewed the specimens. We performed whole-exome sequencing (WES) in samples from 24 patients with relapsed/high-grade meningiomas (18 WHO grade II and 6 de novo grade III tumors from 10 females and 14 males) (Supplementary Table 1/Online Resource 2). This initial cohort included eight paired samples from seven WHO grade II and one WHO grade III meningiomas at diagnosis that progressed as a grade III meningioma at recurrence (Figs. 1, 2). Patient tumor samples analyzed in the current study were obtained from Departments of Neurosurgery at the University Hospital Dresden in Germany and the Massachusetts General Hospital in Boston, USA. The samples were preserved either as fresh frozen or formalin-fixed paraffin-embedded (FFPE) tissue. Blood was obtained when available (n = 48) for germline analysis. All samples (blood and tumor tissue) were collected with informed consent and after approval by the Dana-Farber/Harvard Cancer Center and the local ethics committee of the Medical Faculty Carl Gustav Carus Dresden.

Schematic representation of the methodological flow used in this study with 169 meningioma samples from 53 patients

Autosomal/allosomal alterations and genome-wide SCNA significance levels. Mutations and copy number alterations in autosomes and allosomes in samples with whole-exome sequencing (n = 32 samples from 24 patients)
DNA/RNA purification
DNA and RNA were purified using AllPrep® DNA/RNA/miRNA Universal Kit for fresh frozen tissue, AllPrep DNA/RNA FFPE kit for FFPE tissue, and DNeasy Blood and Tissue Kit for blood samples (Qiagen, Germantown MD) following the manufacturer’s instructions.
Whole-exome sequencing
The Illumina exome targets approximately 37.7 Mb, focusing on coding regions of Gencode V11 genes, and coding regions of RefSeq and KnownGene tracks from the UCSC genome browser (http://genome.ucsc.edu), comprising Agilent SureSelect All Exon V2. The Illumina exome uses an in-solution DNA probe-based hybrid selection method to generate sequencing libraries [17, 19]. See Online Resource 1 for a more detailed description. Data were analyzed using the Broad Picard Pipeline which includes de-multiplexing and data aggregation. Additionally, samples were assessed for possible pre-sequencing contamination using ContEst [13].Somatic mutation calling of paired tumor/normal samples was performed using MuTect (v1.1.6) [12] and Strelka (v1.0.11) [47]. The somatic mutations that passed base- and read-mapping quality and evidence thresholds were annotated with information from various public genomic databases using Oncotator [41]. A table of all synonymous and non-synonymous mutations identified using these criteria are available in the supplemental materials (Supplementary Table 2/Online Resource 4).
Copy number analysis
Somatic copy number aberrations were inferred by normalizing samples’ raw read depths against a panel of normals (blood) using the tools CalculateTargetCoverage and GermlineCNVCaller from the Genome Analysis Toolkit (GATK) (v4) [2]. The resulting normalized copy ratios for each patient’s samples (normal and tumors) were jointly segmented on total copy ratio and variant allele fraction (VAF) of heterozygous loci using multi-sample circular binary segmentation (MSCBS) [63]. Then, we called contiguous segments of total copy ratio (TCR) as deleted/amplified if their log2(TCR) estimates were outside the interval (− 0.1, 0.1). Tumor purity and ploidy were estimated using ABSOLUTE (v1.4) [8]. See Online Resource 1 for a more detailed description.
RNA sequencing
RNA-seq and fusion detection were based on previously described methods [51]. Raw FASTQ files were mapped to the human genome (hg19) with STAR (v2.5.3) [15]. Mapped reads were filtered and de-duplicated using sambamba (v0.6.6) [54]. Feature quantification was performed using featureCounts (v1.5.0-p1) [34] against the RefSeq database (downloaded from the UCSC genome browser on 01/05/2017). See Online Resource 1 for a more detailed description.
To assess specific myogenic transcriptomic signature in meningioma samples with available RNA-seq data, we used the GTEx portal (https://www.gtexportal.org/home/) to identify genes (n = 20) that are exclusively expressed in mesodermal tissue (skeletal or heart muscles). Seven of these 20 genes showed significant expression in at least three samples in the RNA-seq cohort. The seven genes were: ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2 + Transporting 1 (ATP2A1), Desmin (DES), Enolase 3 (ENO3), Myosin light chain, phosphorylatable, fast skeletal muscle (MYLPF), Titin-cap (TCAP), Tropomyosin (TPM2) and Troponin T1 (TNNT1). We compared the expression of the seven genes between samples with and without DMD inactivation after scaling and centering the raw FPKM data within each gene of interest.
Reverse transcriptase PCR
cDNA was synthesized using 1 μg of total RNA with a High Capacity cDNA Kit (Applied Biosystems, Foster City) according to the manufacturer’s instructions. DMD exons 10–15 and 63–69 were amplified using primer set DMD-1 and DMD-2 with GAPDH as a control. In addition, TERT was amplified using primer sets TERT-1 and TERT-2 with 18S as a control. The primer sequences are listed in Supplementary Table 3/Online Resource 5.
Multiplex ligation-dependent probe amplification (MLPA)
MLPA analysis was carried out according to the manufacturer’s instructions (MRC Holland, Amsterdam, Netherlands) [32]. MLPA analysis software (Coffalyser.Net—provided by MRC-Holland) was used to generate and analyze the data.
Western blotting [55]
Western blotting was performed with the primary antibodies to dystrophin NCL-DYS1 (1:125) (Leica Biosystems Newcastle, UK) at 1 µg/ml and ß-actin (sc-47778, Santa Cruz). See Online Resource 1 for a more detailed description.
Immunohistochemistry
Immunohistochemistry (IHC) used to assess the expression of dystrophin in tumor cells was performed using NCL-DYS1 (Leica Biosystems Newcastle, UK), an antibody that reacts with the central rod domain of the full-length predominant isoform of dystrophin isoform 427 (1:7 dilution). The intensity of cytoplasmic signal was evaluated using semi-quantitative binning: 0 negative, 1 + weak, 2 + moderate, and 3 + strong. The H-score, determined by the formula [1 × (% cells 1 +) + 2 × (% cells 2 +) + 3 × (% cells 3 +)], with a maximum score of 300, was used as an expression of overall signal. Samples that were given an H-score of 75 or less by two independent neuropathologists (M.M-L. and E.A.W.) were classified as “reduced” staining. The smooth muscle in the tunica media of leptomeningeal vessels was used as internal positive control.
Cell lines
The human arachnoid cells (AC007-hTERT) and the benign meningioma cell line (Ben-Men-1) were provided by Dr. Vijaya Ramesh (Massachusetts General Hospital) [23]. IOMM-Lee was courtesy of Dr. Randy Jensen (University of Utah).
Electron microscopy
Electron microscopic analyses were carried out on all three meningioma cell lines, on three meningioma tissue samples of DMD-deleted cases and on two cases with no DMD inactivation. See Online Resource 1 for a more detailed description.
Immunofluorescence
Immunofluorescence was performed on cells from the IOMM-Lee and Ben-Men-1 cell lines using the primary antibody (1:100, Dystrophin, Ab15277, Abcam). See Online Resource 1 for a more detailed description.
Archer® FusionPlex®
Samples were interrogated for fusions by the Archer® FusionPlex® Solid Tumor (AK0034) kit. This technology utilizes an anchored multiplex polymerase chain reaction (AMP) technique that detects gene rearrangements agnostic of fusion partners [64]. FASTQ data analysis, including fusion calling, was performed by ArcherDx Analysis software v5.0.6 using default parameters.
Junction confirmation
Specific primers were designed to anneal around the fusion junctions. Each junction fragment was PCR amplified from tumor cDNA, with corresponding cDNA from fusion negative patients as a negative control. The primer sequences are listed in Supplementary Table 3/Online Resource 5.
TERT promoter mutation detection
The TERTp was assessed using Sanger sequencing as previously mentioned [24]. Mutations between the residues of − 124 (1295228) to − 146 (1295250) bp from the ATG start site in the TERTp were scored.
Statistical analysis
The Mann–Whitney U and Fisher’s exact tests were used to test for association of clinical variables with DMD inactivation or TERT alterations. The median point estimate and 95% confidence interval for progression-free survival (PFS) and overall survival (OS) were estimated by the Kaplan–Meier method. Survival curves were compared using a non-stratified log-rank test. Multivariate Cox regression models were applied to assess the impact of DMD inactivation, TERT alterations and NF2 mutations on patient’s survival data. The analysis included the following factors: patients’ age over 65, WHO grade at first diagnosis, sex, NF2 mutation status, TERT promoter status and DMD inactivation. A logistic regression model was used to estimate the odds of detecting under different conditions. Association was deemed significant if the p value was less than 0.05. All p values are two-sided, and all analyses were conducted using the SPSS software package (Version 21.0 SPSS Inc., Chicago, IL, USA). Additional statistical analyses were performed using R (v3.3.0).
Results
Mutation and copy number landscape in progressive/higher-grade meningioma
We identified a median of 1.24 and 1.14 somatic non-synonymous mutations per Mb in the initial tumor and recurrent samples, respectively, among patients with matched samples. None of the patients harbored germline NF2-mutations. We noted mutations in previously reported meningioma genes including NF2 (n = 11), TP53 (n = 1), PTEN (n = 1), CDKN2C (n = 2) (Fig. 2). A genome-wide significance analysis of recurrent somatic copy number events, using the computational algorithm GISTIC [3], similarly found previously reported deletions of CDKN2A (n = 5) and monosomy of chromosome 22 (n = 19) [6, 21, 42, 59] (Fig. 2 and Supplementary Fig. 1/Online Resource 3).
Loss of allosomal/sex chromosomes is common in progressive/high-grade meningiomas
While the female predominance in Grade I meningiomas is widely appreciated, it is also recognized that males have a tendency for more biologically aggressive tumors. We therefore performed a detailed copy number analysis of the sex chromosomes. We observed that 9/14 (64%) of male patients had somatic Y chromosome loss, while 4/10 (33.3%) of female patients had a somatic loss of one or both arms of the X chromosome, as previously reported [62] (Fig. 2 and Supplementary Fig. 1/Online Resource 3). In recurrent cases, we found genetic alterations in many known cancer-related genes on the X chromosome: KDM6A (one nonsense mutation and one focal deletion, n = 2), DDX3X (n = 1), RBM10 (n = 1), and STAG2 (n = 1) (Fig. 2).
DMD focal deletions are frequent structural alterations in progressive/high-grade meningiomas
We identified Xp21.1 as the most significant focal deletion scored by GISTIC within the cohort. Examining cohort samples across the focally deleted region on Xp21.1, there was convergence of these copy loss events around the Duchenne muscular dystrophy (DMD) gene, which encodes the protein dystrophin (Fig. 3a). We discovered focal DMD deletions in 5/24 patients (20.8%, 3 de novo, not previously irradiated meningiomas and two recurrences that received radiation) (Fig. 3a), that were not detectable in the matched blood DNA, providing evidence for the somatic nature of this alteration. While three male patients had focal DMD deletions spanning exons 2–30, two female patients had a larger somatic deletion spanning the entire gene (Fig. 3a).

Intragenic DMD deletions and mRNA expression loss. a Exon deletion pattern of DMD in five patients according to segmentation of normalized copy ratios. b Detection of copy number changes by Multiplex Ligation-dependent Probe Amplification (MLPA) in meningioma samples. Capillary electrophoresis pattern from a reference sample in comparison with a DMD-deleted meningioma (MGH011) shows the loss of exons 1–10 and exon 44, represented by approximately 50% reduced signal of the affected probes (red arrows). c mRNA expression in DMD in fragments per kilobase of transcript per million mapped reads (FPKM)
Since DMD resides in a late-replicating common fragile site (CFS), we considered whether Xp21.1 deletions might represent nonspecific genome-instability in progressive tumors. First, we examined genome-wide common fragile sites as well as all 40 known genes that are larger than 1 Mb across the genome and did not observe recurrent alterations within our cohort, except for DMD (Supplementary Table 2/Online Resource 4). Furthermore, we performed a pan-cancer analysis of separate TCGA samples (n = 10844), which identified DMD as a focal peak region of deletion in only a small fraction of diverse cancer types (Supplementary Fig. 1/Online Resource 3), consistent with a tissue-specific tumor-suppressor function of DMD. In addition, DMD was not focally amplified in any of the 33 individual cancer types analyzed in the TCGA dataset. Taken together, these findings provide evidence that deletions in DMD, regardless of the mechanism, may not simply be a reflection of general genomic instability in these tumors.
Evaluation of DMD deletions using MLPA confirms WES findings
To confirm and extend these findings with an orthogonal method, we used the widely available Multiplex Ligation-dependent Probe Amplification (MLPA) copy number assessment on 35 samples (all tumors from the initial cohort plus 11 validation samples) [32] (Fig. 3b). MLPA confirmed the presence of the DMD deletions in the identical genomic regions predicated by WES in four of five patients. In the fifth case, tumor purity was low [8] (Fig. 3a), potentially limiting the sensitivity of MLPA-based detection. Furthermore, we uncovered four additional DMD deletions in the 11 validation samples. In contrast, MLPA of seven benign meningiomas (five NF2-mutant and two NF2 wild-type) did not detect any deletions.
Corresponding loss of DMD RNA expression is seen in cases with focal deletion
To further evaluate DMD mRNA expression, we performed RNA sequencing of six samples (three with known DMD deletion and three wild-type controls), which confirmed loss of mRNA expression in all cases with DMD deletions (Fig. 3c).
Moreover, to analyze if meningiomas with DMD deletions may harbor a specific mesodermal transcriptomic signature compared to DMD-retained meningiomas, we analyzed the RNA expression data using the GTEx portal to identify genes that are predominantly expressed in the myogenic tissue (skeletal or heart muscles). Seven genes with detectable expression in at least three samples were identified and we compared their expression between samples with and without DMD inactivation. We observed a markedly higher expression level of these genes in the samples with DMD deletions, consistent with a myogenic (mesodermal) signature (Supplementary Fig. 3/Online Resource 3). Finally, we analyzed the transcript levels of DMD using reverse transcriptase PCR (RT-PCR) for mRNA array profiling in 126 progressive/high-grade meningioma samples from 44 patients, combining the initial and expansion cohorts. RT-PCR revealed loss of DMD mRNA expression in 13 patients, including the eight patients with focal DMD deletions detected per WES and MLPA (Supplementary Fig. 5/Online Resource 3).
Assessment of DMD inactivation with Western blotting and immunohistochemistry
We next performed Western blot analysis to assess the expression of dystrophin, the protein product of DMD, in seven samples with DMD deletion and five samples with retained DMD. Six of seven DMD-deleted samples exhibited loss of dystrophin expression, whereas all cases with intact DMD expressed the full-length isoform of dystrophin with the molecular weight of 427 kDa (isoform 427) (Fig. 4a and Supplementary Fig. 5/Online Resource 3). Furthermore, we performed immunohistochemistry (IHC) using an antibody that recognizes the dystrophin central rod domain in 88 available specimens from all 53 relapsed/high-grade meningioma patients, along with an additional control group of 21 grade I and 10 non-relapsed grade II meningiomas (Fig. 4b). As expected, intense and diffuse cytoplasmic dystrophin expression was present in all DMD-intact samples tested, whereas reduced expression was exclusively seen in samples with DMD deletions or loss of mRNA expression (n = 17 patients; 6 females and 11 males; 13 WHO grade II and 4 grade III).

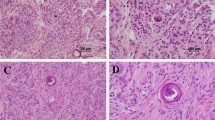
Validation of DMD inactivation using Western blot analysis and immunohistochemistry. a Representative Western blotting data showing loss of dystrophin 427 kDa expression in two samples with DMD deletion (MGH005 and MGH011) using DYS1 (reacts with the rod domain of dystrophin). PC positive control. b Immunohistochemistry with DYS1 (×400 magnification) demonstrates intense and diffuse cytoplasmic dystrophin expression in DMD-intact meningioma samples (I and II). In some cases, IHC demonstrated a patchy loss pattern suggestive of intratumoral heterogeneity (III, MGH012). Loss of dystrophin expression was predominantly seen in samples with DMD deletions (IV–VI) (cases MGH011 and MGH025). The smooth muscle in the tunica media of leptomeningeal vessels (arrow) and the skeletal muscle tissue (asterisk) were used as internal positive controls
In some cases, IHC demonstrated a patchy loss pattern suggestive of intratumoral heterogeneity, which is consistent with the emerging model of spatial heterogeneity in progressive meningiomas (Fig. 4b and Supplementary Table 1/Online Resource 2). Importantly, dystrophin inactivation was significantly more common in progressive grades II/III than the control cohort of grade I or non-progressive grade II meningiomas (17/53 vs. 2/31 patients; p = 0.0069, Fisher’s exact test).
DMD inactivation in meningiomas is associated with subcellular architecture changes
To characterize phenotypic changes associated with DMD inactivation, we profiled three cell meningioma lines for DMD: the human TERT-immortalized human arachnoid cells (AC007-hTERT), the Ben-Men-1 and the IOMM-Lee. We found DMD inactivation (loss of protein expression in immunofluorescence and immunoblotting) in the malignant IOMM-Lee meningioma cell line. This finding was consistent with the RNA expression data from this cell line provided by the CCLE (Supplementary Fig. 4/Online Resource 3). We then carried out electron microscopic (EM) analyses on all three meningioma cell lines, on three meningioma tissue samples of DMD-deleted cases and on two cases with no DMD inactivation. A consistent phenotypic association in the DMD-inactivated cases was a lower density of cytoskeleton filaments compared with the DMD-retained specimen (Fig. 5 and Supplementary Fig. 5/Online Resource 3). These EM findings suggest that loss of dystrophin in meningiomas may be associated with alterations in the cytoskeletal architecture.

Electron microscopic appearance of DMD-deleted and DMD-retained patient meningioma samples and cell lines. Representative electron microscopic imaging demonstrates a consistent phenotype in the DMD-inactivated cases (IOMM-Lee cell line, cases MGH005 and MGH025) with an apparently lower density of cytoskeleton filaments (arrows) compared with the DMD-retained specimen (Ben-Men-1 cell line and case MGH036)
Presence of DMD inactivation in progressive/higher-grade meningiomas confers a poor prognosis
To examine if DMD inactivation is associated with worse clinical outcome in patients, we examined the clinical outcomes in progressive/high-grade meningiomas (Fig. 6a). Strikingly, patients with DMD inactivation (assessed by IHC, n = 17) had a median OS of 5.1 years (95% CI 1.3–9.0), whereas progressive/high-grade patients in the dystrophin-retained cohort had a median OS that was not reached (95% CI 2.9–not reached, p = 0.006) (Fig. 6b). NF2 status was not associated with outcome [8.8 years in the NF2 wild-type cohort (95% CI 6.1–11.5 years) vs. 10.1 years in NF2-mutant patients (95% CI 5.3–14.8 years), p = 0.58].

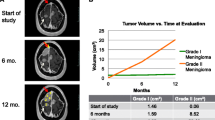
Outcome of meningioma patients with DMD inactivation. a Representative MRI of a patient (MGH011) with a WHO grade II cavernous sinus meningioma that acquired a DMD deletion during progression to WHO grade III. b Patients with a DMD inactivation had a shorter progression-free survival (1.6 years, 95% CI 1.1–2.2 vs. 2.6 years, 95% CI 2.3–2.8, p = 0.038). Likewise, patients with DMD inactivation had a median OS of 5.1 years (95% CI 1.3–9.0), which is significantly shorter than patients in the dystrophin-retained cohort (median not reached, 95% CI 2.9–not reached, p = 0.006). c The independent clinical association of DMD inactivation was substantiated by multivariate analyses, predicting unfavorable outcome in a model inclusive of NF2 mutations, TERT alterations, Age > 60 years, gender and initial grade
DMD inactivation and TERT alterations are mutually independent in predicting unfavorable outcomes in progressive/higher-grade meningiomas
Other genomic alterations are associated with poor outcome in progressive meningiomas, with the most common alterations noted in the TERT promoter (TERTp) [24, 39]. To assess whether DMD deletions are independently associated with unfavorable outcome, we sequenced TERTp in the recurrent sample (n = 53) of each patient in our cohort using quantitative PCR and identified seven TERTp mutations. DMD deletions and TERTp mutations were identified in both NF2-mutant and wild-type samples. In addition, we determined the frequency of TERT mRNA overexpression in this cohort using RT-PCR (Fig. 7a, d). We identified three patients (5.6%) with highly amplified TERT mRNA expression (Fig. 7a, b). Subsequent RNA sequencing revealed TERT rearrangements in all three patients with an amplified TERT mRNA expression: a novel fusion RETREG1-TERT that was present in two patients, along with a previously reported LPCAT1-TERT fusion in a third case [24] (Fig. 7c and Supplementary Table 3/Online Resource 5). In all cases, the TERT rearrangements began in either exon 2 or 3, upstream of the reverse transcriptase domain that begins in exon 4 (Fig. 7c–e and Supplementary Table 3/Online Resource 5). As expected, patients harboring TERT alterations (n = 10, 18.8%) in our cohort had a significantly worse OS (5.1 years, 95% CI 3.1–7.2) compared to TERT wild-type patients (18.5 years, 95% CI 14.6–22.4, p < 0.001) (Fig. 7f). The independent clinical association of DMD inactivation with unfavorable outcome was substantiated by multivariate analysis in a model inclusive of NF2 mutations and TERT alterations (OS, p = 0.033, HR = 2.6, 95% CI 1.0–6.6, Fig. 6c).

TERT alterations in progressive/high-grade meningiomas. a An example for a reverse transcriptase PCR demonstrating four samples from patients MGH030 and MGH032 with TERT mRNA overexpression. b Representative MRIs of a patient with a progressive meningioma harboring TERT rearrangements (case MGH032). c Schematic representation of the TERT rearrangement. Exons involved in the rearrangement are represented by colored boxes: LPCAT1 is reported in gray, RETREG1 in green, while TERT is reported in red. In all cases the rearrangements began in either exon 2 or 3, upstream of the reverse transcriptase domain that begins in exon 4, consistent with a proposed activating mechanism-of-action. d mRNA expression of TERT exons in FPKM confirming a TERT overexpression in the same samples (MGH030, MGH032 and MGH005). e RETREG1 (Exon 2)—TERT (Exon 3) fusion validation PCR in all four samples of patient MGH032 in comparison with a negative control (NC). f Patients whose meningiomas harbored TERT alterations (n = 10) had a significantly worse overall survival (5.1 years, 95% CI 3.1–7.2) compared to TERT wild-type patients (18.5 years, 95% CI 14.6–22.4, p < 0.001)
Discussion
While the female predominance in WHO grade I meningiomas is widely appreciated, it is also recognized that males have a tendency for more biologically aggressive tumors [21, 25, 26, 29, 39]. The mechanisms by which sex can affect meningioma grade and growth are not fully understood yet [5, 61]. In this comprehensive genomic profiling of progressive/high-grade meningiomas, we discovered genomic alterations on the sex chromosomes that could partly explain these phenomena. Notably, due to the difficulties of normalization and segmentation, the sex chromosomes have been typically excluded from copy number analyses of previous meningioma studies. We detected frequent inactivation of the X-linked DMD gene, as well as the emergence of genetic alterations in several known X-linked tumor-suppressor genes that occur in cancers with a skewed gender distribution toward male patients, including: KDM6A in T-cell acute lymphoblastic leukemia, DDX3X in medulloblastomas, and STAG2 in myeloid and CNS neoplasms [44, 52, 56].
DMD, a large gene at approximately 2.4 Mb, is primarily known due to its inactivation in the germline of patients with Duchenne muscular dystrophy [30]. The gene encodes dystrophin, a protein that regulates cytoskeleton remodeling, cell motility and cell proliferation in response to extracellular signals and interacts with adherens junctions in cells [16, 40]. In addition to its well-established function as a myogenic protein, emerging evidence supports the candidacy of DMD as a cancer gene subject to intragenic deletions in neoplasms of neural crest and mesodermal origin [1, 11, 31, 57, 58]. Indeed, DMD has a proven tumor-suppressive function in mouse models, with its inactivation serving as a frequent driver for sarcoma development in mdx mice [9].
Similar to merlin, the protein product of the NF2 meningioma tumor suppressor, dystrophin is expressed in the meninges (which are thought to be developmental derivatives of the mesoderm or the neural crest) and links the cytoskeleton to the cellular membrane, stabilizing cells by linking actin filaments, intermediate filaments, and further multiple components of the cellular cytoskeleton to the transmembrane dystroglycan complex [16, 33, 40, 49, 50]. Multiple independent groups have functionally linked DMD inactivation to cancer progression and metastases, implicating defects in cytoskeletal structure and signaling in mesodermal tumors harboring these genomic events, as subsequently facilitating tumor dissemination [57, 58]. Notably, dystrophin inactivation in cancers has been described as a relatively late event during tumor evolution, arising within subclonal populations, and detectable in all subsequent metastases [31, 57, 58].
Importantly, however, there is controversy about the functional role of DMD deletion in cancer, since it resides in a late-replicating fragile site [37]. Germline genomic deletion events are predominantly caused by truncated mutations located in the genomic region between exons 45–55 [18, 60].These fragile site-associated events result in phenotypic muscular dystrophy [30, 37, 60]. In contrast, we demonstrate that long-segment intragenic deletions of DMD are common in progressive/higher-grade meningiomas and cluster in the region of exons 5–15, distant from the germline fragile site target region. It is interesting to observe many structural aberrations in the cohort but no truncating alterations; however, a recent study analyzed a dataset of next-generation sequencing data from various tumor types for somatic mutations in the DMD gene, and found that only 57 out of 8052 (0.7%) cases harbored truncated mutations (nonsense or small indel frame shift alterations) [36]. Our data are therefore consistent with this finding that truncating mutations in DMD are rare events in neoplasia [36]. Nevertheless, the size of our cohort limits our power to analyze events that happen infrequently, and further studies with significantly more patients with progressive/high-grade meningiomas would be required to understand the mutational spectrum of DMD.
Bi-allelic NF2 inactivation is an early event in the initiation of sporadic meningiomas and is found frequently in both WHO grade I tumors and high-grade tumors [21, 38]. Importantly, not all NF2-mutant meningiomas are progressive; suggesting that additional acquired genetic alterations may be involved in progression. It is well-established that NF2-inacivated meningiomas are genomically unstable. This chromosomal instability has been defined as one of two main pathways for progression that may be causative in the pathogenesis of meningioma progression [21]. It is possible that DMD intragenic deletions may represent a consequent manifestation of the genomic instability at an early or late stage in NF2-inactivated progressive meningioma, which would explain the frequent co-occurrence of both alterations in progressive/high-grade meningiomas. In keeping with this logic, one would expect DMD alterations to be rarer than NF2 alterations and to be associated with worse outcome, both of which we observe within our cohort of high-grade/progressive meningiomas. Importantly, these findings argue that DMD deletion is not merely a proxy for NF2 status.
We further show that DMD-inactivated meningiomas manifest a myogenic (mesodermal) transcriptomic signature, and by electron microscopy we observe disrupted architecture of the cytoskeleton in DMD-deleted meningioma cells, when compared with DMD-retained cells. In addition, the focal clustering of DMD deletions in mesodermal cancers in the region of exons 5–15 (including our results here in meningiomas), which contains the actin-binding domain [43], is suggestive of a disrupted dystrophin–actin interaction. However, it is possible that NF2 inactivation could be related to the lower density of cytoskeleton filaments. Additional studies are needed to more fully characterize the functional role of DMD loss in meningioma pathogenesis.
Regardless, the clinical relevance of DMD inactivation is reflected by our observation of its association with unfavorable outcomes. Together with TERT promoter mutations, we discovered a novel RETREG1–TERT fusion that emerged in two different patients, expanding upon our prior findings [24]. As expected, TERT gene alterations were associated with a significantly worse survival. Importantly, DMD inactivation was independent of TERT alterations, in predicting unfavorable outcomes in progressive/higher-grade meningiomas. Thus, stratification for both DMD inactivation and TERT alterations can improve the design of progressive meningioma clinical trials and help improve patient management.
References
Arbajian E, Puls F, Antonescu CR, Amary F, Sciot R, Debiec-Rychter M et al (2017) In-depth genetic analysis of sclerosing epithelioid fibrosarcoma reveals recurrent genomic alterations and potential treatment targets. Clin Cancer Res 23:7426–7434. https://doi.org/10.1158/1078-0432.CCR-17-1856
Babadi Mehrtash DIB, Lee Samuel K, Andrey Smirnov AC, Lichtenstein Lee et al (2017) Abstract 3580: gATK CNV: copy-number variation discovery from coverage data. Am Assoc Cancer Res 77(13 Supplement):3580. https://doi.org/10.1158/1538-7445.AM2017-3580
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J et al (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463:899–905. https://doi.org/10.1038/nature08822
Bi WL, Greenwald NF, Abedalthagafi M, Wala J, Gibson WJ, Agarwalla PK et al (2017) Genomic landscape of high-grade meningiomas. NPJ Genom Med. https://doi.org/10.1038/s41525-017-0014-7
Blitshteyn S, Crook JE, Jaeckle KA (2008) Is there an association between meningioma and hormone replacement therapy? J Clin Oncol 26:279–282. https://doi.org/10.1200/JCO.2007.14.2133
Boström J, Meyer-Puttlitz B, Wolter M, Blaschke B, Weber RG, Lichter P et al (2001) Alterations of the tumor suppressor genes CDKN2A (p16(INK4a)), p14(ARF), CDKN2B (p15(INK4b)), and CDKN2C (p18(INK4c)) in atypical and anaplastic meningiomas. Am J Pathol 159:661–669. https://doi.org/10.1016/S0002-9440(10)61737-3
Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G et al (2013) Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet 45:285–289. https://doi.org/10.1038/ng.2526
Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T et al (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30:413–421. https://doi.org/10.1038/nbt.2203
Chamberlain JS, Metzger J, Reyes M, Townsend D, Faulkner JA (2007) Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J 21:2195–2204. https://doi.org/10.1096/fj.06-7353com
Chamberlain MC (2012) The role of chemotherapy and targeted therapy in the treatment of intracranial meningioma. Curr Opin Oncol 24:666–671. https://doi.org/10.1097/CCO.0b013e328356364d
Cheng J, Demeulemeester J, Wedge DC, Vollan HKM, Pitt JJ, Russnes HG et al (2017) Pan-cancer analysis of homozygous deletions in primary tumours uncovers rare tumour suppressors. Nat Commun 8:1221. https://doi.org/10.1038/s41467-017-01355-0
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C et al (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31:213–219. https://doi.org/10.1038/nbt.2514
Cibulskis K, McKenna A, Fennell T, Banks E, DePristo M, Getz G (2011) ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics 27:2601–2602. https://doi.org/10.1093/bioinformatics/btr446
Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K et al (2013) Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 339:1077–1080. https://doi.org/10.1126/science.1233009
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Ervasti JM (2007) Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta 1772:108–117. https://doi.org/10.1016/j.bbadis.2006.05.010
Fisher S, Barry A, Abreu J, Minie B, Nolan J, Delorey TM et al (2011) A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol 12:R1. https://doi.org/10.1186/gb-2011-12-1-r1
Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT et al (2009) Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 30:1657–1666. https://doi.org/10.1002/humu.21114
Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W et al (2009) Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 27:182–189. https://doi.org/10.1038/nbt.1523
Goutagny S, Nault JC, Mallet M, Henin D, Rossi JZ, Kalamarides M (2014) High incidence of activating TERT promoter mutations in meningiomas undergoing malignant progression. Brain Pathol 24:184–189. https://doi.org/10.1111/bpa.12110
Goutagny S, Yang HW, Zucman-Rossi J, Chan J, Dreyfuss JM, Park PJ et al (2010) Genomic profiling reveals alternative genetic pathways of meningioma malignant progression dependent on the underlying NF2 status. Clin Cancer Res 16:4155–4164. https://doi.org/10.1158/1078-0432.CCR-10-0891
Harmancı AS, Youngblood MW, Clark VE, Coşkun S, Henegariu O, Duran D et al (2017) Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat Commun 8:14433. https://doi.org/10.1038/ncomms14433
James MF, Stivison E, Beauchamp R, Han S, Li H, Wallace MR et al (2012) Regulation of mTOR complex 2 signaling in neurofibromatosis 2-deficient target cell types. Mol Cancer Res 10:649–659. https://doi.org/10.1158/1541-7786.MCR-11-0425-T
Juratli TA, Thiede C, Koerner MVA, Tummala SS, Daubner D, Shankar GM et al (2017) Intratumoral heterogeneity and TERT promoter mutations in progressive/higher-grade meningiomas. Oncotarget 8:109228–109237. https://doi.org/10.18632/oncotarget.22650
Jääskeläinen J, Haltia M, Servo A (1986) Atypical and anaplastic meningiomas: radiology, surgery, radiotherapy, and outcome. Surg Neurol 25:233–242
Kane AJ, Sughrue ME, Rutkowski MJ, Shangari G, Fang S, McDermott MW et al (2011) Anatomic location is a risk factor for atypical and malignant meningiomas. Cancer 117:1272–1278. https://doi.org/10.1002/cncr.25591
Katz LM, Hielscher T, Liechty B, Silverman J, Zagzag D, Sen R et al (2018) Loss of histone H3K27me3 identifies a subset of meningiomas with increased risk of recurrence. Acta Neuropathol 135:955–963. https://doi.org/10.1007/s00401-018-1844-9
Koelsche C, Sahm F, Capper D, Reuss D, Sturm D, Jones DT et al (2013) Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol 126:907–915. https://doi.org/10.1007/s00401-013-1195-5
Krayenbühl N, Pravdenkova S, Al-Mefty O (2007) De novo versus transformed atypical and anaplastic meningiomas: comparisons of clinical course, cytogenetics, cytokinetics, and outcome. Neurosurgery 61:495–503. https://doi.org/10.1227/01.neu.0000290895.92695.22 (discussion 503–494)
Kunkel LM, Hejtmancik JF, Caskey CT, Speer A, Monaco AP, Middlesworth W et al (1986) Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature 322:73–77. https://doi.org/10.1038/322073a0
Körner H, Epanchintsev A, Berking C, Schuler-Thurner B, Speicher MR, Menssen A et al (2007) Digital karyotyping reveals frequent inactivation of the dystrophin/DMD gene in malignant melanoma. Cell Cycle 6:189–198. https://doi.org/10.4161/cc.6.2.3733
Lalic T, Vossen RH, Coffa J, Schouten JP, Guc-Scekic M, Radivojevic D et al (2005) Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet 13:1231–1234. https://doi.org/10.1038/sj.ejhg.5201465
Lallemand D, Curto M, Saotome I, Giovannini M, McClatchey AI (2003) NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev 17:1090–1100. https://doi.org/10.1101/gad.1054603
Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. https://doi.org/10.1093/bioinformatics/btt656
Louis DN OH, Wiestler OD, Cavenee WK, Ellison DW, Figarella-Branger D, Perry A et al (2016) WHO classification of tumours of the central nervous system, vol 1. Revised 4th Edition
Luce LN, Abbate M, Cotignola J, Giliberto F (2017) Non-myogenic tumors display altered expression of dystrophin (DMD) and a high frequency of genetic alterations. Oncotarget 8:145–155. https://doi.org/10.18632/oncotarget.10426
McAvoy S, Ganapathiraju S, Perez DS, James CD, Smith DI (2007) DMD and IL1RAPL1: two large adjacent genes localized within a common fragile site (FRAXC) have reduced expression in cultured brain tumors. Cytogenet Genome Res 119:196–203. https://doi.org/10.1159/000112061
Perry A, Scheithauer BW, Stafford SL, Lohse CM, Wollan PC (1999) “Malignancy” in meningiomas: a clinicopathologic study of 116 patients, with grading implications. Cancer 85:2046–2056
Peyre M, Gauchotte G, Giry M, Froehlich S, Pallud J, Graillon T et al (2017) De novo and secondary anaplastic meningiomas: a study of clinical and histomolecular prognostic factors. Neuro Oncol. https://doi.org/10.1093/neuonc/nox231
Prins KW, Humston JL, Mehta A, Tate V, Ralston E, Ervasti JM (2009) Dystrophin is a microtubule-associated protein. J Cell Biol 186:363–369. https://doi.org/10.1083/jcb.200905048
Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G et al (2015) Oncotator: cancer variant annotation tool. Hum Mutat 36:E2423–E2429. https://doi.org/10.1002/humu.22771
Rempel SA, Schwechheimer K, Davis RL, Cavenee WK, Rosenblum ML (1993) Loss of heterozygosity for loci on chromosome 10 is associated with morphologically malignant meningioma progression. Cancer Res 53:2386–2392
Reuss DE, Piro RM, Jones DT, Simon M, Ketter R, Kool M et al (2013) Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol 125:351–358. https://doi.org/10.1007/s00401-013-1093-x
Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L et al (2012) Novel mutations target distinct subgroups of medulloblastoma. Nature 488:43–48. https://doi.org/10.1038/nature11213
Sahm F, Schrimpf D, Olar A, Koelsche C, Reuss D, Bissel J et al (2016) TERT promoter mutations and risk of recurrence in meningioma. J Natl Cancer Inst. https://doi.org/10.1093/jnci/djv377
Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S et al (2017) DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol 18:682–694. https://doi.org/10.1016/S1470-2045(17)30155-9
Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK (2012) Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28:1811–1817. https://doi.org/10.1093/bioinformatics/bts271
Shankar GM, Abedalthagafi M, Vaubel RA, Merrill PH, Nayyar N, Gill CM et al (2017) Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas. Neuro Oncol 19:535–545. https://doi.org/10.1093/neuonc/now235
Shaw RJ, McClatchey AI, Jacks T (1998) Localization and functional domains of the neurofibromatosis type II tumor suppressor, merlin. Cell Growth Differ 9:287–296
Shaw RJ, McClatchey AI, Jacks T (1998) Regulation of the neurofibromatosis type 2 tumor suppressor protein, merlin, by adhesion and growth arrest stimuli. J Biol Chem 273:7757–7764
Smith KM, Fagan PC, Pomari E, Germano G, Frasson C, Walsh C et al (2018) Antitumor activity of entrectinib, a Pan-TRK, ROS1, and ALK inhibitor, in. Mol Cancer Ther 17:455–463. https://doi.org/10.1158/1535-7163.MCT-17-0419
Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT et al (2011) Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333:1039–1043. https://doi.org/10.1126/science.1203619
Strickland MR, Gill CM, Nayyar N, D’Andrea MR, Thiede C, Juratli TA et al (2016) Targeted sequencing of SMO and AKT1 in anterior skull base meningiomas. J Neurosurg. https://doi.org/10.3171/2016.8.jns161076
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P (2015) Sambamba: fast processing of NGS alignment formats. Bioinformatics 31:2032–2034. https://doi.org/10.1093/bioinformatics/btv098
Taylor LE, Kaminoh YJ, Rodesch CK, Flanigan KM (2012) Quantification of dystrophin immunofluorescence in dystrophinopathy muscle specimens. Neuropathol Appl Neurobiol 38:591–601. https://doi.org/10.1111/j.1365-2990.2012.01250.x
Van der Meulen J, Sanghvi V, Mavrakis K, Durinck K, Fang F, Matthijssens F et al (2015) The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood 125:13–21. https://doi.org/10.1182/blood-2014-05-577270
Wang Y, Fletcher JA (2015) Cell cycle and dystrophin dysregulation in GIST. Cell Cycle 14:2713–2714. https://doi.org/10.1080/15384101.2015.1064676
Wang Y, Marino-Enriquez A, Bennett RR, Zhu M, Shen Y, Eilers G et al (2014) Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat Genet 46:601–606. https://doi.org/10.1038/ng.2974
Weber RG, Boström J, Wolter M, Baudis M, Collins VP, Reifenberger G et al (1997) Analysis of genomic alterations in benign, atypical, and anaplastic meningiomas: toward a genetic model of meningioma progression. Proc Natl Acad Sci USA 94:14719–14724
White SJ, den Dunnen JT (2006) Copy number variation in the genome; the human DMD gene as an example. Cytogenet Genome Res 115:240–246. https://doi.org/10.1159/000095920
Wigertz A, Lönn S, Mathiesen T, Ahlbom A, Hall P, Feychting M et al (2006) Risk of brain tumors associated with exposure to exogenous female sex hormones. Am J Epidemiol 164:629–636. https://doi.org/10.1093/aje/kwj254
Zankl H, Seidel H, Zang KD (1975) Cytological and cytogenetical studies on brain tumors. V. Preferential loss of sex chromosomes in human meningiomas. Humangenetik 27:119–128
Zhang NR, Siegmund DO, Ji H, Li JZ (2010) Detecting simultaneous changepoints in multiple sequences. Biometrika 97:631–645. https://doi.org/10.1093/biomet/asq025
Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD et al (2014) Anchored multiplex PCR for targeted next-generation sequencing. Nat Med 20:1479–1484. https://doi.org/10.1038/nm.3729
Acknowledgements
This work is supported by US National Institutes of Health (NIH) 1R21NS099844 (to Drs. D. Cahill and P. Brastianos); the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Projektnummer: 401837860 and the Max Kade Foundation (to Dr. T. Juratli); the Meningioma Mommas (to Dr. H. Wakimoto); the Burroughs Wellcome Fund Career Award (to Dr. D. Cahill.); the Brain Science Foundation, the American Brain Tumor Association, and the Damon Runyon Research Foundation (to Dr. P. Brastianos).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
I.M.S. and H.A.E. are employees in Ignyta, and Inc. J.C. is an employee and owns stocks in Ignyta, Inc. All other authors have no conflicts of interest to report with regard to this manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1:
Detailed method and materials description: Whole-exome sequencing, copy number analysis, RNA sequencing, Multiplex Ligation-dependent Probe Amplification (MLPA), Western blotting, Electron Microscopy and Immunofluorescence (PDF 540 kb)
Online Resource 2: Supplementary Table
1: patients’ and tumor characteristics (XLSX 19 kb)
Online Resource 3:
Supplementary Fig. 1 | Copy number variations in progressive/high-grade meningiomas Deletions are depicted in blue, whereas amplifications are shown in red. Supplementary Fig. 2 | An overview of DMD deletions across multiple GISTIC analyses performed on the entire dataset (all_cancers) DMD is significantly focally deleted in 13 of 33 independent cancer types analyzed in the dataset. Among these, it is located within a focal peak region of deletion in 10 cancer types. For reference, 11.221% of all genes are significantly focally deleted in at least 13 cancer types and 5.877% of all genes are present in focal deletion peaks in at least 10 cancer types. The q-value calculated of focal DMD deletions in our cohort is 10−9.(http://portals.broadinstitute.org/tcga/gistic/browseGisticByGene). Supplementary Fig. 3 | Myogenic transcriptomic signature in higher-grade/progressive meningiomas RNA expression data was analyzed using the GTEx portal (https://www.gtexportal.org/home/) to identify genes that are predominantly expressed in the mesodermal tissue (skeletal or heart muscles). The expression of these genes was compared between samples with and without DMD inactivation. Of the seven mesodermal genes with detectable expression in at least three samples, a markedly higher expression level of these genes was observed in meningioma samples with DMD deletions.The seven genes shown in the heat map are:- ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2 + Transporting 1 (ATP2A1): encodes one of the SERCA Ca(2 +)-ATPases, which are intracellular pumps located in the sarcoplasmic or endoplasmic reticula of muscle cells.- Desmin (DES): encodes a muscle-specific class III intermediate filament.- Enolase 3 (ENO3): encodes an isoenzyme in skeletal muscle cells that may play a role in muscle development and regeneration.- Myosin light chain, phosphorylatable, fast skeletal muscle (MYLPF).- Titin-cap(TCAP): encodes a protein found in striated and cardiac muscle that binds to the titin Z1–Z2 domains and is a substrate of titin kinase, interactions thought to be critical to sarcomere assembly. Mutations in this gene are associated with limb-girdle muscular dystrophy type 2G- Tropomyosin (TPM2): encodes beta-tropomyosin, a member of the actin filament-binding protein family, and mainly expressed in slow, type 1 muscle fibers.- Troponin T1 (TNNT1): encodes a protein that is a subunit of troponin, which is a regulatory complex located on the thin filament of the sarcomere. Supplementary Fig. 4 | DMD inactivation in the malignant IOMM-Lee meningioma cell line(a) Immunohistochemistry with DYS1 demonstrates loss of dystrophin expression in IOMM-Lee cells implanted into the mouse brain. (b) Immunofluorescence using DMD ab15227 (Abcam) revealed the cytoplasmic loss of expression in the IOMM-Lee cell line when compared with the Ben-Men-1 (BM1) cell line. (c) Screenshot of DMD mRNA expression in three anaplastic meningioma cell lines (IOMM-Lee, CH157 and F5) as shown by the CCLE dataset. A significant reduction of DMD mRNA expression is seen in the IOMM-Lee cell line (RPKM 0.000712), whereas CH157 and F5 cell lines showed a higher expression (RPKM 0.404 and 0.510, respectively). https://portals.broadinstitute.org/ccle/data Supplementary Fig. 5 | Additional Western blotting, RT-PCR and electron microscopic data(a) Representative Western blotting using DYS1 (reacts with the rod domain of dystrophin) shows loss of dystrophin 427 kDa expression in two patient samples with DMD deletion (MGH001 and MGH025) and in the malignant IOMM-Lee meningioma cell line. (b) An example for a reverse transcriptase PCR demonstrating loss of DMD mRNA expression of exons 10–15 and 63–68 in case MGH031 (DMD-deleted sample), compared to samples of patients with intact DMD. (c) Representative electron microscopic imaging demonstrates the DMD-inactivated IOMM-Lee cell line displayed predominantly rounded cell morphology with an apparently lower density of cytoskeleton filaments compared with the human arachnoid cells (AC007-hTERT). (PDF 6619 kb)
Online Resource 4: Supplementary Table
2: Sheet 1: List of all synonymous and non-synonymous mutations identified using the criteria for somatic mutation calling Sheet 2: List of genes residing in late-replicating common fragile sites that were assessed for deletions in the current study Sheet 3: List of genes larger than 1 Mb evaluated for alterations in the current study (XLSX 498 kb)
Online Resource 5: Supplementary Table
3: Sheet 1: TERT fusion data Sheet 2: List of used RT-PCR primers in the study Sheet 3: List of used RT-PCR primers to validate the TERT fusions (XLSX 14 kb)
Rights and permissions
About this article

Cite this article
Juratli, T.A., McCabe, D., Nayyar, N. et al. DMD genomic deletions characterize a subset of progressive/higher-grade meningiomas with poor outcome. Acta Neuropathol 136, 779–792 (2018). https://doi.org/10.1007/s00401-018-1899-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-018-1899-7