
Abstract
In G93A mice, the most popular model of amyotrophic lateral sclerosis (ALS), neuronal Lewy-body-like hyaline inclusions (LBHIs) and mitochondria-derived vacuoles are observed in addition to motor neuron loss. Although LBHIs are thought to be toxic, the significance of the mitochondria-derived vacuoles has not been fully investigated. In this study, the relationship between the formation of these vacuoles and LBHIs was clarified statistically in the lumbar segment from two phyletic lines of G93A mice (G1L, G1H), using immunohistochemical methods. Furthermore, the distributions of vacuoles and LBHIs were examined in the pons including the facial nucleus, where pathological changes occur in ALS patients and G93A mice. Numerous vacuoles 2–3 μm in diameter were detected in the neuropil of the lumbar segment from G1L mice euthanatized approximately 3.5 months prior to the onset of the disease. Most of the vacuoles disappeared, but some became larger as the disease progressed. The number of vacuoles with a diameter exceeding 5 μm began to decrease after disease onset, while that of intra-neuritic LBHIs increased rapidly. There was a strong inverse correlation between the numbers of vacuoles and LBHIs in symptomatic mice (P<0.01; G1L, r=−0.91; G1H, r=−0.93). In the facial nucleus of G1L mice, where the number of motor neurons was significantly reduced, only a few LBHIs were detected along with prominent vacuole formation. In contrast, significantly more LBHIs with little vacuole formation were evident around the facial nucleus in G1L mice. Furthermore, the SOD1 immunoreactivity in vacuoles initially increased and then decreased after disease onset. Taken together, the present findings suggest that the mitochondria-derived vacuoles might prevent the formation of LBHIs by sequestering mutated SOD1 from the cytoplasm.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal motor neuron disease whose pathogenesis remains unknown. About 10% of ALS cases are familial and approximately 15–20% of familial ALS patients possess the copper/zinc superoxide dismutase (SOD1) gene mutation [8, 40]. Since transgenic mice or rats carrying the human mutated SOD1 gene (SOD1 mice or rats) develop progressive motor deficits caused by loss of anterior horn cells [3, 12, 16, 32, 39, 56], they have been used by many researchers as a model of ALS. SOD1 mice or rats carrying different mutated SOD1 genes have been reported to show different pathologic features: in G93A mice or rats, many Lewy-body-like hyaline inclusions (LBHIs) and mitochondria-derived vacuoles are observed [6, 7, 32]; in G85R or G86R mice, many LBHIs, but almost no vacuoles, appear long before the onset of the disease [3, 39]; in H46R rats, many LBHIs and very few vacuoles are found [32]; in G37R mice, there is prominent vacuole formation and almost no LBHIs [55, 56]. Although in G85R mice the level of mutant G85R SOD1 protein expressed is only 20% of endogenous SOD1, these mice show very progressive motor deficits [3]. Other SOD1 mice or rats express a mutant protein level approximately 10 times higher than that in murine SOD1 [12, 16, 32, 56]. Several lines of G93A or G37R mice with different levels of mutant protein expression show different pathologic features [6, 7, 56]. Thus, the differences in neuropathology observed among SOD1 mice or rats appear to depend on the character of the mutant protein and its level of expression.
SOD1-positive LBHIs in neurons are neuropathological hallmarks of familial ALS linked with SOD1 mutation [15, 17, 22, 44]. In cell lines transfected with mutated SOD1, aggregations of mutated SOD1 or LBHI-like structures are formed [9, 21, 46]. In SOD1 mice or rats, such aggregates of SOD1 or LBHIs appear before onset of the disease [3, 4]. These aggregates of SOD1 or LBHIs, which are found specifically in the affected spinal cord or brainstem [15, 44, 54], are resistant to strong detergents or reducing agents. Although formation of aggresomes [21] or inclusions such as Lewy bodies in Parkinson’s disease might be considered the result of cell-protective responses to various forms of stress [36], aggregation of mutant SOD1 or the formation of LBHIs in SOD1-mutated ALS is reported to have a toxic effect [4, 5, 23] due to sequestration of the components that are essential for maintaining cell functions [24, 25], induction of repetitive misfolding and reduction of chaperone function [2], or reduction of the activity of the proteasome integral for protein turnover [20, 21, 53].
The most important function of mitochondria in cells is the production of ATP, which is indispensable for sustaining life. Cytochrome c oxidase (CCO), which is located in the inner membrane of the mitochondrion and, in mammals, is composed of 13 different subunits, participates in electron transport within mitochondria [50]. Cytochrome c (cyt c) is oxidised by CCO through electron transport in the intermembrane space of the mitochondrion [38]. The large amount of energy required for action potentials in neurons depends on the ATP produced by mitochondria through electron transport. In G93A mice, the most widely used animal model of ALS, one of the very early pathologic features is the appearance of vacuoles followed by that of abnormal mitochondria [6, 14, 28, 42, 43], suggesting that the vacuoles are derived from mitochondria [14, 28].
The formation of LBHIs has been thought to have toxic effects, but the significance of the vacuoles remains unclear. Since the vacuoles appear far earlier than the LBHIs, an investigation of vacuoles would be important for clarifying the pathogenesis of the disease in G93A mice. In the study presented here, we carried out a quantitative examination of the vacuoles using an immunohistochemical method and analyzed the relationship between the vacuoles and the formation of LBHIs in the lumbar segment or facial nucleus of G93A mice in order to clarify the significance of mitochondria-derived vacuoles.
Materials and methods
Animals
Transgenic mice expressing the G93A mutated human SOD1 gene at a low (B6SJL-TgN[SOD1-G93A]1Gurdl, G1L) or high (B6SJL-TgN[SOD1-G93A]1Gur, G1H) level were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). These mice were bred and maintained as hemizygotes by mating with wild-type B6SJL mice. Non-transgenic littermates were used as controls. All animals were genotyped using polymerase chain reaction amplification of the tail DNAs under conditions that have been described previously [33]. All animals were handled in accordance with the Guidelines for the Care and Use of Laboratory Animals at Osaka University Graduate School of Medicine. We evaluated the animals clinically, examining their hindlimb extension when they were suspended in the air by the tail [1, 33, 34, 49].
Tissue preparation
We examined control (260±6 days old), G1L, and G1H mice (n=3 in each group). G1L mice were euthanatized at the age of 90, 140 (the asymptomatic stage), 180 (the presymtomatic stage), 230 days (the symptomatic stage) or at the end stage when they could hardly move or drink water because of severe paralysis (259±6 days old, the moribund state). G1H mice were euthanatized at the age of 66 (the asymptomatic stage), 100 (the early symptomatic stage), or 115 days (the late symptomatic stage). They were deeply anesthetized with sodium pentobarbital, and perfused with phosphate-buffered saline (PBS, pH 7.4) followed by 4% paraformaldehyde. The brainstem and spinal cord were removed, immersed in the same fixative overnight at 4°C, and then cryoprotected. Ten-micrometer-thick frozen sections were prepared and stained with hematoxylin and eosin (HE). Small pieces of lumbar segments were fixed with 2.5% glutaraldehyde in 0.1 M PB (pH 7.4) for 2 h at 4°C, followed by 1% osmium tetroxide in 0.1 M PB at 4°C, dehydrated in an ethanol series and embedded in Epon; LUVEAK-812 (glycerol triglycidyl ether, Nakarai Tesque, Kyoto, Japan)/LUVEAK-DDSA (dodecylsuccinic anhydride, Nakarai Tesque)/LUVEAK-MNA (methylnadic anhydride, Nakarai Tesque)/LUVEAK-DMP-30 (2,4,6-Tris (dimethylaminomethyl) phenol, Nakarai Tesque). Transverse sections 1 μm thick were stained with toluidine blue.
Immunohistochemical analysis of vacuoles in lumbar segment and brainstem
For immunohistochemical evaluations, frozen or deparaffinized sections were incubated for 30 min with 0.3% H2O2 to quench endogenous peroxidase activity and then washed with PBS. Normal goat serum was used as a blocking reagent. Mouse monoclonal antibodies against cyt c (1:100 in 1% PBS containing bovine serum albumin, clone 6H2.B4, BD PharMingen, CA, USA), CCO subunit I (1:400 clone 1D6, Molecular Probes Inc, OR, USA), human SOD1 (0.5 μg/ml, clone 1G2, MBL, Aichi, Japan) or GFAP (ready to use, DAKO, Glostrup, Denmark) were used as primary antibodies. Tissue sections were incubated with each primary antibody for 18 h at 4°C. The avidin–biotin–immunoperoxidase complex (ABC) method was employed according to the manufacturer’s instructions to detect each bound antibody using the appropriate Vectastain ABC kits (Vector Laboratories, Burlingame, CA, USA). 3,3′-Diaminobenzidine tetrahydrochloride (DAKO, Glostrup, Denmark) was used as the final chromogen. Hematoxylin was used to counterstain cell nuclei. In control experiments, primary antibodies were omitted from the incubation medium.
Quantitative evaluation of motor neurons, LBHIs and cyt c-positive vacuoles in the lumbar segment
To estimate the number of neurons in the gray matter (VII, VIII, IX Rexed areas) showing clear nucleoli and cell bodies with a diameter greater than 25 μm [26, 28, 48], presumed to be alpha motoneurons [10, 30, 31], video images of the anterior horns were obtained with a digital camera (KEYENCE VB-7010, KEYENCE, Osaka, Japan) attached to a light microscope (ECLIPSE E800, Nikon, Tokyo, Japan) for each section, and the areas of motor neurons showing clear nucleoli and cell bodies were measured using image analysis software (VH-H1A5, KEYENCE). The number of neurons with a diameter greater than 25 μm were counted in HE-stained sections. LBHIs with a halo and core and cyt-c-positive vacuoles in the gray matter were also counted (×100 objective). LBHIs were also confirmed using antibodies against human SOD1 and GFAP. Since the vacuoles are contained exclusively in neuronal processes [19, 43], and not in astrocytes, only neuronal LBHIs were counted to examine the relationship between the vacuoles and LBHIs in neurons. LBHIs in cells possessing glial nuclei were omitted. To establish how the size and number of vacuoles in the lumbar segments of G1L mice changes chronologically over the clinical course of the disease, vacuoles were divided according to size into small (<5 μm) or large (>5 μm). Every fifth section (40 μm interval) was obtained, and three sections from each mouse were used to obtain the total number of neurons, LBHIs or vacuoles. The quantitative evaluation was analyzed statistically. Moreover, the relationship between the number of vacuoles and LBHIs was estimated in each symptomatic mouse (G1L, 230 days and the moribund state 259±6 days; G1H, 100 and 115 days).
Quantitative analysis of LBHIs in the pons including the facial nucleus
For histological analyses of the facial nucleus (nVII), we examined non-transgenic littermates (n=3, 264±7 days) and G1L mice in the moribund state (n=7, 258±7 days). Each carefully hemisected brainstem was embedded in paraffin and sectioned transversely. The pons including the nVII was identified in these sections, with reference to the mouse brain atlas of Paxinos and Franklin [37]. Six-micrometer-thick paraffin sections were prepared and stained with HE. In the nVII, large neurons with clear nucleoli and cell bodies were counted. LBHIs located in three subregions of the pons (vacuole-rich area = intra-nVII, the border zone, and vacuole-poor area, as delineated in Fig. 1) were also counted (×40 objective). To estimate the number of LBHIs per unit area in each subregion, video images of the pons were obtained with a digital camera (KEYENCE VB-7010, KEYENCE, Osaka, Japan) attached to a light microscope (ECLIPSE E800, Nikon, Tokyo, Japan) for each section, and the areas of three subregions were measured using image analysis software (VH-H1A5, KEYENCE). Every fifth section (at 24-μm intervals) was obtained, and three sections from each mouse were used to obtain the total number of neurons and the density of LBHIs. The number of motor neurons and the density of LBHIs were analyzed statistically. All quantitative investigations were performed independently by three neuropathologists (HS, HF, SK).

Schematic diagram of the three subregions in the pons; vacuole-rich area (=intra-nVII), the border zone and vacuole-poor area. The boundary of massive vacuole formation limited to the facial nucleus (nVII) is shown at low (a) and high (b) magnification as a yellow line, using actual microscopic fields digitized for illustrative purposes. The definition of the vacuole-rich area (=intra-nVII), border zone and vacuole-poor area is as follows: vacuole-rich area (light blue) corresponds to the inner area outlined in yellow; the border zone (white) corresponds to the surrounding area outlined in yellow and green, 120 μm exterior to the yellow line; the vacuole-poor area (pink) corresponds to the outer area of the pons excluding the other two regions. N neuron
Statistics
Data are expressed as the mean ± standard error of the mean (SEM). All statistical analyses of histopathological data were carried out using the Statview for Macintosh software package (Ver5.0, SAS Institute Inc, CA, USA). A nonparametric test, Mann–Whitney U test, was used to analyze the number of neurons, LBHIs and vacuoles in the lumbar segment or the number of neurons and the density of LBHIs in the pons. The relationship between the number of LBHIs and vacuoles in the lumbar segment was estimated by regression analysis.
Results
Morphological changes of vacuoles in the lumbar segment of G1L or G1H mice
Many tiny vacuoles 2–3 μm in diameter were found in neurites mainly at the edge of the anterior horn in G1L mice at 90 days (Fig. 2a). Some of the vacuoles were hardly distinguishable from capillary vessels. At 140 days, larger but fewer vacuoles than those at 90 days were observed in the same area (Fig. 2b). Vacuoles were also scattered diffusely in the anterior horn. At 180 days, round vacuoles larger than those at 140 days (Fig. 2c) were frequently found throughout the gray matter. At the later stages in G1L mice, the large vacuoles appeared slightly deformed and were reduced in number (Fig. 2d). Many LBHIs were scattered in the gray matter. In G1H mice, more prominent vacuole formation was observed (Fig. 4a, d) than that in G1L mice. The vacuoles became larger and their number was reduced by 115 days (Fig. 4d) in comparison with those at 100 days (Fig. 4a). Although at 100 days the vacuoles were observed mainly in the anterior horn, by 115 days their distribution had also extended to the whole of the gray matter.

Morphological changes in vacuoles in the anterior horn of G1L mice at different stages. (Epon sections, toluidine blue, a 90 days, b 140 days, c 180 days, d end stage). a Many small vacuoles (<5 μm, arrow heads) are evident at the edge of the anterior horn. b The number of vacuoles has decreased. Vacuoles (arrows) at the edge of the anterior horn have become larger. c The number of vacuoles (arrows) is smaller than in b. Note the large vacuoles in neurites including axons (clear arrow). d Only a few vacuoles (arrows) are evident. LBHIs (clear arrow) appear in the anterior horn. N neuron; V vessels. Scale bar a (also for b–d) 20 μm
Immunohistochemical analyses of vacuoles in the lumbar segment of G1L mice
As a negative control, sections were incubated without the primary antibody; this resulted in no staining in the lumbar segments of normal and G93A low-copy transgenic mice (G1L mice). As expected, the cytoplasm of motor neurons was clearly stained for cyt c or CCO in the lumbar segment of normal mice (Fig. 3a, b). The staining pattern with the two antibodies was similar, showing a fine granular pattern. The neuropil was also weakly stained for these antibodies. The immunohistochemical analysis revealed many tiny vacuoles, strongly positive for cyt c at the edge of the anterior horn in G1L mice (90 days; Fig. 3c–e). Some vacuoles were scattered in the neuropil of the anterior horn. Axons were lined with tiny vacuoles that appeared to be attached together. The rim of the vacuoles in the neuropil was stained for cyt c (Fig. 3d, e). The structures lying interior to the vacuolar rim were CCO-positive, although the rim itself was CCO-negative. At 140 days, the number of vacuoles was lower than that at 90 days. In contrast, large vacuoles (>5 μm), which were never seen at 90 days, were frequently observed at the edge of the anterior horn (Fig. 3f). The cyt c immunoreactivity was reduced within the rim of these large vacuoles (Fig. 3f, g). Small vacuoles (<5 μm) did not fuse to form a large vacuole. Some small and large vacuoles were found in the dorsal horn. Each vacuole had CCO-positive structures lying interior to the rim (Fig. 3h). At 180 days, there was a reduction in the number of small vacuoles (<5 μm), and many large vacuoles (>5 μm) were found scattered not only at the edge of the anterior horn, but also throughout the neuropil. These vacuoles had highly CCO-positive structures lying interior to the rim (Fig. 3i). At later stages, the tiny, strongly cyt c-positive vacuoles were seen only rarely (Fig. 3j, l), and some large vacuoles were observed scattered throughout the neuropil. The CCO-positive structures lying interior to the vacuole rim became atrophic (Fig. 3k), especially in moribund mice. Cyt c-positive vacuoles were never seen in the soma of neurons from G1L mice.


Immunohistochemistry of vacuoles in the anterior horn of G1L mice (a, b normal control, c–l G1L mice; a, b 263 days, c–e 90 days, f–h 140 days, i 180 days, j, k 230 days, l end stage; a, c–g, j, l cyt c, b, h, i, k CCO). a A fine granular staining pattern is evident in the cytoplasm of motor neurons (N, arrows). The neuropil is also weakly stained. b Motor neurons (N, arrows) show well-defined cytoplasmic staining for CCO. Diffusely scattered fine dots can be seen in the neuropil. c–e Tiny, densely immunolabeled vacuoles within the rim (arrow heads) are prominent at the edge of the anterior horn. d is a high magnification view of c. e is a high magnification view of d. f The number of vacuoles is lower than in d. Large vacuoles (>5 μm, arrows), which were never seen in d. The immunoreactivity for cyt c is reduced within the rim of large vacuoles (arrows) compared to that of small vacuoles (<5 μm, arrow heads). In axons, tiny vacuoles are tightly packed to form a columnar shape (clear arrow). g is a high magnification view of f. h Abnormal structures lying interior to the rim of vacuoles (arrows, arrow heads) are strongly positive for CCO. Irregular CCO-positive dots (arrow heads) are scattered in the neuropil. i Large CCO-positive complexes lying interior to the rim of large vacuoles. j The number of vacuoles (arrow, arrow head) is lower than that in i. Cyt-c-positive small vacuoles (arrow head) are seen only rarely. k CCO-positive complexes (arrows) lying interior to the vacuolar rim have become atrophic in comparison with those in i. l Small vacuoles are not evident. Some large vacuoles (arrows) remain. N neurons; V vessels. Scale bars: a (also for b)50 μm, c 100 μm, d (also for f, h–l) 20 μm, and e (also for g) 10 μm
Immunohistochemical analyses of vacuoles in the lumbar segment of G1H mice
In symptomatic G1H mice (100 and 115 days), prominent vacuole formation was observed (Fig. 4b, c, e, f). The vacuolar rims were cyt-c-positive and there were CCO-positive structures lying interior to them, as observed in G1L mice. Cyt c immunoreactivity was reduced in the large vacuoles (Fig. 4b, e). The structures lying interior to the vacuolar rim, evident at 100 days (Fig. 4c), became rather atrophic by 115 days (Fig. 4f). At 100 days, the ratio of the number of large vacuoles (>5 μm) to the total number of vacuoles was more than 50% (Fig. 4b); this figure had reached almost 90–100% by 115 days (Fig. 4e). The vacuoles in G1H mice tended to be larger than those in G1L mice.

More prominent vacuole formation with similar immunostaining patterns for cyt c and CCO in the anterior horn of G1H mice (a–c; 100 days, b–f; 115 days, a, d; Epon sections, toluidine blue, b, e; cyt c, c, f; CCO). a Abundant small (<5 μm, arrow heads) or large (>5 μm, arrows) vacuoles are present in neurites. b Cyt-c-positive staining in the rim of small (arrow heads) and large (arrows) vacuoles. The immunoreactivity of the large vacuoles (arrows) is reduced, as in G1L mice. c CCO-positive structures are evident in small (arrow heads) or large vacuoles (arrows). d The number of vacuoles is reduced. Larger vacuoles than those in a are frequently evident. e Large vacuoles about 15–20 μm in diameter (arrows) are observed more frequently than in the G1L lumbar segment. f Somewhat atrophic CCO-positive structures (arrows) lying interior to the rim of large vacuoles. N neurons; V vessels. Scale bar, a (also for b–f) 20 μm
Quantitative analysis of the vacuoles, LBHIs and motor neurons in the lumbar segment of G1L and G1H mice
Figure 5 shows the number of small (<5 μm) and large (>5 μm) vacuoles, and LBHIs and motor neurons (>25 μm) in the lumbar segment during the clinical course of the disease in G1L mice. Numerous small vacuoles were already evident at 90 days, and their number decreased as the disease progressed (Fig. 5a, Table 1, P<0.05). The number of large vacuoles, which appeared at around 140 days, were increased significantly by 180 days but then decreased significantly by 230 days (Fig. 5a, Table 1, P<0.05). The ratio of the number of large vacuoles to the total number of vacuoles was 10–20% at 140 and 180 days, and ~50% in moribund mice (Fig. 5a). In G1H mice, the total number of vacuoles showed a tendency to decrease, but not to a significant degree (Table 1, P=0.1266). The number of neurons declined significantly after 180 days in G1L mice, and after 66 days in G1H mice (Table 1, P<0.05). LBHIs were restricted to the anterior horn at 180 and 100 days in G1L and G1H mice, respectively, becoming more widespread at the later stages. The number of LBHIs increased significantly in G1L or G1H mice as the disease progressed (Fig. 5b, Table 1, P<0.05). Most of the LBHIs were intra-neuritic.

Time course of changes in the number of vacuoles (a), LBHIs and motor neurons (b) in the anterior horn of G1L mice. a Numerous small vacuoles are evident at 90 days. The number of small vacuoles decreases significantly with disease progression (*P<0.05). The large vacuoles are present from 140 days, and their number then increases significantly by 180 days, followed by a significant decrease by 230 days. b The number of neurons (>25 μm) declines significantly after 180 days (*P<0.05) and, conversely, the number of LBHIs increases significantly after 180 days (*P<0.05). Data are presented as mean ± SEM (n=3 for each group). c Representative hematoxylin and eosin (HE) staining of the anterior horn in control (1) and G1L mice at the end stage (2). The number of neurons (>25 μm, arrows) is reduced in (2). Scale bar c 25 μm
Correlation between the numbers of vacuoles and LBHIs in symptomatic G1L and G1H mice
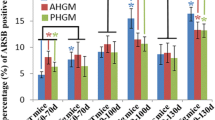
Figure 6 shows the correlation between the numbers of vacuoles and LBHIs observed in symptomatic G1L (230 days and in the moribund state) and G1H (100 and 115 days) mice. The number of vacuoles in G1H mice was greater than in G1L mice. Regression analysis revealed a statistically significant inverse correlation between the numbers of vacuoles and LBHIs in both G1L (r=−0.91, P<0.01) and G1H (r=−0.93, P<0.01) mice.

Inverse correlation between the numbers of mitochondria-derived vacuoles and LBHIs. A statistically significant inverse correlation was observed between the numbers of LBHIs and vacuoles in both G1L (green) and G1H (orange) mice. Numbers shown in italics represent the age of the G93A mice at perfusion
Quantitative analysis of LBHIs in the nVII and the differential localization of LBHIs and vacuoles
HE staining revealed atrophic motor neurons with prominent vacuole formation in the nVII of moribund G1L mice. Most of the vacuoles in the nVII (Fig. 7b) were larger than those observed in the lumbar segment. The number of motor neurons in the nVII was significantly reduced (Table 2, P<0.05), and LBHIs were found only rarely in the vacuole-rich area (=intra-nVII). In marked contrast, LBHIs were found frequently in the border zone (see Materials and methods), but there were very few vacuoles (Fig. 7c). Most of the vacuoles were located in the neuropil, and not in the soma of neurons (Fig. 7d). Most of the LBHIs were also intra-neuritic. The LBHIs in the border zone were larger than those in the vacuole-rich area (=intra-nVII). Quantitative analysis of data from the three subregions of the pons demonstrated a preferential localization of LBHIs to the border zone, the area surrounding the nVII (Table 2, P<0.01).

Representative hematoxylin and eosin staining in the nVII of control (a; 261 days) and G1L (b–d; end stage) mice. a, b The number of motor neurons in the G1L nVII is reduced, and all of the residual neurons are atrophic in G1L mice as compared with the controls. Note the prominent vacuolization (arrow heads) and lack of LBHIs inside the G1L nVII. c Many LBHIs are present (arrows, white arrow) in the area surrounding the nVII. The yellow line delineates the vacuole-rich area (=intra-nVII). d A large LBHI (white arrow), more than 10 μm in diameter, is located in a neurite, and not in the soma. Scale bar a (also for b) 50 μm, c 100 μm, and d 20 μm
Immunohistochemical analysis of vacuoles in the facial nucleus of G1L mice
Immunohistochemical analysis of vacuoles in the facial nucleus (nVII) showed that abundant tiny cyt-c-positive vacuoles appeared at the edge of the nVII at an early stage (Fig. 8a), similar to those in the lumbar segments. As the disease progressed, the vacuoles became larger (Fig. 8b–f) and were distributed throughout the nVII. Thereafter, although their number decreased progressively, a considerable amount of large vacuoles remained in the nVII even at the end stage (Figs. 7b, 8f). The immunoreactivity for cyt-c was weaker in the larger vacuoles than in the tiny vacuoles (Fig. 8b). Immunohistochemical staining for mutant SOD1 showed that the rim of the vacuoles was stained at an early stage (Fig. 8c, d). The SOD1 immunoreactivity of the rim increased at 140 days (Fig. 8d). By 180 days, not only the rim, but also the core of the vacuoles became SOD1-positive (Fig. 8e). At the end stage, the SOD1-positive structures inside the vacuoles decreased. Most of the vacuoles, except for those present in axons, showed weak immunoreactivity for SOD1 in their rim. Vacuoles that were almost SOD1-negative were frequently observed (Fig. 8f).

Immunohistochemical analysis of vacuoles in the nVII of G1L mice (a, c; 90 days, b, d; 140 days, e; 180 days, f; end stage, a, b; cyt-c, c–f; SOD1). a Many tiny cyt-c-positive vacuoles (arrow heads) are evident. b Larger vacuoles (arrows) appear, showing lower immunoreactivity than in a. c Tiny vacuoles (arrow heads) are SOD1-positive. d SOD1 immunoreactivity becomes stronger at the rim of larger vacuoles (arrows) than that of tiny vacuoles observed in c. e SOD1-positivity is evident not only at the rim, but also the core of the vacuoles (arrows). The immunoreactivity has become stronger than that in d. f Only a few large vacuoles (arrows, clear arrows) remain. The SOD1 immunoreactivity of vacuoles has become weaker than that in e. Some of the vacuoles (clear arrows) are almost immunonegative. Scale bar a (also for b–f) 20 μm
Discussion
In the G93A mouse, the most commonly studied model of ALS, the characteristic neuropathological features are LBHIs and vacuoles [6, 7]. Although scarcely found in FALS patients associated with SOD1 gene mutation [41], it has been considered important to investigate vacuoles [18, 19, 28, 42, 43] because the model mice overexpressing the mutant SOD1 protein could provide clues for understanding the characteristics of mutant SOD1. Also, since evaluation of LBHIs, which are the hallmarks of FALS linked with SOD1 gene mutation, is indispensable for pathological analysis, it is necessary to clarify the factors that can influence their formation.
In control mice, the cytoplasm of the motor neurons showed granular staining with antibodies against subunit I of CCO, a component of the mitochondrion-specific enzyme that is localized in the inner membrane [50], and against cyt c, a mediator of CCO that is localized in the intermembrane space [38]. The staining with these antibodies in the motor neurons of control mice corresponds to the localization of mitochondria [11, 19]. In G93A mice, the rims of vacuoles were cyt-c-positive and CCO-negative, and the structures lying interior to the vacuolar rim were CCO-positive and cyt-c-negative. The latter structures are probably non-functional mitochondrial membrane remnants [14, 28, 43]. Since cyt c is released into the cytosol from the permeability transition pores of injured mitochondria [11, 27, 29, 45, 47, 57, 58], the presence of a cyt-c-positive rim might imply that cyt c is released from the intermembrane space of injured mitochondria into vacuoles to accumulate along the vacuolar rim. Since both small (<5 μm) and large (>5 μm) vacuoles in the neuropil exhibited the same immunostaining patterns for cyt c and CCO, and since small vacuoles did not appear to fuse to form large ones, it is thought that the small vacuoles grow to become the large vacuoles [14].
According to a previous report, the appearance of vacuoles in the lumbar segments might be related to the onset, rather than progression of the disease, since the number of vacuoles was found to be maximal at disease onset, and declined thereafter [28]. Using antibodies against mitochondrial components, we have shown here that numerous tiny vacuoles, which are distinguishable from cyt-c-negative capillary blood vessels, appear in the neuropil approximately 3.5 months before disease onset [14, 19]. In G1L mice, the number of vacuoles in both the lumbar segment and brainstem decreased progressively thereafter, irrespective of disease onset. In contrast with the findings of the previous study [28], therefore, we have found that the presence of these vacuoles appears to be unrelated to disease onset. Although the number of large vacuoles (>5 μm) increased until disease onset and decreased thereafter, they appeared to be unrelated to disease onset because the degree of neuron loss in the nVII, exhibiting prominent large vacuoles, was much less marked than in the lumbar segment. If the presence of large vacuoles had been related to disease onset, which is determined by neuron loss, then the degree of neuronal loss in the nVII would have been more severe.
In the lumbar segment, intra-neuritic LBHIs appeared just before disease onset and increased rapidly [55] with the progressive reduction in the number of vacuoles in neurites [28, 42, 43]. Although the number of vacuoles was greater in G1H mice than in G1L mice, there was a statistically significant inverse correlation between the number of vacuoles and LBHIs in symptomatic G1L and G1H mice. In the G1L brainstem with prominent vacuole formation, the number of motor neurons was significantly reduced, in agreement with previous reports [13, 35], but only a few LBHIs were found even at the end stage. A notable observation was the presence of many intra-neuritic LBHIs in the border zone where there were few vacuoles. As LBHI formation is known to occur in affected neurons [15, 44, 54], the LBHIs around the nVII would be contained within the neurites of motor neurons in that area. Therefore, there was an inverse correlation between these two neuropathological changes in both the lumbar segment and the nVII.
Immunohistochemistry using anti-SOD1 antibody revealed that mutant SOD1 accumulated in the rim of tiny vacuoles at an early stage, and was increased in both the rim and the core of the vacuoles [19, 43] until onset. After onset, the intensity of SOD1 immunostaining decreased in both the rim and the core, and SOD1-negative large vacuoles were frequently observed. Since the amount of mutant SOD1 protein increases with age [19, 51], the increased SOD1 immunoreactivity in vacuoles would suggest that more mutant SOD1 was retained in them before onset. The fact that the SOD1 immunoreactivity in vacuoles decreased after onset suggests that vacuoles would not be able to sequester mutant SOD1, despite its higher level of expression in neurons. It is possible that only well-functioning vacuoles at the early stage might be able to retain mutant SOD1. Since mutant SOD1 does not disappear, even when it leaks from vacuoles after onset, it would still remain in the cell [52]. Mutant SOD1 that increased outside vacuoles but remained within the cell would be related to the formation of LBHIs, followed by aggregation of mutant SOD1 in the cell.
In a study using electron and immunofluorescence microscopy, Higgins et al. have reported that the vacuoles originate through expansion of the mitochondrial intermembrane space and extension of the outer mitochondrial membrane and lack lysosomal signals [14]. This suggests that the vacuoles would be produced by a nonautophagic, but still uncharacterized and unique mechanism. In this study, we found a negative correlation between the mitochondria-derived vacuoles and LBHIs, although this does not necessarily mean that the former prevents the latter. However, considering the results of SOD1 immunostaining, it is not unlikely that the mitochondria-derived vacuoles might prevent the formation of LBHIs by sequestering mutated SOD1 from the cytoplasm.
References
Barneoud P, Lolivier J, Sanger DJ, Scatton B, Moser P (1997) Quantitative motor assessment in FALS mice: a longitudinal study. Neuroreport 8:2861–2865
Bruening W, Roy J, Giasson B, Figlewicz DA, Mushynski WE, Durham HD (1999) Up-regulation of protein chaperones preserves viability of cells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amyotrophic lateral sclerosis. J Neurochem 72:693–699
Brujin LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18:327–338
Brujin LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281:1851–1854
Cleveland DW, Liu J (2000) Oxidation versus aggregation—how do SOD1 mutants cause ALS? Nat Med 6(12):1320–1321
Dal Canto MC, Gurney ME (1994) Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol 145:1271–1279
Dal Canto MC, Gurney ME (1997) A low expressor line of transgenic mice carrying a mutant human Cu, Zn superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis. Acta Neuropathol 93:537–550
Deng H-X, Hentati A, tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getsoff ED, Hu P, Herzfeldt B, Roos RP, Warner C, Deng G, Soriano E, Smyth C, Parge HE, Ahmed A, Roses AD, Hallwell RA, Pericak-Vance MA, Siddique T (1993) Amyotrophic lateral sclerosis and structural defects in Cu/Zn superoxide dismutase. Science 261:1047–1051
Durham HD, Roy J, Dong L, Figlewicz DA (1997) Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol 56(5):523–530
Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185:232–240
Guegan C, Vila M, Rosoklija G, Hays AP, Przedborski S (2001) Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci 21:6569–6576
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775
Haenggeli C, Kato AC (2002) Differential vulnerability of cranial motoneurons in mouse models with motor neuron degeneration. Neurosci Lett 335:39–43
Higgins CMJ, Jung C, Xu Z (2003) ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci 4:16
Hirano A, Kurland LT, Sayre GP (1967) Familial amyotrophic lateral sclerosis. A subgroup characterized by posterior and spinocerebellar tract involvement and hyaline inclusions in the anterior horn cells. Arch Neurol 16:232–243
Howland DS, Jiu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA 99:1604–1609
Inoue K, Fujimura H, Ogawa Y, Satoh T, Shimada K, Sakoda S (2002) Familial amyotrophic lateral sclerosis with a point mutation (G37R) of the superoxide dismutase 1 gene: a clinicopathological study. Amyotroph Lateral Scler Other Motor Neuron Disord 3:244–247
Jaarsma D, Haasdijk ED, Grashorn JAC, Hawkins R, Dujin WV, Verspaget HW, London J, Holstege JC (2000) Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis 7:623–643
Jaarsma D, Rognoni F, Duijn WV, Verspaget HW, Haasdijk ED, Holstege JC (2001) CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol 102:293–305
Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143:1883–1898
Johnston JA, Dalton MJ, Gurney ME, Kopito RR (2000) Formation of high molecular weight complexes of mutant Cu,Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 97:12571–12576
Kato S, Takikawa M, Nakashima K, Hirano A, Cleveland DW, Kusaka H, Shibata N, Kato M, Nakano I, Ohama E (2000) New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: inclusions containing SOD1 in neurons and astrocytes. Amyotroph Lateral Scler Other Motor Neuron Disord 1:163–184
Kato S, Horiuchi S, Liu J, Cleveland DW, Shibara N, Nakashima K, Nagai R, Hirano A, Takikawa M, Kato M, Nakano I, Ohama E (2000) Advanced glycation endproduct-modified superoxide dismutase-1 (SOD1)-positive inclusions are common to familial amyotrophic lateral sclerosis patients with SOD1 gene mutations and transgenic mice expressing human SOD1 with a G85R mutation. Acta Neuropathol 100:490–505
Kato S, Sumi-Akamaru H, Fujimura H, Sakoda S, Kato M, Hirano A, Takikawa M, Ohama E (2001) Copper chaperone for superoxide dismutase co-aggregates with superoxide dismutase 1 (SOD1) in neuronal Lewy body-like hyaline inclusions: an immunohistochemical study on familial amyotrophic lateral sclerosis with SOD1 gene mutation. Acta Neuropathol 102:233–238
Kato S, Saeki Y, Aoki M, Nagai M, Ishizaki A, Itoyama Y, Kato M, Asayama K, Awaya A, Hirano A, Ohama E (2004) Histological evidence of redox system breakdown caused by superoxide dismutase 1 (SOD1) aggregation is common to SOD1-mutated motor neurons in humans and animal models. Acta Neuropathol 107:149–158
Klivenyi P, Ferrante RJ, Mathews RT, Bogdanov MB, Klein AM, Andreassen OA, Mueller G, Wermer M, Kaddurah-Daouk R, Flint Beal M (1999) Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med 5:347–351
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD (1997) The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275:1132–1136
Kong J, Xu Z (1998) Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci 18(9):3241–3250
Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86:147–157
McHanwell S, Biscoe TJ (1981) The sizes of motoneurons supplying hindlimb muscles in the mouse. Proc R Soc Lond 213:201–216
Mohajeri MH, Figlewicz DA, Bohn MC (1998) Selective loss of α motoneurons innervating the medial gastrocnemius muscle in a mouse model of amyotrophic lateral sclerosis. Exp Neurol 150:329–336
Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, Brown RH Jr, Itoyama Y (2001) Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci 21(23):9246–9254
Nagano S, Ogawa Y, Yanagihara T, Sakoda S (1999) Benefit of a combined treatment with trientine and ascorbate in familial amyotrophic lateral scledosis model mice. Neurosci lett 265:159–162
Nagano S, Satoh M, Sumi H, Fujimura H, Tohyama C, Yanagihara T, Sakoda S (2001) Reduction of metallothioneins promotes the disease expression of familial amyotrophic lateral sclerosis mice in a dose-dependent manner. Eur J Neurosci 13:1363–1370
Nimchinsky EA, Young WG, Yeung G, Shah RA, Gordon JW, Bloom FE, Morrison JH, Hof PR (2000) Differential vulnerability of oculomotor, facial, and hypoglossal nuclei in G86R superoxide dismutase transgenic mice. J Comp Neurol 416:112–125
Olanow CW, Perl DP, DeMartino GN, McNaught KSP (2004) Lewy-body formation is an aggresome-related process: a hypothesis. Lancet Neurol 3:496–503
Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates second edition. Academic Press, Figs 79–85
Reed JC (1997) Cytochrome c: can’t live with it-can’t live without it. Cell 91:559–562
Ripps ME, Huntley GW, Hoff PR, Morrison JH, Gordon JW (1995) Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 92:689–693
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, Mckenna-Yasck D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pricak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horritz HR, Brown RH (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Sasaki S, Ohsawa Y, Yamane K, Sakuma H, Shibata H, Nakano R, Kikugawa K, Mizutani T, Tsuji S, Iwata M (1998) Familial amyotrophic lateral sclerosis with widespread vacuolation and hyaline inclusions. Neurology 51:871–873
Sasaki S, Warita H, Abe K, Iwata M (2004) Slow component of axonal transport is impaired in the proximal axon of transgenic mice with G93A mutant SOD1 gene. Acta Neuropathol 107:452–460
Sasaki S, Warita H, Murakami T, Abe K, Iwata M (2004) Ultrastructural study of mitochondria in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol 107:461–474
Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, Hung WY, Kato T, Asayama K (1996) Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol 55:481–490
Shimizu S, Narita M, Tsujimoto Y (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399:486–487
Stathopulos PB, Rumfeldt JAO, Scholzs GA, Irani RA, Frey HE, Hallewell RA, Lepock JR, Meiering EM (2003) Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc Natl Acad Sci USA 100(12):7021–2026
Steiber A, Gonatas JO, Gonatas NK (2000) Aggregation of ubiquitin and a mutant ALS-linked SOD1 protein correlate with disease progression and fragmentation of the Golgi apparatus. J Neurol Sci 173:53–62
Stephens B, Navarrete R, Guiloff RJ (2001) Ubiquitin immunoreactivity in presumed spinal interneurons in motor neurone disease. Neuropathol Appl Neurobiol 27:352–361
Sugai F, Yamamoto Y, Miyaguchi K, Zhou Z, Sumi H, Hamasaki T, Goto M, Sakoda S (2004) Benefit of valproic in suppressing disease progression of ALS model mice. Eur J Neurosci 20:3179–3183
Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S (1996) The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8A. Science 272:1136–1144
Turner BJ, Lopes EC, Cheema SS (2003) Neuromuscular accumulation of mutant superoxide dismutase 1 aggregates in a transgenic mouse of familial myotrophic lateral sclerosis. Neurosci Lett 350:132–136
Turner BJ, Atkin JD, Farg MA, Zang DW, Rembach A, Lopes EC, Patch JD, Hill AF, Cheema SS (2005) Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J Neurosci 25:108–117
Urushitani M, Kurisu J, Takahashi R (2002) Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem 83:1030–1042
Wang J, Xu G, Borchelt DR (2002) High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue specific accumulation. Neurobiol Dis 9:139–148
Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD (2001) Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis 8:933–941
Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14:1105–1116
Yamashita S, Mita S, Kato S, Okado H, Ohama E, Uchino M (2003) Bcl-2 expression using retrograde transporter of adenoviral vectors inhibits cytochrome c-release and caspase-1 activation in motor neurons of mutant superoxide dismutase 1 (G93A) transgenic mice. Neurosci Lett 350:17–20
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275:1129–1132
Acknowledgment
This study was supported in part by a Health and Labour Sciences Research Grant, Research on Measures for Incurable Disease, Ministry on Health, Labour and Welfare of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sumi, H., Nagano, S., Fujimura, H. et al. Inverse correlation between the formation of mitochondria-derived vacuoles and Lewy-body-like hyaline inclusions in G93A superoxide-dismutase-transgenic mice. Acta Neuropathol 112, 52–63 (2006). https://doi.org/10.1007/s00401-006-0056-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-006-0056-x