
Abstract
Pediatric glioblastoma (pedGBM) is an extremely aggressive pediatric brain tumor, accounting for ~6% of all central nervous system neoplasms in children. Approximately half of pedGBM harbor recurrent somatic mutations in histone 3 variants or, infrequently, IDH1/2. The remaining subset of pedGBM is highly heterogeneous, and displays a variety of genomic and epigenetic features. In the current study, we aimed to further stratify an H3-/IDH-wild type (wt) pedGBM cohort assessed through genome-wide molecular profiling. As a result, we identified three molecular subtypes of these tumors, differing in their genomic and epigenetic signatures as well as in their clinical behavior. We designated these subtypes ‘pedGBM_MYCN’ (enriched for MYCN amplification), ‘pedGBM_RTK1’ (enriched for PDGFRA amplification) and ‘pedGBM_RTK2’ (enriched for EGFR amplification). These molecular subtypes were associated with significantly different outcomes, i.e. pedGBM_RTK2 tumors show a significantly longer survival time (median OS 44 months), pedGBM_MYCN display extremely poor outcomes (median OS 14 months), and pedGBM_RTK1 tumors harbor an intermediate prognosis. In addition, the various molecular subtypes of H3-/IDH-wt pedGBM were clearly distinguishable from their adult counterparts, underlining their biological distinctiveness. In conclusion, our study demonstrates significant molecular heterogeneity of H3-/IDH-wt pedGBM in terms of DNA methylation and cytogenetic alterations. The recognition of three molecular subtypes of H3-/IDH-wt pedGBM further revealed close correlations with biological parameters and clinical outcomes and may therefore, be predictive of response to standard treatment protocols, but could also be useful for stratification for novel, molecularly based therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pediatric glioblastoma (pedGBM) accounts for ~6% of all brain tumors in children aged 0–18 years (or ~15% when considering all high-grade gliomas together) [17, 19]. These aggressive malignancies remain incurable with current treatment strategies, which are similar to those applied for treatment of adult GBM and typically consist of surgical tumor removal followed by radiotherapy in combination with concurrent and maintenance temozolomide (TMZ) [6, 7]. Recently, however, it has become apparent that childhood and adult GBM represent molecularly distinct entities with differing biological backgrounds which, in turn, require different treatment strategies [11, 18, 21, 27]. Moreover, molecular studies conducted over the past few years have transformed our understanding of the extensive heterogeneity of pedGBM, with distinct subgroups correlating with specific genetic and epigenetic alterations and clinical patterns [4, 5, 10, 16, 27, 29].
Approximately, half of pedGBM harbor recurrent somatic mutations in H3F3A (encoding the histone variant H3.3) or H3.1/H3.2-encoding genes (HIST1H3B, HIST1H3C, HIST2H3C). In addition, IDH1 and IDH2 mutations, which are frequent in adult gliomas, are found in a handful of their pediatric counterparts [4, 5, 8, 26, 30, 33, 34]. The remaining pedGBM, lacking H3 or IDH mutations, can thus be termed H3-/IDH-wild type (wt) tumors. It is clear that this remaining subset of pedGBM is also heterogeneous, and includes molecularly distinct subgroups with a variety of genomic and epigenetic profiles, and perhaps, different clinical behavior. As an example, we recently identified a set of H3-/IDH-wt tumors originally diagnosed as “GBM” upon histological evaluation, but which disclosed the molecular signatures and more favorable clinical course of either pleomorphic xanthoastrocytomas (PXA) or low-grade gliomas (LGG) [16]. In the current study, we aimed to further stratify H3-/IDH-wt pedGBM through genome-wide molecular profiling, a strategy which has recently proved fruitful in other pediatric brain tumors, such as medulloblastoma [9], ependymoma [20], and tumors previously grouped under the term ‘CNS-PNET’ [28]. As a result, we identified three molecular subtypes of these tumors, differing in their genomic and epigenetic signatures, as well as in their associated clinical characteristics.
Materials and methods
Patient population
Tissue samples (77 formalin-fixed paraffin-embedded (FFPE), 10 frozen) were obtained from 87 pediatric patients (age 2–18 years; only two cases <3 years) with a histological diagnosis of “glioblastoma, WHO grade IV” and methylome signatures corresponding to the molecular diagnosis “glioblastoma”. Tumors with a histological appearance of pedGBM but clearly displaying molecular signatures of other tumor types, e.g. PXA or LGG, were excluded [10]. Thirty-nine of the 87 cases were previously reported [16]. Histological diagnosis was based on the current WHO criteria for GBM—an astrocytic glioma with brisk mitotic activity, microvascular proliferation and/or necrosis [17]. Details of the cohort are given in Supplementary Table 1. All tumors were considered H3-/IDH-wt pedGBM because they showed no mutations of genes encoding histone 3 variants (H3F3A, HIST1H3A, HIST1H3B, HIST1H3C, HIST2H3C) or IDH1, which were screened by direct sequencing. The majority of samples were collected from the NN Burdenko Neurosurgical Institute in Moscow, and the H3-/IDH-wt samples represented about one third of all pediatric cases histologically diagnosed as high-grade glioma during the period of collection.
Molecular analysis
DNA was extracted from tumors and analyzed for genome-wide DNA methylation patterns using the Illumina HumanMethylation450 BeadChip (450 k) array. Processing of DNA methylation data was performed with custom approaches as previously described [9, 29], and copy number profiles were generated using the ‘conumee’ package for R (https://www.bioconductor.org/packages/release/bioc/html/conumee.html). Clustering was performed using the beta values of the 10,000 most variably methylated probes as measured by standard deviation. Samples were clustered using Pearson correlation coefficient as the distance measure and average linkage (x-axis). Methylation probes were reordered by hierarchical clustering using Euclidean distance and average linkage (y-axis). Additional analysis of tumor subgroups was performed using a t-distributed stochastic neighbor embedding (t-SNE)-based approach [31]. None of the samples showed any signatures resembling H3 or IDH mutant groups, further supporting that they are wildtype for these genes and not harboring mutations at allele frequencies below the detection of Sanger sequencing.
Gene expression data for selected genes from the Affymetrix Human Genome U133 Plus2.0 array were collated from previously published sources [3, 29, 32].
To evaluate the methylation status of the MGMT promoter region, we used the MGMT_STP27 logistic regression model [2]. In addition, mutational analysis for TP53 gene and the TERT promoter region was performed using targeted Sanger sequencing. FISH analyses with commercially available probes to human oncogenes for confirmation of selected copy number aberrations (CNAs), as well as immunohistochemical analysis for ATRX protein expression, were performed as previously described [15, 25]. Scoring of amplifications for PDGFRA, EGFR and MYCN was consistent between FISH and array-based analysis for all cases.
Statistics
The distribution of overall survival (OS) was calculated according to the Kaplan–Meier method. OS was calculated from the date of diagnosis until death of patient from disease or last contact for patients who were still alive. Cox proportional hazards regression models were used to estimate hazard ratios, which are provided with 95% confidence intervals and a p value from the Wald test. Tests with a p value below 0.05 were considered significant. The multivariate analysis considered all variables showing significance on a univariate level.
Results
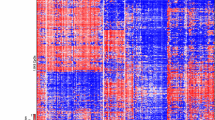
Hierarchical clustering of genome-wide DNA methylation data from 87 H3-/IDH-wt tumors (excluding those with PXA- or LGG-like patterns) showed three clearly demarcated clusters (Fig. 1a). These clusters were robustly reproducible when using various numbers of differentially methylated CpG sites (5000 or 2500; not shown). This grouping was also supported by multiple additional analysis using t-SNE (Fig. 1b). Although additional epigenetic sub-clusters could potentially be inferred from the clustering data, combined assessment of clustering and t-SNE supported three major subtypes, as described below.

a Unsupervised hierarchical clustering analysis of 87 H3-/IDH-wt pediatric GBM samples (based on the 10,000 most variably methylated probes). Three epigenetic subtypes of these tumors are clearly distinguishable, and enriched for specific oncogene amplifications: RTK1 (PDGFRA), RTK2 (EGFR) and MYCN (p < 0.005). b Grouping of tumor methylation profiles according to t-SNE confirms the presence of three distinct tumor subtypes
Tumors within these three clusters disclosed significant differences in their cytogenetic profiles (Fig. 1a; Table 1) and we thus designated these pedGBM subtypes further according to an enrichment for specific oncogene amplifications (representative copy number plots shown in Fig. 2). The largest pedGBM subtype, termed ‘pedGBM_MYCN’ (n = 36), disclosed a high frequency of MYCN amplification (50%; p < 0.0001; Fisher’s exact test), often together with co-amplification of the nearby ID2 gene on 2p (seen in 12/18 MYCN amplified tumors, 66%). Three further tumors without MYCN amplification displayed an amplification of MYC on Chr8q. Other recurrent amplifications in this subgroup included CDK4/6 (22%). The second largest subtype, designated as ‘pedGBM_RTK1’ (n = 33), showed an enrichment for PDGFRA amplification (33%; p < 0.005; Fisher’s exact test), whereas other high-level CNAs were rarer. The third, comparably smaller tumor subtype (n = 18) was designated ‘pedGBM_RTK2’, and showed frequent amplification of EGFR (50%; p < 0.0001; Fisher’s exact test). This group also displayed some other CNAs that are prototypic of adult GBM, most notably CDKN2A/B homozygous deletions (72%), and also losses involving Chr10q (50%) and gain of chromosome 7 (28%). Examination of gene expression data for a subset of independent cases with discernible subgroup affiliation confirmed high expression of EGFR, PDGFRA and MYCN in the respective subgroups (as well as partially across subgroups), including samples without an obvious amplification event (Supplementary Fig. 2), supporting a wider oncogenic role for these candidates.

Cytogenetic patterns of H3-/IDH-wt pediatric GBM. Amplifications (arrows) of MYCN/ID2 (a), PDGFRA (b), and EGFR (c) were detected by 450 k analysis as prototypic aberrations for each of three tumor variants
Subtype-specific differences of H3-/IDH-wt pedGBM were also supported by integration of additional molecular data. In contrast to IDH- and H3-mutant pedGBM, ATRX alterations (as measured by loss of immunopositivity) were found in only 7% of these tumors. In contrast, TP53 mutations were common (56% of the total samples analyzed), but displayed differences in subtype-specific frequency—being most frequent in pedGBM_MYCN tumors (67%) but rarer in the PDGFRA and EGFR groups (48 and 50%, respectively). Hotspot mutations in the TERT promoter, previously described as a rare event in pediatric GBM as a whole [14], were also assessed. Interestingly, while no pedGBM_RTK1 samples displayed this alteration and a relatively low proportion of pedGBM_MYCN tumors carried a mutation (26%), it was a highly recurrent event in the pedGBM_RTK2 subgroup (64%; approaching the rate observed in adult GBM [13]). MGMT promoter methylation was found in only 10/87 H3-/IDH-wt pedGBM (11%), and also displayed variability amongst the molecular subtypes, being found in 18% of pedGBM_RTK1 cases but only occasionally in the MYCN and pedGBM_RTK2 groups (11 and 0%, respectively). Pairwise comparisons showed no significant differences in the age distribution of each subtype (not shown).
To compare the biological nature of our newly outlined H3-/IDH-wt pedGBM subtypes with adult GBM also lacking these hotspot mutations, we additionally performed a combined unsupervised clustering and t-SNE analysis with a cohort of H3-/IDH-wt adult GBM (adGBM, n = 75) displaying previously described molecular signatures (RTKI, RTKII and Mesenchymal) [27, 29]. The pedGBM and adGBM samples clustered separately, and the pedGBM molecular subtypes remained apparent in this larger extended analysis (Fig. 3 and Supplementary Fig. 1). We therefore, did not observe any close relationship between the molecular subtypes of H3-/IDH-wt adGBM and pedGBM. Furthermore, comparison with MYC/MYCN associated medulloblastoma (Group 3 and Group 4, respectively) and other malignant pediatric brain tumors such as the four new entities previously subsumed under ‘CNS-PNET’ [28] confirmed the distinct nature of the GBM subgroups (Supplementary Fig. 3). The MYCN tumors with GBM histology were, however, similar to a group of MYCN tumors with more PNET-like histology [28], suggesting that this is one biological group with varying histological features.

Analysis of methylation patterns of pediatric and adult glioblastoma by t-SNE indicates that the three pedGBM subtypes are clearly distinct from the adult tumors
Correlation of pedGBM subtypes and other clinical and/or molecular parameters with patient outcomes revealed additional notable associations. The three subtypes showed specific patterns of tumor location in the CNS; 14 and 18% of pedGBM_MYCN and pedGBM_RTK1 tumors, respectively, were located infratentorially (pedGBM_MYCN all in the brain stem; pedGBM_RTK1—three brain stem and three cerebellar tumors) whereas only 1/18 pedRTK2 tumor (6%) was located in the cerebellum. No tumors displayed neuroradiological patterns of tumor dissemination at initial presentation, although a subset presented with disseminated relapsed disease. Treatment details were available for 80/87 patients (92%), with most (n = 75) being managed in the Burdenko Neurosurgical Institute (Moscow; Russia) between 2000 and 2014. All these patients were treated on standard therapeutic protocols according to the histopathological diagnosis “glioblastoma, WHO grade IV” including surgery (gross total or subtotal tumor resection) followed by radiotherapy (limited field fractionated external beam radiotherapy with a dose 54–59.4 Gy on the tumor bed) and adjuvant chemotherapy with TMZ. Follow-up data were available for 74 patients, demonstrating that 62% of patients died within the follow-up period (median overall survival (OS) of 22 months). Tumors from the pedGBM_RTK2 group were associated with a significantly longer overall survival time (median OS 44 months); pedGBM_MYCN tumors displayed extremely poor outcomes (median OS 14 months), and pedGBM_RTK1 tumors were associated with an intermediate prognosis (median OS 21 months; survival differences are statistically significant; log rank test, p < 0.0001; Fig. 4). In contrast, there was no survival difference when only comparing tumors with or without PDGFRA or EGFR amplification (p = 0.265 and p = 0.136, respectively). The presence of a MYCN amplification is a poor prognostic marker, but does not show as significant a difference in survival curves as the three-group structure (p = 0.003). Univariate OS analysis for various clinical and molecular parameters across the cohort revealed that presence of homozygous CDKN2A/B deletion was also significantly associated with poor outcome, whereas other parameters were not. Multivariate analysis identified molecular tumor subtype as the only independent significant prognostic marker for clinical outcome across the H3-/IDH-wt pedGBM cohort (p < 0.0001), highlighting the prognostic impact of our proposed molecular subtypes in comparison with other variables (Table 2).

Overall survival for the molecular subtypes of H3-/IDH-wt pediatric GBM. All inter-group differences are statistically significant (log rank test; p < 0.0001)
Discussion
Changing the current paradigm of pediatric high-grade glioma treatment will require a better understanding of the key molecular mechanisms driving these lethal neoplasms. Given that pedGBMs are clinically and biologically heterogeneous, with distinct subtypes driven by various molecular events, any attempts at improving therapy will require patient stratification based upon molecular hallmarks. In contrast to well-established subgroups with known driving events such as H3 mutation, which have recently become a major focus of international research efforts, the remaining H3-/IDH1-wt tumors have not yet received the same attention.
Applying an integrated approach using genome-wide DNA methylation profiling and other targeted methods for mutation detection and verification of copy number changes, we delineated three biological subtypes of H3-/IDH1-wt pedGBM based on their global DNA methylation patterns. The absence of any clear overlap with their adult counterparts suggests a marked age-related difference between not just histone-mutant, but also H3-/IDH-wt adult and pediatric GBM subgroups. The spectra of GBM in these different age groups, therefore, truly appear to be rather ‘distant cousins’ than ‘close relatives’ [11]. Further evaluation of features such as gene expression patterns and mutational spectrum is now warranted to verify the true extent of this separation and the distinct nosologic classes within the wide-spectrum of GBM. A distinct origin of pedGBM is also further suggested by the low-frequency of MGMT promoter methylation across all H3-/IDH-wt pedGBM subtypes, implying a likely low efficacy of TMZ-based therapy (or other alkylating agents) and thus a need for alternative treatment strategies.
Our study revealed pedGBM_MYCN tumors as the most biologically aggressive molecular subtype of H3-/IDH-wt pedGBM, with an average OS comparable with that of the extremely unfavorable K27-mutant diffuse midline high-grade gliomas [12, 16, 29]. This “MYCN group” has previously been identified as a distinct molecular variant of diffuse intrinsic pontine glioma (DIPG) [4]. Here, we find that many of these tumors are located outside of the brainstem. These tumors disclosed recurrent high-level amplifications of the MYCN oncogene, frequently co-amplified with the nearby ID2, and recurrent cooperating TP53 mutation. In addition, a tangible proportion of tumors diagnosed as “CNS-PNET” were recently found to share the same pedGBM_MYCN epigenetic signature as described here [28], making a broader biologically defined group of supratentorial tumors with GBM and/or PNET-like histopathology (Supplementary Fig. 3). The molecular background of their biological aggressiveness is still unclear, although most display various oncogene amplifications. Targeted therapy matched to the particular genetic aberrations may therefore, represent an alternative to standard TMZ-based protocols in pedGBM_MYCN tumors [18]. For example, patients from this pedGBM subgroup with MYCN amplification may benefit from therapies targeting MYCN, such as indirect inhibition of MYCN by BRD4 inhibitors [24].
The pedGBM_RTK1 subgroup was characterized by a high frequency of PDGFRA amplifications and scarcity of other typical GBM-associated cytogenetic aberrations. These tumors occupy an intermediate prognostic niche. PDGFRA amplification has previously been identified as a frequent feature of pedGBM/DIPG, and was recently characterized by GEP analysis as being associated with a “proneuronal” expression pattern [21, 22], which in DIPG was associated with resistance to therapy and dismal prognosis [22, 23]. It is interesting to note that H3 K27-mutant diffuse midline gliomas also show a high frequency of PDGFRA amplification and an enrichment of genes from the “proneuronal” expression signature [27, 29]. PDGFRA amplification may represent an attractive treatment target given the availability of several inhibitors acting against this receptor tyrosine kinase [1]. Although there have not yet been convincing reports of efficacy of these drugs in high-grade glioma, it could be proposed that stratification based on true genetic PDGFRA alteration (and possibly excluding K27-mutant tumors) may provide a rationale for further attempts at such targeted therapy.
The third molecular subgroup, pedGBM_RTK2, disclosed a cytogenetic similarity to adult GBM, but unexpectedly showed relatively favorable outcomes—5-year OS was close to 50%, and over 80% of patients survived more than 2 years. Consistent with this, the epigenetic profile of pedGBM_RTK2 was clearly different from all known adGBM variants, stressing their biological distinctiveness and a necessity of further molecular clarification in future studies. In particular, the relatively favorable outcomes may suggest responsiveness of pedGBM_RTK2 to the standard treatment protocols applied, although MGMT methylation (a marker for response to TMZ in adult GBM) was absent in this group. On the other hand, targeted use of RTK/MAPK inhibitors as an alternative or in addition to TMZ-based therapy may be a logical option for this pedGBM subtype.
In conclusion, this study demonstrates the significant molecular heterogeneity of H3-/IDH-wt pedGBM on a global DNA methylation level and in terms of cytogenetic aberrations. Whilst the true extent of this heterogeneity will likely continue to evolve with increasing cohort sizes and improved understanding (i.e. further rare subsets), our delineation of three molecular subtypes of H3-/IDH-wt pedGBM revealed correlations with clinical outcomes, which may be predictive of response to standard treatment protocols and could provide a rationale for molecularly informed therapeutic strategies. Practical and reliable prognostication of pedGBM through global DNA methylation assessment will be essential for rational stratification in future clinical trials for these aggressive, treatment-resistant tumors.
References
Arrondeau J, Huillard O, Tlemsani C, Cessot A, Boudou-Rouquette P, Blanchet B, Thomas-Schoemann A, Vidal M, Tigaud JM, Durand JP et al (2015) Investigational therapies up to Phase II which target PDGF receptors: potential anti-cancer therapeutics. Expert Opin Investig Drugs 24:673–687. doi:10.1517/13543784.2015.1005736
Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C, Dietrich PY, Weller M, Mariani L, Heppner FL et al (2012) MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol 124:547–560. doi:10.1007/s00401-012-1016-2
Bender S, Gronych J, Warnatz HJ, Hutter B, Groebner S, Ryzhova M, Pfaff E, Hovestadt V, Weinberg F, Halbach S et al (2016) Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat Med 22:1314–1320. doi:10.1038/nm.4204
Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U et al (2014) Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46:451–456. doi:10.1038/ng.2936
Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, Pages M, Taylor KR, Saulnier P, Lacroix L et al (2015) Histone H3F3A and HIST1H3B K27 M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 130:815–827. doi:10.1007/s00401-015-1478-0
Cohen KJ, Pollack IF, Zhou T, Buxton A, Holmes EJ, Burger PC, Brat DJ, Rosenblum MK, Hamilton RL, Lavey RS et al (2011) Temozolomide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro Oncol 13:317–323. doi:10.1093/neuonc/noq191
Duffner PK, Horowitz ME, Krischer JP, Burger PC, Cohen ME, Sanford RA, Friedman HS, Kun LE (1999) The treatment of malignant brain tumors in infants and very young children: an update of the Pediatric Oncology Group experience. Neuro Oncol 1:152–161
Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA et al (2014) Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 46:462–466. doi:10.1038/ng.2950
Hovestadt V, Remke M, Kool M, Pietsch T, Northcott PA, Fischer R, Cavalli FM, Ramaswamy V, Zapatka M, Reifenberger G et al (2013) Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol 125:913–916. doi:10.1007/s00401-013-1126-5
Jones C, Baker SJ (2014) Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer. doi:10.1038/nrc3811
Jones C, Perryman L, Hargrave D (2012) Paediatric and adult malignant glioma: close relatives or distant cousins? Nat Rev Clin Oncol 9:400–413. doi:10.1038/nrclinonc.2012.87
Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L et al (2012) K27 M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439–447. doi:10.1007/s00401-012-0998-0
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA 110:6021–6026. doi:10.1073/pnas.1303607110
Koelsche C, Sahm F, Capper D, Reuss D, Sturm D, Jones DT, Kool M, Northcott PA, Wiestler B, Bohmer K et al (2013) Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol 126:907–915. doi:10.1007/s00401-013-1195-5
Korshunov A, Capper D, Reuss D, Schrimpf D, Ryzhova M, Hovestadt V, Sturm D, Meyer J, Jones C, Zheludkova O et al (2016) Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol 131:137–146. doi:10.1007/s00401-015-1493-1
Korshunov A, Ryzhova M, Hovestadt V, Bender S, Sturm D, Capper D, Meyer J, Schrimpf D, Kool M, Northcott PA et al (2015) Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol 129:669–678. doi:10.1007/s00401-015-1405-4
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellison DW, Figarella-Branger D, Perry A, Reifenberger G, Von Deimling A (2016) WHO classification of tumours of the central nervous system, revised, 4th edn. IARC, City
Northcott PA, Pfister SM, Jones DT (2015) Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies. Lancet Oncol 16:e293–e302. doi:10.1016/S1470-2045(14)71206-9
Ostrom QT, de Blank PM, Kruchko C, Petersen CM, Liao P, Finlay JL, Stearns DS, Wolff JE, Wolinsky Y, Letterio JJ et al (2015) Alex’s Lemonade stand foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol 16(Suppl 10):x1–x36. doi:10.1093/neuonc/nou327
Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27:728–743. doi:10.1016/j.ccell.2015.04.002
Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D et al (2010) Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 28:3061–3068. doi:10.1200/JCO.2009.26.7252
Phillips JJ, Aranda D, Ellison DW, Judkins AR, Croul SE, Brat DJ, Ligon KL, Horbinski C, Venneti S, Zadeh G et al (2013) PDGFRA amplification is common in pediatric and adult high-grade astrocytomas and identifies a poor prognostic group in IDH1 mutant glioblastoma. Brain Pathol 23:565–573. doi:10.1111/bpa.12043
Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, Andreiuolo F, Carvalho D, Reis R, Guerrini-Rousseau L et al (2012) Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One 7:e30313. doi:10.1371/journal.pone.0030313
Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P et al (2013) Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov 3:308–323. doi:10.1158/2159-8290.CD-12-0418
Reuss DE, Sahm F, Schrimpf D, Wiestler B, Capper D, Koelsche C, Schweizer L, Korshunov A, Jones DT, Hovestadt V et al (2015) ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol 129:133–146. doi:10.1007/s00401-014-1370-3
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231. doi:10.1038/nature10833
Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF et al (2014) Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer 14:92–107. doi:10.1038/nrc3655
Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DT, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I et al (2016) New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164:1060–1072. doi:10.1016/j.cell.2016.01.015
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. doi:10.1016/j.ccr.2012.08.024
Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D et al (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46:457–461. doi:10.1038/ng.2925
van der Maarten L, Hinton G (2008) Visualizing high-dimensional data using t-SNE. J Mach Learn Res 9:2579–2605
Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag A, Boudalil M, Previti C, Wolf S, Schmidt S et al (2016) Next-generation personalised medicine for high-risk paediatric cancer patients—the INFORM pilot study. Eur J Cancer 65:91–101. doi:10.1016/j.ejca.2016.06.009
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44:251–253. doi:10.1038/ng.1102
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46:444–450. doi:10.1038/ng.2938
Acknowledgements
We thank the Microarray Unit of the German Cancer Research Center (DKFZ) Genomics and Proteomics Core Facility for excellent technical support. This work was principally supported by the PedBrain Tumor Project contributing to the International Cancer Genome Consortium, funded by German Cancer Aid (109252) and by the German Federal Ministry of Education and Research (BMBF, grant #01KU1201A and the e:Med Joint Research Project SYS-GLIO #031A425A). Additional support came from the German Cancer Research Center—Heidelberg Center for Personalized Oncology (DKFZ-HIPO) and the German Cancer Consortium (DKTK). CJ and AB acknowledge NHS funding to the NIHR Biomedical Research Centre at The Royal Marsden and the ICR.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Korshunov, A., Schrimpf, D., Ryzhova, M. et al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol 134, 507–516 (2017). https://doi.org/10.1007/s00401-017-1710-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1710-1