
Abstract
Olfactory impairment is a common feature of neurodegenerative diseases such as Parkinson’s disease (PD), Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB). Olfactory bulb (OB) pathology in these diseases shows an increased number of olfactory dopaminergic cells, protein aggregates and dysfunction of neurotransmitter systems. Since cholinergic denervation might be a common underlying pathophysiological feature, the objective of this study was to determine cholinergic innervation of the OB in 27 patients with histological diagnosis of PD (n = 5), AD (n = 14), DLB (n = 8) and 8 healthy control subjects. Cholinergic centrifugal inputs to the OB were clearly reduced in all patients, the most significant decrease being in the DLB group. We also studied cholinergic innervation of the OB in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated monkeys (n = 7) and 7 intact animals. In MPTP-monkeys, we found that cholinergic innervation of the OB was reduced compared to control animals (n = 7). Interestingly, in MPTP-monkeys, we also detected a loss of cholinergic neurons and decreased dopaminergic innervation in the horizontal limb of the diagonal band, which is the origin of the centrifugal cholinergic input to the OB. All these data suggest that cholinergic damage in the OB might contribute, at least in part, to the olfactory dysfunction usually exhibited by these patients. Moreover, decreased cholinergic input to the OB found in MPTP-monkeys suggests that dopamine depletion in itself might reduce the cholinergic tone of basal forebrain cholinergic neurons.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Olfactory dysfunction is a common feature in patients with neurodegenerative diseases and has been well characterized in Parkinson’s disease (PD) [20, 21, 30], Alzheimer’s disease (AD) [6, 52] and dementia with Lewy bodies (DLB) [51, 62, 84, 85]. However, the mechanisms involved are not fully understood. The presence of Tau, β-amyloid [6, 43] and α-synuclein [37, 60, 81] protein aggregates in the olfactory bulb (OB), olfactory tract and cortex, increased number of olfactory dopaminergic neurons [34, 35, 60] and atrophy [64, 80] or dysfunction of higher olfactory related structures such as hippocampus or amygdala [12] have all been suggested as possible origins of smell loss. The OB receives heavy inputs from cholinergic, noradrenergic, and serotonergic subcortical systems that seem to exert robust effects on odor processing and odor memory by acting either on inhibitory local interneurons or on output neurons [19, 24, 65]. Deficits in noradrenergic, serotonergic and cholinergic systems [22] and decrease in the number of neurons in the locus coeruleus [27, 78], raphe nuclei [36, 59] and nucleus basalis of Meynert (NBM) are frequently observed in PD, AD, and DLB diseases [5, 40, 44, 46, 70], and they might actively contribute to the olfactory dysfunction seen in these disorders.
The main objective of this study was to investigate the cholinergic innervation of the OB in patients with AD, PD, DLB and age-matched controls using immunohistochemistry against choline acetyl transferase (CHAT). OBs from control and 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP)-treated monkeys were also studied following the same protocol. Correlations between CHAT immunoreactivity (CHAT-ir), protein aggregates and number of dopaminergic neurons in the OB were also performed in a group of cases already included in our previous publication [60]. In order to determine the impact of dopaminergic innervation on the olfactory cholinergic system, we also analyzed in control and MPTP-treated monkeys, the dopaminergic innervation and the number of cholinergic neurons in the horizontal limb of the diagonal band of Broca (HLDB) in the basal forebrain, the nucleus that gives rise to cholinergic centrifugal projections to the OB [53–55].
Materials and methods
Human tissue information
Human cases
The study was based on 35 human brains collected from the Brain Bank of Navarra (Spain). The Brain Donation Program was approved by the Government of Navarra (Foral decree 23/2001). Subjects received standardized neuropathological examination and were distributed into different groups according to the histological post-mortem diagnosis. Specific consensus diagnostic criteria were used for AD [1, 2, 14, 15], PD [3, 16] and DLB [3, 49]. Fourteen cases exhibited neuropathological findings compatible with the diagnosis of AD at different stages (7 cases VIC, 2 cases VIB, 3 cases VC, 1 case VB, 1 case IVB); five subjects met the diagnostic criteria for PD (1 with a Braak stage 3, 2 stage 4 and 2 stage 5); 8 subjects fulfilled diagnosis for DLB [2 cases diffuse neocortical/VC; 1 case brainstem predominant/VC; 5 cases limbic transitional (including 2 cases VC, one case VIC, one case VB, and one case IIIB)]. Brains from eight elderly subjects with no history or histological findings of any neurological disease were used as a control group (Table 1). Patients and controls were matched for age. No gender differences were observed. The clinical diagnosis of all cases included in the study was done according to the established criteria (for AD, [4, 50]; for PD, [33]; for DLB, [49]).
Neuropathological study
The neuropathological study was carried out according to the procedure previously described [23]. Post-mortem delay between death and tissue sampling in all cases was between 2.5 and 5.5 h, which is optimal for histological studies. After fixation in 10 % formalin (21–25 days), representative brain areas from olfactory bulb, cortical and subcortical areas, brainstem, cerebellum, and spinal cord were taken and embedded in paraffin in order to make a neuropathological diagnosis. Specific anatomical brain regions were studied in a standardized neuropathological manner based on staging procedures commonly applied for histological diagnosis of PD [3, 16], DLB [3, 49] and AD [1, 2, 14, 15]. Three micrometer thick sagittal sections were processed for immunohistochemical analysis. The deparaffinized and rehydrated sections were pretreated in a heat-induced epitope retrieval device for 20–35 min at 98 °C. Immunohistochemistry was performed with a DAKO Autostainer (DAKO, Glostrup, Denmark). Sections were treated with hydrogen peroxide to block endogenous peroxidase activity, followed by normal serum to block nonspecific antibody binding. Sections were treated with anti-phosphorylated Tau (mouse monoclonal antibody, Novocastra, NCL-Tau-2, clone Tau 2; 1:100), anti-α-synuclein (liquid mouse monoclonal antibody, NCL-L-ASIN, clone KN51; 1:50), anti-β amyloid (mouse monoclonal antibody, Novocastra, NCL-B- amyloid, clone 6F-3D; 1:200) and anti-TARDBP/TDP-43 (mouse monoclonal antibody, Abnova Corporation, clone 2E2-D3; 1:1,500), anti-PrP (mouse monoclonal antibody, Dako Cytomation, clone 3F4; 1:100), anti-ubiquitin (lyophilized monoclonal antibody, Novocastra, NCL-UBICm, clone FPM1; 1:700), and anti-crystallin (mouse monoclonal antibody, Novocastra, clone ABCrys-512; 1:100). A biotinylated secondary antibody (goat anti-mouse) was used in all cases. Antibodies were visualized using the avidin–biotin-peroxidase complex and 3,3′-diaminobenzidine tetrahydrocloride (DAB) as the chromate. After immunostaining, the sections were hematoxylin–eosin counterstained. Definitive post-mortem diagnosis was performed in all cases included in the study. The other hemisphere (left) was freshly sectioned, and stored at −80 °C for later biochemical and molecular studies. The corresponding OBs were fixed by immersion in phosphate-buffered 4 % PFA for 5 days and cryoprotected in 30 % sucrose in PBS. After the tissue sank in sucrose, 12 parallel series of 40 μm coronal sections were collected. One of these series was immunohistochemically processed to reveal CHAT.
CHAT immunohistochemistry
Free floating sections were treated for 15 min with 1 % H2O2 in PBS to inactivate the endogenous peroxidase and then incubated for 45 min in 5 % normal donkey serum diluted in PBS–0.25 % triton. Subsequently, the sections were incubated overnight with a primary goat polyclonal antibody raised against CHAT (diluted 1:300 in blocking solution; AB144P, Millipore), and then rinsed three times for 5 min in PBS and incubated for 45 min at room temperature with biotinylated donkey anti-goat Ig diluted 1:500 in PBS. The sections were then processed using the avidin–biotin-peroxidase complex (Vectastain kit, Vector laboratories) and reacted with 0.05 % 3,3′-diaminobenzidine tetrahydrochloride and 0.015 % H2O2 in 50 mM Tris HCl, pH 7.2 (DAB). The sections were mounted on gelatinized slides, Nissl stained and coverslipped with DPX mounting medium. Staining specificity was confirmed by incubating the primary antibody with Acetyl Cholinesterease (AG220, Millipore) for 3 h at 4 °C diluted 3 μg/ml. We found nonspecific endothelial cell staining in the human OB. No labeling was detected in the monkey tissue after this treatment.
Quantification of CHAT immunoreactivity (CHAT-ir) in the OB
Samples were viewed and digitalized with an Olympus BX-51 microscope equipped with an Olympus DP-70 digital camera at 40× using CAST grid software package (Olympus, Denmark). For CHAT-ir quantification, bulbar layers were analyzed individually as glomerular layer (GL), external plexiform layer (EPL), and anterior olfactory nucleus (AON) while granule cell layer (GCL) and the internal plexiform layer (IPL) were quantified together. In human cases, eight 40× images of each layer were randomly digitalized and analyzed using ImageJ software as follows. Images were converted to 8 bit-RGB stacked (blue channel) to process only the dark brown CHAT labeling and to remove the blue Nissl staining, and a background subtraction procedure was performed. Consequently, the perimeter of each layer was outlined manually for each image excluding any unwanted immunostained structures (i.e., capillary endothelia in human cases). Then, the same threshold limits were defined for each image to select CHAT-ir structures and in human cases we quantified the number of CHAT-ir bouton-like structures per μm2 (CHAT-density). The same investigator (I.C.M.) performed all quantifications in a blinded manner.
Correlation studies
Since we previously found an increased number of protein aggregates and dopaminergic cells in the OB of patients with neurodegenerative diseases, we searched for any correlation between CHAT-ir innervation, the number of protein aggregates [Tau neuropil threads (NT), Tau pretangles (PT), Tau neurofibrillary tangles (NFT), Tau neuritic plaques (NP), amyloid senile plaques (SP), amyloid matures senile plaques, synuclein Lewy neurites (LN) and bodies (LB), total estimated number and density (number of neurons per mm3) of dopaminergic periglomerular neurons in the OB. For this purpose we used data from 21 patients (7 controls, 9 AD and 5 PD) previously studied [60]. Three additional cases with histological diagnosis of DLB were also included for correlation studies. In these three cases, the analysis of OB for specific protein deposits, TH immunohistochemistry and cell counting was performed as described previously [60]. Briefly, for immunohistochemistry against a tyrosine hydroxylase (TH) free floating sections were treated with H2O2 for 45 min, then 45 min in 5 % normal goat serum. After being 24 h incubated with primary antibody (rabbit polyclonal anti-TH, 1:2,000 in PBS; AB152, Millipore, CA), sections were rinsed in several times in PBS and incubated in a secondary goat anti-rabbit Ig, 1:500 in PBS for 1 h. Reaction was visualized using ABC kit (Vectastain kit, Vector Laboratories) and DAB technique. Stereological quantification of total dopaminergic periglomerular neurons was performed by using an optical fractionator unbiased sample design as described previously [60].
Animal tissue information
Animals
Fourteen male cynomolgus monkeys (Macaca fascicularis), weighing 2.5–3.4 kg and aged 3.8–4 years, were included in the study. Animals were housed in an animal room under standard conditions of humidity (50 %), air exchange (16 l/min) and dark/light cycles (8 a.m.–8 p.m.). They were fed fresh fruits and commercial pellets and had free access to water. The attending veterinarian monitored their health according to the recommendations of the Weatherall Report. The animals were euthanized following deep anesthesia, and all efforts were made to minimize suffering. The experimental protocol was in accordance with the European Communities Council Directive of November 24th 1986 (86/609/EEC) and it was approved by the Ethics Committee for Animal Experimentation of the University of Navarra. Seven animals were rendered parkinsonian by systemic administration of MPTP (one weekly intravenous injection of 0.25–0.8 mg/kg; cumulative doses 13–25 mg/kg) and seven animals served as controls. Motor deficits induced by MPTP were assessed according to a non-human primate disability rating scale, which independently scores parkinsonian features such as tremor (intensity and duration), balance, feeding and freezing from 0 (normal) to 3 (maximum disability); bradykinesia and posture from 0 (normal) to 4 (maximum disability), and the reduction in spontaneous activity from 0 (normal) to 5 (maximum disability), thus giving a total maximum score of 28 [48]. These animals have participated in another study and the remaining brain tissue from all of them is being used in other studies in our laboratory. None of the animals received L-DOPA treatment at any time during induction of parkinsonism.
The animals were killed 5 months after discontinuing MPTP treatment. After deep sedation with a mixture of ketamine (10 mg/kg) and midazolam (1 mg/kg), animals were transcardially perfused with 0.01 M phosphate buffer saline (PBS) and 4 % paraformaldehyde (PFA, Sigma) in 0.01 M PBS. Brains were immediately removed, blocked and post-fixated for 2 days in 4 % PFA. They were then cryoprotected in a 30 % sucrose solution in 0.01 M PBS until processing. Brains were sliced into 40-μm-thick coronal sections along the rostral axis with a freezing microtome (Leica, Germany) and collected in 0.125 M PBS containing 2 % dimethylsulphoxide (Sigma), 20 % glycerin (Panreac) and 0.05 % sodium azide and stored at −20 °C until their subsequent analysis.
Immunohistochemistry/immunofluorescence
Monkey coronal sections of OB and basal forebrain were processed for immunohistological detection of CHAT as described previously for human tissue. In all animals, brain tissue sections containing the substantia nigra (SN) were processed in the same way for tyrosine hydroxylase (TH) immunohistochemistry (primary rabbit polyclonal anti-TH, 1:1,000 in PBS; AB152, Millipore, CA; donkey anti-rabbit Ig, 1:500 in PBS) to confirm the degeneration of nigral dopaminergic cells. In three controls and three MPTP-treated animals, double-labeling immunofluorescence was performed to detect possible abnormalities either in the number of cholinergic neurons or in the dopaminergic innervation of HLDB. Analysis was performed on five coronal sections of each animal. Tissue sections were rinsed and permeabilized following the protocol used for peroxidase immunohistochemistry (see above). Subsequently, they were incubated overnight at 4 °C in a solution containing a mixture of primary rabbit polyclonal anti-TH (dilution 1:1,000; AB152, Millipore, CA) and goat anti-CHAT (dilution 1:300; AB144P, Millipore, CA). After rinsing with PBS, sections were incubated for 2 h in 0.01 M PBS containing normal donkey serum (1:20) and a combination of secondary antibodies coupled to fluorescent markers, donkey anti-rabbit Alexa Fluor 488 and donkey anti-goat Alexa Fluor 568 (dilution 1:500; Molecular Probes). Finally, sections were counterstained with a nucleic acid stain (TO-PRO-3 iodide, Molecular Probes, Netherlands) and coverslipped with mounting medium (Immu-mount, Thermo-Shandon). Double labeling was detected in confocal images by using a laser scanning microscope 510, equipped with three lasers (LSM 510/Meta; Zeiss, Germany).
Quantification methods
Quantification of CHAT-ir in the monkey OBs was performed in the same way as in human tissue, but for each layer the percentage of the area labeled by CHAT immunolabeling was analyzed.
TH immunoreactivity (TH-ir) in the HLDB of monkeys was performed as described above for CHAT-ir in the OB, with some modifications. Twenty samples per animal were digitalized with a 20× objective and the percentage of the total HLDB area showing TH-ir innervation was quantified.
The total number of CHAT-ir neurons was estimated in the HLDB of intact and MPTP-monkeys by using an optical fractionator unbiased sample design [29]. Stereological analysis was performed with an Olympus BX51 microscope with a monitored x-y-z stage linked to the CAST Grid software package (Olympus, Denmark). The HLDB was outlined using a 4× objective on each of the double CHAT/TH labeled sections. In each animal, a series of five coronal sections of the commissural basal forebrain, regularly spaced at intervals of 480 μm and covering the entire HLDB, was used for cell counting. From a random start position a counting frame was superimposed on the image and cholinergic neurons were sampled using a 20× objective. CHAT-ir neurons were included when the soma came into focus and did not touch the exclusion lines of the counting frame. The sampling frames were spaced 555.9 μm along the x and y axis from each other, based on the shape of the region. We used a sampling frame size of 92,690 μm2. The dissector z-step was 30 μm and a guard zone of 2 μm at both the upper and lower surface of the section was used. A minimum of 100 cells were sampled according to the rules of the optical dissector method and the coefficient of error was <0.1.
Behavioral manifestations and loss of dopaminergic cells in SN of MPTP-monkeys
All seven monkeys developed a mild stable parkinsonian syndrome after being exposed to MPTP and their disability scores ranged from 10 to 12 [48]. The extent of MPTP-induced dopaminergic cell depletion was confirmed by immunohistochemical detection of TH in the substantia nigra pars compacta (SNpc) and ventral tegmental area (VTA). Although we did not perform a detailed quantification of the number of TH-ir cells in the SNpc, the decreased intensity of TH-labeling in the SNpc and VTA was very similar in all MPTP-monkeys as reflected in Fig. 1.

Coronal sections of the substantia nigra (SN) and ventral tegmental area (VTA) of control (a) and MPTP-monkey (b) immunostained with tyrosine hydroxylase. A marked depletion of TH-ir cells is observed. Scale bar 1 mm
Statistical analysis
Statistical analysis was carried out using SPSS 15.0 software. In human cases multiple comparisons were performed by using a non-parametric Kruskal–Wallis test followed by Mann–Whitney U test (2–2) in order to estimate the overall significance of CHAT-density, immunopositive and button-like structures among control, AD, PD and DLB patients. In monkey OBs, the average percentages of CHAT immunostained area of each OB layer were compared between control and MPTP group using non-parametric Mann–Whitney U test. Changes in the average number of CHAT-ir neurons in the HLDB and percentage of CHAT immunoreactive areas in the OB between control and MPTP-monkeys were compared using non-parametric Mann–Whitney U test. P values <0.05 were considered to be statistically significant. CHAT and TH immunoreactive density and percentage values were presented as mean ± SEM. Non-parametric Spearman’s correlations were used to study relationships between CHAT-ir density, number of protein aggregates, number and density of dopaminergic periglomerular neurons in the human OB.
Results
Labeling and distribution of OB structures expressing CHAT exhibited several differences between humans and monkeys. In human cases, very few fibers were observed and many CHAT-ir bouton-like structures were detected. In monkeys, CHAT immunohistochemistry revealed fibers and terminal-like puncta. These differences might partly be due to the different fixation processes followed in humans and monkeys. Moreover, the distribution of cholinergic centrifugal terminals in human and monkey OBs did not follow the same pattern. In control human OBs, the GL displayed a very weak CHAT-ir, and only scarce labeling was observed in the inner part of the GL, close to the EPL. The EPL exhibited a moderate and homogenous CHAT-ir density. Rich centrifugal CHAT innervation was observed in the IPL and the GCL, with decreased labeling in the inner areas of the GCL. Finally, the AON showed very dense cholinergic innervation. The capillary endothelial cells of human OBs had high immunoreactivity for CHAT antibody.
As previously described by Porteros et al. [67], in control monkeys, cholinergic innervation was located mainly in the GL, with its inner area being most innervated, followed by the periglomerular zone. The EPL displayed homogeneously decreased labeling. The IPL was the second most innervated layer after the GL, while the GCL exhibited the weakest CHAT-ir. White matter showed very sparsely stained elements, while the olfactory nerve layer showed no immunoreactivity. No labeled cell bodies were observed in the OBs of control monkeys.
CHAT-ir quantification in human OBs
As compared to controls, the OB of PD, AD, and DLB brains showed a statistically significant reduction of CHAT-ir in all layers studied except in the GL. Particularly, DLB patients displayed more reduced cholinergic innervation in the AON when compared to the other groups of patients (Fig. 2).

Photomicrographs of the olfactory bulb showing the decreased CHAT-ir bouton-like structures in the glomerular layer (GL) (a–d), external plexiform layer (EPL) (f–i), granule cell and internal plexiform layers (GCL/IPL) (k–n) and anterior olfactory nucleus (AON) (p–s) from patients with neurodegenerative diseases and normal aged-matched subjects. Bar charts showing the density of CHAT-ir bouton-like structures in the different olfactory bulb layers (c, j, o, t). PD Parkinson’s disease, AD Alzheimer’s disease, DLB dementia with Lewy bodies. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Data are expressed as mean values ± SEM. Scale bar 50 μm
CHAT-ir density in the GL showed a tendency to decrease in patients with neurodegenerative diseases, but did not reach significant differences (P = 0.074): control cases (6,439.3 ± 1,294.5); PD cases (4,983 ± 419.9); AD cases (3,879.6 ± 394.3) and DLB (3,337.5 ± 658.3) (Fig. 2a–e).
In patients with neurodegenerative diseases, the EPL exhibited a significant reduction of CHAT-ir compared to control cases (1.7-fold in PD; 1.7-fold in AD; and 2.8-fold in DLB). Thus, the mean CHAT-ir density in controls was 18,304.9 ± 1,797.6, while in PD, AD, and DLB it was 10,724.6 ± 1,493.1 (P = 0.019 vs controls; 10,744.2 ± 882.8 (P = 0.004 vs controls) and 6,515.8 ± 622.4 (P ≤ 0.001 vs controls; P = 0.006 vs AD; P = 0.019 vs PD), respectively (Fig. 2f–j).
The highest cholinergic denervation was observed in the IPL/GCL, where CHAT-density was found to be reduced by 2.24-fold in PD (9,338 ± 1,381; P ≤ 0.001); 2.45-fold in AD (8,543.5 ± 802.9; P ≤ 0.001) and 3.95-fold in DLB (5,300.1 ± 708; P ≤ 0.001 vs controls; P = 0.008 vs AD) when compared to controls (20,942.6 ± 1,643.7) (Fig. 2k–o).
As compared to controls, the AON of patients with PD, AD, and DLB displayed a significant reduction of olfactory cholinergic innervation, 1.76-fold (P = 0.005); 1.69-fold (P = 0.002); and 3.52-fold (P = 0.001), respectively. Mean density values were: 24,795.8 ± 2,696.2 in control cases, 14,041.1 ± 668.9 in PD cases; 14,649 ± 1,091 in AD cases and 7,032.9 ± 887.5 in DLB cases. Moreover, OBs from DLB patients exhibited significant CHAT-ir reduction in the AON as compared to AD (P ≤ 0.001) and PD (P ≤ 0.001) patients (Fig. 2p–t).
Correlation between CHAT-ir, protein deposits and number of dopaminergic neurons in the human OB
The number of amyloid senile plaques was negatively correlated with the density of CHAT-ir in all bulbar layers and with the averaged OB CHAT-ir. In addition, CHAT-ir density in the GCL correlated negatively with the amount of olfactory Tau neuropil threads, pretangles, neurofibrillary tangles, amyloid senile plaques and mature senile plaques. The density of dopaminergic periglomerular neurons also correlated negatively with CHAT-ir in the whole OB and in the GCL (Table 2). In other words, in the human OB the increased number of protein aggregates and dopaminergic neurons seems to be associated with decreased cholinergic innervation.
CHAT-ir quantification in monkey OBs
In MPTP-treated monkeys the distribution pattern of cholinergic inputs was similar to that in controls, but showed reduced fiber and button density. In two of the seven MPTP-monkeys included, some labeled cells were found mainly in the mitral cell layer and the EPL. The OBs of MPTP-treated monkeys showed a significantly decreased CHAT-ir as compared to control animals (1.7-fold in the GL; 1.55-fold in the EPL). In the GL, the average percentage of CHAT-ir area was 5.4 ± 0.3 in control animals and 3.1 ± 0.4 in MPTP-monkeys (P = 0.006). CHAT-ir in the EPL of control animals was 0.7 ± 0.1, while in MPTP-monkeys it was reduced to 0.4 ± 0.1 (P = 0.028). No significant differences were observed in the GCL between control animals (1.2 ± 0.4) and MPTP-monkeys (0.9 ± 0.1) (P = 0.487) (Fig. 3).

Photomicrographs of the olfactory bulb (OB) of control (a, c, e) and MPTP-monkey (b, d, f) showing the cholinergic centrifugal innervation in the olfactory bulb. In the glomerular layer (GL) (a, b) and the external plexiform layer (EPL) (c, d) MPTP-treated monkeys showed decreased immunoreactivity against CHAT. In the granule cell layer (GCL) (e, f) CHAT-ir was similar in both groups. Note CHAT-ir mitral cells in f. Scale bar 50 μm. g Bar charts. The percentage of CHAT-ir area in the olfactory bulb of MPTP-treated monkeys is significantly reduced in the glomerular layer (GL) and the external plexiform layer (EPL). No differences were detected in the granule cell layer (GCL)
CHAT-ir neurons in monkey HLDB
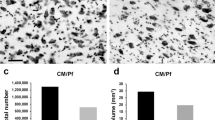
The total estimated number of cholinergic neurons in the HLDB of MPTP-monkeys was 1.72-fold reduced (1,968 ± 101.05; P = 0.05) with respect to control animals (3,396 ± 280.9) (Fig. 4a–c).

CHAT and TH immunohistochemistry in the horizontal limb of the diagonal band of Broca (HLDB). a Some CHAT-ir neurons (arrows) in one control monkey. b In the MPTP-treated monkey CHAT-ir neurons were clearly reduced. c Graph showing the stereological estimation of the total CHAT-it cell number in the HLDB of control and MPTP-treated monkeys. Tyrosine hydroxylase (TH) immunofluorescence in the horizontal limb of the diagonal band of Broca of control (d) and MPTP-treated monkeys (e). Dopaminergic innervation (f) of this nucleus is significantly reduced in the MPTP-treated animals. Scale bars in b and d 500 μm. Data are expressed as mean values ± SEM. *P ≤ 0.05
TH-ir innervation in the monkey HLDB
Dopaminergic innervation of the HLDB in MPTP-treated monkeys was 1.82-fold reduced as compared to controls (P = 0.05). Thus, the average percentage of TH-ir of HLDB of control animals was 8.48 ± 0.94, while in MPTP-monkeys it was reduced to 4.64 ± 0.56 (Fig. 4d–f). In control animals, but not in MPTP-monkeys, cholinergic neurons in the HLDB seemed to have dopaminergic synaptic contacts (Fig. 5).

Double immunofluorescence images against choline acetyl transferase (CHAT) and tyrosine hydroxylase (TH) in the horizontal limb of the diagonal band of Broca of one control monkey (a–d) and one MPTP-treated monkey (e–h). The number of CHAT-ir neurons and the density of TH innervation are reduced in the MPTP-treated monkey. d In a higher magnification image the control animal shows dopaminergic possible synaptic contacts on CHAT-ir neurons. h In the MPTP-monkey no synaptic contacts can be observed. Scale bars a–cp, e–g 500 μm, d, h 125 μm
Discussion
This study is the first to demonstrate that cholinergic centrifugal olfactory inputs are severely reduced in patients with AD, PD and DLB, and this decrease is most important in OBs from DLB patients. Interestingly, cholinergic OB innervation was also decreased in MPTP parkinsonian monkeys. In addition, a significant loss of cholinergic neurons and dopaminergic innervation was observed in HLDB, the origin of centrifugal input to the OB. In previous studies, we described how the number of dopaminergic periglomerular neurons is increased in the OB of patients with PD, AD and frontotemporal dementias [60] and also in MPTP-monkeys [8]. These results were interpreted as a compensatory mechanism created by the lack of centrifugal inputs to the OB in an attempt to preserve olfactory function. Interestingly, the decreased cholinergic innervation of the OB in patients with neurodegenerative diseases here described correlates with the increased number of olfactory dopaminergic neurons, thus supporting our previous hypothesis. Moreover, decreased CHAT innervation in the GCL correlated with the extent of Tau and β-amyloid deposits in the OB. On the other hand, the loss of centrifugal cholinergic innervation observed in MPTP-monkeys suggests that dopamine itself might contribute to maintaining functional integrity of the cholinergic systems in the OB.
CHAT distribution in the human OB
To our knowledge, this is the first description focusing on the study of cholinergic centrifugal innervation of the OB using CHAT immunohistochemistry. The cholinergic centrifugal olfactory system had previously been explored in control and AD patients by histochemical methods (acetylcholinesterase histochemistry) [40, 44, 45]. Using this technique, the authors detected no cholinergic structures in the GL and described scattered CHAT labeling in the EPL of control cases, which clearly contrasts with our results. Although they failed to detect any cholinergic labeling in the EPL, MCL and IPL in AD patients, they described diffuse labeling in the GCL and white matter and a low CHAT-density in the AON. The lack of specificity of acetylcholinesterase for CHAT activity [47] along with the long post-mortem interval (3–24 h) of the tissue samples might account for the divergent results. In agreement with our results, Simpson et al. [73] have described reduced CHAT activity in the olfactory tubercle of patients with AD, Down’s syndrome and Huntington disease using radiochemical methods to assess CHAT activity.
CHAT-ir decreases in the OB of patients with neurodegenerative diseases
One of the most relevant finding of this study is the demonstration that CHAT-ir is markedly reduced in the OB of patients with neurodegenerative disorders. What could be the clinical significance of this finding? Might it account for the olfactory dysfunction of these patients? Loss of cholinergic neurons in the NBM and decreased cholinergic innervation of temporal lobes correlates significantly with cognitive deficits in PD and AD [5, 17, 42, 70]. In vivo and post-mortem studies indicate that the magnitude of forebrain cholinergic deficits increases over the course of the disease [41, 71, 72]. However, significantly decreased AChE activity has also been reported in the novo PD patients indicating that cholinergic denervation occurs at very early stages of the disease and may precede the motor manifestations [11]. As cholinergic afferents to cerebral cortex and OB arise from the same forebrain structures, it could be hypothesized that olfactory cholinergic denervation also occurs early in the course of the disease and might contribute to the hyposmia.
Severe olfactory dysfunction is risk factor and a prodromal symptom of dementia associated with PD [7] indicating that both features might share a similar pathological substrate and suggesting that cholinergic dysfunction might account for hyposmia [12, 58, 68, 75]. Accordingly, the OB of our patients with LBD exhibited the most severe cholinergic denervation, and interestingly, in these patients AChE activity levels in cerebral cortices are equal to those in PD patients with dementia and larger than AD patients indicating a close relationship between cortical and olfactory cholinergic activity [17, 51, 84, 85]. However, although the hypothesis of cholinergic olfactory dysfunction is attractive, it should be taken into account that volumetric magnetic resonance imaging analysis has demonstrated a narrow correlation between olfactory dysfunction and atrophy of focal brain structures, including the amygdala and limbic structures [82, 83].
Although the patients here included were at an advanced stage of the disease and olfactory tests were not performed, our results clearly demonstrate a defective cholinergic innervation of the OB in patients with neurodegenerative diseases. Whether this alteration is already present at the beginning of the disease when patients first complain of hyposmia cannot be inferred from this study.
Another interesting result of this work is the negative correlation found between CHAT-ir, protein deposits and the number of dopaminergic cells in the OB. The negative correlation between protein deposits (β-amyloid and Tau) and CHAT-ir in the OB might merely reflect the advanced stage of the disease of our patients in whom this would convey simultaneously the marked progressive loss of cholinergic neurons in the forebrain structures and the spread of protein deposits within the brain. So, it is also possible that as the disease progresses both the number of protein deposits and the degeneration of cholinergic ascending pathways increases. However, we cannot rule out the possibility that the deposition of anomalous proteins in the OB, especially β-amyloid senile plaques, can provoke first a synaptic disruption in the olfactory cholinergic synapses and subsequently its degeneration. In fact, over the past decade much evidence has appeared that β-amyloid [63, 66] and Tau [79] oligomers contribute to degeneration of synaptic structure and function [18]. Moreover, interactions between β-amyloid and cholinergic deficits seem to be clear [38, 61, 69]. We also found a negative correlation between cholinergic innervation and the number of dopaminergic cells. In other words, the greater the decrease of olfactory cholinergic innervation the greater the increase in the number of dopaminergic cells. These results are consistent with our previous hypothesis, indicating that the increase in the number of dopaminergic cells might be a compensatory mechanism created to compensate for the lack of cholinergic inhibitory influence. Additionally, our results would also explain why the number of olfactory dopaminergic cells is increased in all neurodegenerative diseases, since as described in this study, cholinergic innervation of the OB is defective in all of these as a consequence of the degeneration of ascending cholinergic projections.
CHAT/TH deficits in the MPTP-monkey olfactory system
Although the majority of PD patients report hyposmia, some of them do not complain of olfactory dysfunction and not all PD patients with hyposmia become demented, indicating that dopamine itself can alter the functional integrity of the cholinergic system in the OB [7]. To address this point, we included the OB and HDLB of monkeys chronically exposed to MPTP that exhibited moderate parkinsonism. Interestingly, in these animals, we found cholinergic neuronal loss in the HDLB. The effect of MPTP on the survival of cholinergic neurons [25, 31, 32] is not conclusive. For instance, in the mouse, MPTP causes a reduction of cholinergic neurons in the NBM and in the vertical limb of the diagonal band, and also damages their respective projections to the cortex and hippocampus [76, 77]. In aged monkeys exposed to MPTP [39], the number of cholinergic neurons, degree of metabolic activity, and levels of CHAT mRNA and substance P [28] are reduced in the pedunculo pontine nucleus. These data and our results suggest that MPTP can directly affect the viability of cholinergic neurons in the HLDB and consequently reduce the cholinergic input to the OB. However, whether MPTP affects olfactory projecting neurons in the locus coeruleus or raphe nuclei in parkinsonian monkeys deserves further investigation. Likewise, the integrity of serotonergic and noradrenergic centrifugal inputs should be further studied in patients with neurodegenerative disorders.
MPTP-monkeys showed marked dopaminergic denervation in the HLDB. As the VTA projects to the HLDB [26, 74], reduced dopaminergic activity in these structures would affect their excitability and the acetylcholine efflux to their terminal fields [56, 57]. Therefore, the abnormal mesolimbic-basal forebrain dopaminergic transmission created by MPTP might disrupt the functionality of cholinergic neurons in the basal forebrain nuclei and might be involved in cognitive deficits, olfactory loss or both. Given that VTA also send dopaminergic innervation to the hippocampus [9], these data are consistent with the results from Bohnen et al. [10, 13] showing that dopaminergic hippocampal denervation in PD patients is closely related to olfactory deficits. These results would also explain why mild dopamine deficiency, as seen in early PD stages, could induce hyposmia by reducing the cholinergic tone in basal forebrain cholinergic and hippocampal neurons.
Abbreviations
- AD:
-
Alzheimer’s disease
- AON:
-
Anterior olfactory nucleus
- CHAT:
-
Choline acetyl transferase
- CHAT-ir:
-
Choline acetyl transferase immunoreactivity/immunoreactive
- DLB:
-
Dementia with Lewy bodies
- EPL:
-
External plexiform layer
- GCL:
-
Granule cell layer
- GL:
-
Glomerular layer
- HLDB:
-
Horizontal limb of the diagonal band of Broca
- IPL:
-
Internal plexiform layer
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NBM:
-
Nucleus basalis of Meynert
- OB:
-
Olfactory bulb
- PBS:
-
Phosphate buffer saline
- PD:
-
Parkinson’s disease
- PFA:
-
Paraformaldehyde
- SEM:
-
Standard error of the mean
- SN:
-
Substantia nigra
- TH:
-
Tyrosine hydroxylase
- TH-ir:
-
Tyrosine hydroxylase immunoreactivity/immunoreactive
- Tris HCl:
-
2-Amino-2-hydroxymethyl-propane-1,3-diol hydrochloride
- VTA:
-
Ventral tegmental area
References
Alafuzoff I, Thal DR, Arzberger T, Bogdanovic N, Al-Sarraj S, Bodi I et al (2009) Assessment of beta-amyloid deposits in human brain: a study of the BrainNet Europe Consortium. Acta Neuropathol 117:309–320. doi:10.1007/s00401-009-0485-4
Alafuzoff I, Arzberger T, Al-Sarraj S, Bodi I, Bogdanovic N, Braak H et al (2008) Staging of neurofibrillary pathology in Alzheimer’s disease: a study of the BrainNet Europe Consortium. Brain Pathol 18:484–496. doi:10.1111/j.1750-3639.2008.00147.x
Alafuzoff I, Parkkinen L, Al-Sarraj S, Arzberger T, Bell J, Bodi I et al (2008) Assessment of alpha-synuclein pathology: a study of the BrainNet Europe Consortium. J Neuropathol Exp Neurol 67:125–143. doi:10.1097/nen.0b013e3181633526
American-Psychiatric-Association (1987) Diagnostic and statistical manual of mental disorders. Third, revised edn. Washington DC
Arendt T, Bigl V, Arendt A, Tennstedt A (1983) Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s Disease. Acta Neuropathol 61:101–108
Attems J, Jellinger KA (2006) Olfactory tau pathology in Alzheimer disease and mild cognitive impairment. Clin Neuropathol 25:265–271
Baba T, Kikuchi A, Hirayama K, Nishio Y, Hosokai Y, Kanno S et al (2012) Severe olfactory dysfunction is a prodromal symptom of dementia associated with Parkinson’s disease: a 3 year longitudinal study. Brain 135:161–169. doi:10.1093/brain/awr321
Belzunegui S, Sebastián WS, Garrido-Gil P, Izal-azcárate A, Vázquez-Claverie M, López B et al (2007) The number of dopaminergic cells is increased in the olfactory bulb of monkeys chronically exposed to MPTP. Synapse 61:1006–1012. doi:10.1002/syn.20451
Björklund A, Dunnett SB (2007) Dopamine neuron systems in the brain: an update. Trends Neurosci 30:194–202. doi:10.1016/j.tins.2007.03.006
Bohnen NI, Muller ML (2012) In vivo neurochemical imaging of olfactory dysfunction in Parkinson’s disease. J Neural Transm. doi:10.1007/s00702-012-0956-y
Bohnen NI, Albin RL (2009) Cholinergic denervation occurs early in Parkinson disease. Neurology 73:256–257. doi:10.1212/WNL.0b013e3181b0bd3d
Bohnen NI, Muller MLTM, Kotagal V, Koeppe RA, Kilbourn MA, Albin RL et al (2010) Olfactory dysfunction, central cholinergic integrity and cognitive impairment in Parkinson’s disease. Brain 133:1747–1754. doi:10.1093/brain/awq079
Bohnen NI, Gedela S, Herath P, Constantine GM, Moore RY (2008) Selective hyposmia in Parkinson disease: association with hippocampal dopamine activity. Neurosci Lett 447:12–16
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. doi:10.1007/s00401-006-0127-z
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Candy JM, Perry RH, Perry EK, Irving D, Blessed G, Fairbairn AF et al (1983) Pathological changes in the nucleus of Meynert in Alzheimer’s and Parkinson’s diseases. J Neurol Sci 59:277–289
Crimins JL, Pooler A, Polydoro M, Luebke JI, Spires-Jones TL (2013) The intersection of amyloid beta and tau in glutamatergic synaptic dysfunction and collapse in Alzheimer’s disease. Ageing Res Rev. doi:10.1016/j.arr.2013.03.002
Devore S, Linster C (2012) Noradrenergic and cholinergic modulation of olfactory bulb sensory processing. Front Behav Neurosci 6:52. doi:10.3389/fnbeh.2012.00052
Doty RL (2012) Olfactory dysfunction in Parkinson disease. Nat Rev Neurol 8:329–339. doi:10.1038/nrneurol.2012.80
Doty RL, Deems DA, Stellar S (1988) Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology 38:1237–1244
Doty RL (2012) Olfaction in Parkinson’s disease and related disorders. Neurobiol Dis 46:527–552. doi:10.1016/j.nbd.2011.10.026
Echavarri C, Caballero MC, Aramendia A, Garcia-Bragado F, Tunon T (2011) Multiprotein deposits in neurodegenerative disorders: our experience in the tissue brain bank of Navarra. Anat Rec (Hoboken) 294:1191–1197. doi:10.1002/ar.21413
Fletcher ML, Chen WR (2010) Neural correlates of olfactory learning: critical role of centrifugal neuromodulation. Learn Mem 17:561–570. doi:10.1101/lm.941510
Garvey J, Petersen M, Waters CM, Rose SP, Hunt S, Briggs R et al (1986) Administration of MPTP to the common marmoset does not alter cortical cholinergic function. Mov Disord 1:129–134. doi:10.1002/mds.870010207
Gaykema RP, Zaborszky L (1996) Direct catecholaminergic-cholinergic interactions in the basal forebrain. II. Substantia nigra–ventral tegmental area projections to cholinergic neurons. J Comp Neurol 374:555–577
German DC, Manaye KF, White CL 3rd, Woodward DJ, McIntire DD, Smith WK et al (1992) Disease-specific patterns of locus coeruleus cell loss. Ann Neurol 32:667–676. doi:10.1002/ana.410320510
Gomez-Gallego M, Fernandez-Villalba E, Fernandez-Barreiro A, Herrero MT (2007) Changes in the neuronal activity in the pedunculopontine nucleus in chronic MPTP-treated primates: an in situ hybridization study of cytochrome oxidase subunit I, choline acetyl transferase and substance P mRNA expression. J Neural Transm 114:319–326. doi:10.1007/s00702-006-0547-x
Gundersen HJ, Bendtsen TF, Korbo L, Marcussen N, Moller A, Nielsen K et al (1988) Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS 96:379–394
Hawkes CH, Shephard BC, Daniel SE (1997) Olfactory dysfunction in Parkinson’s disease. J Neurol Neurosurg Psychiatry 62:436–446. doi:10.1136/jnnp.62.5.436
Heise CE, Teo ZC, Wallace BA, Ashkan K, Benabid AL, Mitrofanis J (2005) Cell survival patterns in the pedunculopontine tegmental nucleus of methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkeys and 6OHDA-lesioned rats: evidence for differences to idiopathic Parkinson disease patients? Anat Embryol (Berl) 210:287–302. doi:10.1007/s00429-005-0053-1
Herrero MT, Hirsch EC, Javoy-Agid F, Obeso JA, Agid Y (1993) Differential vulnerability to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine of dopaminergic and cholinergic neurons in the monkey mesopontine tegmentum. Brain Res 624:281–285
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55:181–184
Huisman E, Uylings HB, Hoogland PV (2008) Gender-related changes in increase of dopaminergic neurons in the olfactory bulb of Parkinson’s disease patients. Mov Disord 23:1407–1413
Huisman E, Uylings HB, Hoogland PV (2004) A 100% increase of dopaminergic cells in the olfactory bulb may explain hyposmia in Parkinson’s disease. Mov Disord 19:687–692. doi:10.1002/mds.10713
Huot P, Fox SH, Brotchie JM (2011) The serotonergic system in Parkinson’s disease. Prog Neurobiol 95:163–212. doi:10.1016/j.pneurobio.2011.08.004
Jellinger KA (2009) Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol 117:215–216; author reply 217–218. doi:10.1007/s00401-008-0454-3
Kar S, Slowikowski SP, Westaway D, Mount HT (2004) Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatry Neurosci 29:427–441
Karachi C, Grabli D, Bernard FA, Tande D, Wattiez N, Belaid H et al (2010) Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J Clin Invest 120:2745–2754. doi:10.1172/JCI42642
Kasa P, Rakonczay Z, Gulya K (1997) The cholinergic system in Alzheimer’s disease. Prog Neurobiol 52:511–535
Kehagia AA, Barker RA, Robbins TW (2010) Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol 9:1200–1213. doi:10.1016/S1474-4422(10)70212-X
Korczyn AD (2001) Dementia in Parkinson’s disease. J Neurol 248(Suppl 3):III-1–III-4. doi:10.1007/PL00022916
Kovács CL (1999) β-Amyloid deposition and neurofibrillary tangle formation in the olfactory bulb in ageing and Alzheimer’s disease. Neuropathol Appl Neurobiol 25:481–491
Kovacs I, Torok I, Zombori J, Kasa P (1998) Cholinergic structures and neuropathologic alterations in the olfactory bulb of Alzheimer’s disease brain samples. Brain Res 789:167–170
Kovacs I, Torok I, Zombori J, Kasa P (1996) Neuropathologic changes in the olfactory bulb in Alzheimer’s disease. Neurobiology (Bp) 4:123–126
Lehericy S, Hirsch EC, Cervera-Pierot P, Hersh LB, Bakchine S, Piette F et al (1993) Heterogeneity and selectivity of the degeneration of cholinergic neurons in the basal forebrain of patients with Alzheimer’s disease. J Comp Neurol 330:15–31. doi:10.1002/cne.903300103
Levey AI, Wainer BH, Mufson EJ, Mesulam MM (1983) Co-localization of acetylcholinesterase and choline acetyltransferase in the rat cerebrum. Neuroscience 9:9–22
Luquin MR, Montoro RJ, Guillen J, Saldise L, Insausti R, Del Rio J et al (1999) Recovery of chronic parkinsonian monkeys by autotransplants of carotid body cell aggregates into putamen. Neuron 22:743–750
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872. doi:10.1212/01.wnl.0000187889.17253.b1
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology 34:939–944
McShane RH, Nagy Z, Esiri MM, King E, Joachim C, Sullivan N et al (2001) Anosmia in dementia is associated with Lewy bodies rather than Alzheimer’s pathology. J Neurol Neurosurg Psychiatry 70:739–743
Mesholam RI, Moberg PJ, Mahr RN, Doty RL (1998) Olfaction in neurodegenerative disease: a meta-analysis of olfactory functioning in Alzheimer’s and Parkinson’s diseases. Arch Neurol 55:84–90. doi:10.1001/archneur.55.1.84
Mesulam MM, Geula C (1988) Nucleus basalis (Ch4) and cortical cholinergic innervation in the human brain: observations based on the distribution of acetylcholinesterase and choline acetyltransferase. J Comp Neurol 275:216–240. doi:10.1002/cne.902750205
Mesulam M-, Mufson EJ, Levey AI, Wainer BH (1983) Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol 214:170–197. doi:10.1002/cne.902140206
Mesulam M, Mufson EJ, Levey AI, Wainer BH (1984) Atlas of cholinergic neurons in the forebrain and upper brainstem of the macaque based on monoclonal choline acetyltransferase immunohistochemistry and acetylcholinesterase histochemistry. Neuroscience 12:669–686. doi:10.1016/0306-4522(84)90163-5
Momiyama T, Sim JA (1996) Modulation of inhibitory transmission by dopamine in rat basal forebrain nuclei: activation of presynaptic D1-like dopaminergic receptors. J Neurosci 16:7505–7512
Momiyama T, Sim JA, Brown DA (1996) Dopamine D1-like receptor-mediated presynaptic inhibition of excitatory transmission onto rat magnocellular basal forebrain neurones. J Physiol 495(Pt 1):97–106
Morley JF, Weintraub D, Mamikonyan E, Moberg PJ, Siderowf AD, Duda JE (2011) Olfactory dysfunction is associated with neuropsychiatric manifestations in Parkinson’s disease. Mov Disord 26:2051–2057. doi:10.1002/mds.23792
Mossner R, Schmitt A, Syagailo Y, Gerlach M, Riederer P, Lesch KP (2000) The serotonin transporter in Alzheimer’s and Parkinson’s disease. J Neural Transm Suppl 345–350
Mundinano IC, Caballero MC, Ordonez C, Hernandez M, DiCaudo C, Marcilla I et al (2011) Increased dopaminergic cells and protein aggregates in the olfactory bulb of patients with neurodegenerative disorders. Acta Neuropathol 122:61–74. doi:10.1007/s00401-011-0830-2
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ et al (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71:362–381. doi:10.1097/NEN.0b013e31825018f7
Olichney JM, Murphy C, Hofstetter CR, Foster K, Hansen LA, Thal LJ et al (2005) Anosmia is very common in the Lewy body variant of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 76:1342–1347. doi:10.1136/jnnp.2003.032003
Parodi J, Sepulveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG (2010) Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J Biol Chem 285:2506–2514. doi:10.1074/jbc.M109.030023
Pearce RK, Hawkes CH, Daniel SE (1995) The anterior olfactory nucleus in Parkinson’s disease. Mov Disord 10:283–287. doi:10.1002/mds.870100309
Petzold GC, Hagiwara A, Murthy VN (2009) Serotonergic modulation of odor input to the mammalian olfactory bulb. Nat Neurosci 12:784–791. doi:10.1038/nn.2335
Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A et al (2010) Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J 277:3051–3067. doi:10.1111/j.1742-4658.2010.07719.x
Porteros A, Gomez C, Valero J, Calvo-Baltanas F, Alonso JR (2007) Chemical organization of the macaque monkey olfactory bulb: III. Distribution of cholinergic markers. J Comp Neurol 501:854–865. doi:10.1002/cne.21280
Postuma R, Gagnon JF (2010) Cognition and olfaction in Parkinson’s disease. Brain 133:e160; author reply e161. doi:10.1093/brain/awq225
Potter PE, Rauschkolb PK, Pandya Y, Sue LI, Sabbagh MN, Walker DG et al (2011) Pre- and post-synaptic cortical cholinergic deficits are proportional to amyloid plaque presence and density at preclinical stages of Alzheimer’s disease. Acta Neuropathol 122:49–60. doi:10.1007/s00401-011-0831-1
Rogers JD, Brogan D, Mirra SS (1985) The nucleus basalis of Meynert in neurological disease: a quantitative morphological study. Ann Neurol 17:163–170. doi:10.1002/ana.410170210
Ruberg M, Rieger F, Villageois A, Bonnet AM, Agid Y (1986) Acetylcholinesterase and butyrylcholinesterase in frontal cortex and cerebrospinal fluid of demented and non-demented patients with Parkinson’s disease. Brain Res 362:83–91
Shimada H, Hirano S, Shinotoh H, Aotsuka A, Sato K, Tanaka N et al (2009) Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 73:273–278. doi:10.1212/WNL.0b013e3181ab2b58
Simpson J, Yates CM, Gordon A, St Clair DM (1984) Olfactory tubercle choline acetyltransferase activity in Alzheimer-type dementia, Down’s syndrome and Huntington’s chorea. J Neurol Neurosurg Psychiatry 47:1138–1139
Smiley JF, Subramanian M, Mesulam MM (1999) Monoaminergic-cholinergic interactions in the primate basal forebrain. Neuroscience 93:817–829
Stephenson R, Houghton D, Sundarararjan S, Doty RL, Stern M, Xie SX et al (2010) Odor identification deficits are associated with increased risk of neuropsychiatric complications in patients with Parkinson’s disease. Mov Disord 25:2099–2104. doi:10.1002/mds.23234
Szego EM, Gerhardt E, Outeiro TF, Kermer P (2011) Dopamine-depletion and increased alpha-synuclein load induce degeneration of cortical cholinergic fibers in mice. J Neurol Sci 310:90–95. doi:10.1016/j.jns.2011.06.048
Szegő ÉM, Outeiro TF, Kermer P, Schulz JB (2013) Impairment of the septal cholinergic neurons in MPTP-treated A30P α-synuclein mice. Neurobiol Aging 34:589–601. doi:10.1016/j.neurobiolaging.2012.04.012
Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA (2006) Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J Neurosci 26:467–478. doi:10.1523/JNEUROSCI.4265-05.2006
Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT (2012) The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin–proteasome system. Am J Pathol 181:1426–1435. doi:10.1016/j.ajpath.2012.06.033
Thomann PA, Dos Santos V, Toro P, Schönknecht P, Essig M, Schröder J (2009) Reduced olfactory bulb and tract volume in early Alzheimer’s disease—a MRI study. Neurobiol Aging 30:838–841
Ubeda-Banon I, Saiz-Sanchez D, de la Rosa-Prieto C, Argandona-Palacios L, Garcia-Munozguren S, Martinez-Marcos A (2010) Alpha-synucleinopathy in the human olfactory system in Parkinson’s disease: involvement of calcium-binding protein- and substance P-positive cells. Acta Neuropathol 119:723–735. doi:10.1007/s00401-010-0687-9
Wattendorf E, Welge-Lussen A, Fiedler K, Bilecen D, Wolfensberger M, Fuhr P et al (2009) Olfactory impairment predicts brain atrophy in Parkinson’s disease. J Neurosci 29:15410–15413. doi:10.1523/JNEUROSCI.1909-09.2009
Westermann B, Wattendorf E, Schwerdtfeger U, Husner A, Fuhr P, Gratzl O et al (2008) Functional imaging of the cerebral olfactory system in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 79:19–24. doi:10.1136/jnnp.2006.113860
Williams SS, Williams J, Combrinck M, Christie S, Smith AD, McShane R (2009) Olfactory impairment is more marked in patients with mild dementia with Lewy bodies than those with mild Alzheimer disease. J Neurol Neurosurg Psychiatry 80:667–670. doi:10.1136/jnnp.2008.155895
Wilson RS, Yu L, Schneider JA, Arnold SE, Buchman AS, Bennett DA (2011) Lewy bodies and olfactory dysfunction in old age. Chem Senses 36:367–373. doi:10.1093/chemse/bjq139
Acknowledgments
This study was supported by the UTE-project/Foundation for Applied Medical Research (FIMA). The authors thank Monica Kurtis for English edition of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mundiñano, IC., Hernandez, M., DiCaudo, C. et al. Reduced cholinergic olfactory centrifugal inputs in patients with neurodegenerative disorders and MPTP-treated monkeys. Acta Neuropathol 126, 411–425 (2013). https://doi.org/10.1007/s00401-013-1144-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-013-1144-3