
Abstract
Living cells produce reactive oxygen species (ROSs). To protect themselves from these ROSs, the cells have developed both an antioxidant system containing superoxide dismutase 1 (SOD1) and a redox system including peroxiredoxin2 (Prx2, thioredoxin peroxidase) and glutathione peroxidase1 (GPx1): SOD1 converts superoxide radicals into hydrogen peroxide (H2O2), and H2O2 is then converted into harmless water (H2O) and oxygen (O2) by Prx2 and GPx1 that directly regulate the redox system. To clarify the biological significance of the interaction of the redox system (Prx2/GPx1) with SOD1 in SOD1-mutated motor neurons in vivo, we produced an affinity-purified rabbit antibody against Prx2 and investigated the immunohistochemical localization of Prx2 and GPx1 in neuronal Lewy body-like hyaline inclusions (LBHIs) in the spinal cords of familial amyotrophic lateral sclerosis (FALS) patients with a two-base pair deletion at codon 126 and an Ala→Val substitution at codon 4 in the SOD1 gene, as well as in transgenic rats expressing human SOD1 with H46R and G93A mutations. The LBHIs in motor neurons from the SOD1-mutated FALS patients and transgenic rats showed identical immunoreactivities for Prx2 and GPx1: the reaction product deposits with the antibodies against Prx2 and GPx1 were localized in the LBHIs. In addition, the localizations of the immunoreactivities for SOD1 and Prx2/GPx1 were similar in the inclusions: the co-aggregation of Prx2/GPx1 with SOD1 in neuronal LBHIs in mutant SOD1-related FALS patients and transgenic rats was evident. Based on the fact that Prx2/GPx1 directly regulates the redox system, such co-aggregation of Prx2/GPx1 with SOD1 in neuronal LBHIs may lead to the breakdown of the redox system itself, thereby amplifying the mutant SOD1-mediated toxicity in mutant SOD1-linked FALS patients and transgenic rats expressing human mutant SOD1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Living cells produce reactive oxygen species (ROSs) during physiological processes and in response to external stimuli such as ultraviolet radiation. To protect itself from potentially destructive ROSs, each cell of living organisms has developed a sophisticated antioxidant enzyme defense system. In this system, there are two groups of enzymes: the enzymes of the first group convert superoxide radicals into hydrogen peroxide (H2O2), and the enzymes of the second group convert H2O2 into harmless water (H2O) and oxygen (O2). For the first antioxidant enzyme group, three isoforms of superoxide dismutase (SOD) [EC 1.15.1.1] have been identified: SOD1, SOD2, and SOD3 [9]. In the second enzyme group, the peroxiredoxin (Prx) and glutathione peroxidase (GPx) families, as well as catalase localized within peroxisomes have been identified. Unlike SOD and catalase, enzymes of the Prx and GPx families require secondary enzymes and cofactors to function at high efficiency. In particular, the enzymes of the Prx- and GPx-families are considered to play a role in directly controlling the redox system. In general, the redox system regulates versatile control mechanisms in signal transduction and gene expression [29]. In mammalian cells, this redox signal transduction is linked to systems such as cellular differentiation, immune response, growth control, and apoptosis [10].
Peroxiredoxin2 (Prx2) is a novel organ-specific antioxidative enzyme that is mainly expressed in mammalian brain [23]. This protein is a member of Prx family, whose members possess reactive cysteine residues [23]. Prx2 requires thioredoxin reductase (TR) as a secondary enzyme as well as thioredoxin and NADPH as cofactors to function at high efficiency; the activity of Prx2 in the thioredoxin/TR/NADPH system is over five times higher than that of Prx2 by itself [5]. In this milieu, Prx2 is also called thioredoxin peroxidase 1 (thioredoxin-dependent peroxide reductase 1) or thiol-specific antioxidant [4, 5, 6]. In addition to controlling the intracellular content of H2O2, Prx2 directly regulates the redox signals of the thioredoxin/TR/NADPH system, because alone the secondary enzyme and cofactors (i.e., thioredoxin/TR/NADPH) can not directly regulate the redox system and can not act on H2O2. Cytosolic GPx [EC 1.11.1.9], a classical selenium-dependent isoform (also assigned as GPx1), was first described as an enzyme that protects hemoglobin from oxidative degradation in red blood cells [25]. The GPx family is composed of at least four GPx isoforms in mammals [7]. Among them, GPx1 is considered as the major enzyme responsible for removing intracytoplasmic H2O2. Like Prx2, GPx1 needs glutathione reductase (GR) as a secondary enzyme as well as glutathione and NADPH as cofactors to work at high efficiency, and this process is also one of the redox signals in living cells [21, 24]. Therefore, Prx2 and GPx1 directly control the redox system.
Approximately 20% of the cases of familial amyotrophic lateral sclerosis (FALS) are caused by a mutant SOD1 [15, 17, 18]. SOD1 is thought to be an essential component of neuronal Lewy body-like hyaline inclusions (LBHIs): neuronal LBHIs in affected anterior horn cells are morphological hallmarks of SOD1-mutated motor neurons of FALS patients [3, 11, 12, 13, 14, 15, 16, 17, 18, 30]. To cope with destructive ROSs, even SOD1-mutated motor neurons induce mutant and wild-type SOD1 as well as Prx2 and GPx1. Considering that Prx2 and GPx1 interact not only with wild-type SOD1 but also with mutant SOD1, the interaction of Prx2/GPx1 with SOD1 has been suggested to contribute to mutant SOD1 aggregation toxicity: Prx2/GPx1 possibly aggregate as LBHIs in SOD1-mutated motor neurons. Furthermore, the aggregation of Prx2/GPx1 might affect the intracytoplasmic redox regulation and amplify mutant SOD1-mediated toxicity. To clarify the biological significance of the interaction of Prx2/GPx1 (redox system) with SOD1 in SOD1-mutated motor neurons in vivo, we first produced an antibody against Prx2, and analyzed the characteristic expressions of both Prx2 and GPx1 in neuronal LBHIs in SOD1-mutated motor neurons of humans and animal models.
Materials and methods
Preparation of polyclonal antibody against Prx2
A synthetic peptide corresponding to the C-terminal region of Prx2 (amino acids 184–198: NH2-KPNVDDSKEYFSKHN-COOH) with or without conjugation to human serum albumin (HSA) at the carboxyl end was supplied by Peptide Institute (Osaka, Japan). This amino acid sequence is homologous with those of the C-terminal region of the human, rat or mouse Prx2. The polyclonal antibody preparation was carried out according to the method of Kato et al. [16]. To prepare immunogen, 6 mg synthesized Prx2 peptide was conjugated with 6 mg keyhole limpet hemocyanin (KLH) in the presence of 50 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide-HCl (Pierce Chemical Co., Rockford, IL) and 2.5 mM N-hydroxysulfosuccinimide (Pierce) in 3 ml phosphate-buffered saline (PBS) pH 7.4 for 1 h at room temperature. The reaction was terminated by adding 2-mercaptoethanol to the concentration of 20 mM and dialyzed against PBS for 24 h. To raise polyclonal antibodies, 500 µg of the immunogen in 50% Freund’s complete adjuvant was inoculated intradermally into a rabbit at 20 skin sites; four booster inoculations of 500 µg immunogen in 50% Freund’s incomplete adjuvant were given at 10, 17, 24 and 31 days after the first inoculation. The serum was taken 10 days after the final immunization. The IgG fraction in the antiserum against the immunogen, the hapten-conjugated KLH, was purified by absorption on a protein G-Sepharose gel column (Pharmacia Biotech, Uppsala, Sweden). Subsequently, the antibodies were further purified on an affinity column of immobilized KLH conjugated with the synthetic Prx2 peptide, as described previously [16].
Enzyme-linked immunosorbent assay
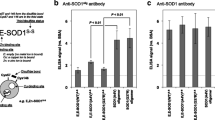
Noncompetitive ELISA was carried out according to the method described by Kato et al. [16]. Each well of a 96-well microtiter plate was coated with 100 µl of 5 µg/ml immunogen in 5 mM sodium carbonate buffer (pH 9.6) and incubated for 60 min. This was followed by triplicate washing with PBS containing 0.05% Tween 20 (buffer A). Each well was blocked with 0.5% gelatin for 60 min and then washed three times with buffer A. Antibody solutions (100 µl) at the concentrations indicated in Fig. 1 (horizontal line) were added to each well and incubated for 60 min. The wells were then washed three times with buffer A. The binding of the horseradish peroxidase-conjugated secondary antibody (Wako Pure Chemical Industries, Osaka, Japan) to the primary antibody was visualized with 2, 2’-azino-bis-(3-ethylbenzothiazoline-6-sulfonate)-(NH3)2. The reaction was terminated with 1 M sulfuric acid, and the absorbance at 415 nm was read on a micro-ELISA plate reader (Tecan, Hombrechtikon, Switzerland).

Specificity of antibody against Prx2. The immunoreactivity of the antibody to HSA-conjugated Prx2 peptide (solid circles) and native HSA (open circles) was determined by noncompetitive ELISA. The anti-Prx2 antibody recognizes the HSA-conjugated Prx2 peptide, but does not react with HAS (Prx2 peroxiredoxin2, ELISA enzyme-linked immunosorbent assay, HAS human serum albumin)
Tissue collection
Histochemical and immunohistochemical studies were performed on archival, buffered 10% formalin-fixed, paraffin-embedded tissues obtained at autopsy from five FALS patients who were members of two different families. The main clinicopathological characteristics of the FALS patients are summarized in Table 1, and have been reported previously [12, 13, 20, 22, 28, 30, 31]. SOD1 analysis revealed that the members of the Japanese Oki family had a two-base pair deletion at codon 126 (frame-shift 126 mutation) [12] and the American C family members had an Ala→Val substitution at codon 4 (A4V) [30]. As human controls, we examined autopsy specimens of the spinal cord from 20 neurologically and neuropathologically normal individuals (11 male, 9 females; aged 37–75 years).
Histochemical and immunohistochemical studies were also carried out on specimens from transgenic rats with the H46R and G93A types of mutations (three rats of each type). The H46R rats used in this study were a transgenic line (H46R-4) in which the level of human SOD1 with the H46R mutation was 6 times the level of that of endogenous rat SOD1 [27]. The G93A rats were a transgenic line (G93A-39) in which the level of human SOD1 with the G93A mutation was 2.5 times the level of endogenous rat SOD1 [27]. These rats were killed at an age of over 180 days; an age corresponding to an advanced stage of disease in these strains. The detailed clinical signs and pathological characteristics of the neuronal LBHIs of the H46R and G93A rats have been demonstrated previously [27]. As rat controls, we investigated the spinal cord specimens of three age-matched littermates of H46R and G93A rats and five age-matched normal Sprague-Dawley rats. Rats were anesthetized with sodium pentobarbital (0.1 ml/100 g body weight). After perfusion of the rats via the aorta with physiological saline at 37°C, they were fixed by perfusion with 4% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.3). The spinal cords were removed and then postfixed in the same solution.
Histochemistry and immunohistochemistry
After fixation, the specimens were embedded in paraffin, cut into 6-µm-thick sections, and examined by light microscopy. Spinal cord sections were stained by the following histochemical methods: hematoxylin and eosin (HE), Klüver-Barrera, Holzer, phosphotungstic acid-hematoxylin, periodic acid-Schiff, alcian blue, Masson’s trichrome, Mallory azan and Gallyas-Braak stains. Representative paraffin sections were used for immunohistochemical assays. The following primary antibodies were utilized: an affinity-purified rabbit antibody against Prx2 (concentration: 1 µg/ml), a polyclonal antibody to GPx1 [diluted 1:2,000 in 1% bovine serum albumin-containing phosphate-buffered saline (BSA-PBS), pH 7.4] [2], and a polyclonal antibody to human SOD1 (diluted 1:10,000 in 1% BSA-PBS, pH 7.4) [1]. Sections were deparaffinized, and endogenous peroxidase activity was quenched by incubation for 30 min with 0.3% H2O2. The sections were then washed in PBS. Normal sera homologous with secondary antibody was used as a blocking reagent. Tissue sections were incubated with the primary antibodies for 18 h at 4°C. PBS-exposed sections served as controls. As a preabsorption test, some sections were incubated with the anti-Prx2 antibody that had been preabsorbed with an excess amount of the synthetic Prx2 peptide. Bound antibodies were visualized by the avidin-biotin-immunoperoxidase complex (ABC) method using the appropriate Vectastain ABC Kit (Vector Laboratories, Burlingame, CA) and 3,3’-diaminobenzidine tetrahydrochloride (DAB; Dako, Glostrup, Denmark) as chromogen.
Results
We successfully produced an affinity-purified rabbit antibody against Prx2 peptide (amino acids 184–198; although this amino acid sequence is homologous with that of each C-terminal region of the human, rat, mouse, Chinese hamster or Bos Taurus Prx2, this peptide does not share homology with other members of the Prx family or any other peptide sequence in GenomeNet), and applied it to stain of paraffin sections from both humans and rats. This anti-Prx2 antibody recognized the HSA-conjugated Prx2 peptide, but did not react with HSA (Fig. 1).
Analysis of the essential changes of five cases of FALS revealed a subtype of FALS with posterior column involvement (PCI). This subtype is characterized by the degeneration of the middle root zones of the posterior column, Clarke nuclei, and the posterior spinocerebellar tracts, in addition to spinal cord motor neuron lesions. A long-term surviving patient with a clinical course of 11 years (case 2 in Table 1) showed multisystem degeneration in addition to the features of FALS with PCI. Neuronal Lewy body-like hyaline inclusions (LBHIs) were present in all five FALS cases. As observed in HE preparations, the neuronal LBHIs in the FALS patients were essentially identical to those in the H46R and G93A transgenic rats; the inclusions were round eosinophilic or paler inclusions and often showed eosinophilic cores with pale peripheral halos. In mutant SOD1-linked FALS patients, the neuronal LBHIs were generally composed of eosinophilic cores with pale peripheral halos and sometimes showed ill-defined forms that consisted of obscure eosinophilic materials. In H46R and G93A transgenic rats, the intracytoplasmic LBHIs with cores and halos were less frequently observed and round or sausage-like LBHIs, which were though to be intradendritic LBHIs, were often seen in the neuropil, although these round or sausage-like LBHIs in the neuropil were not remarkable in the human FALS patients. Histochemically, most of the neuronal LBHIs in the H46R and G93A transgenic rats were argyrophilic in Gallyas-Braak stain, and they were generally blue to violet after Masson’s trichrome or Mallory azan staining, similar to the histochemical findings of the neuronal LBHIs of the human FALS patients. The spinal cords of normal individuals in both humans and rats did not exhibit any distinct histopathological alterations.
When control and representative paraffin sections were incubated with PBS alone (i.e., no primary antibody), no staining was detected. Prx2 immunoreactivity in normal spinal cords was identified in almost all neurons. In addition, Prx2-immunostaining was found throughout the neuropil with considerably lower intensity (Fig. 2A, C). With respect to the intracellular localization of Prx2, immunostaining of the neuronal cytoplasm and proximal dendrites was specifically observed (Fig. 3A, C). Additionally, the nuclei of some neurons were immunostained by the anti-Prx2 antibody, albeit the staining of positively stained nuclei varied (Fig. 3F). Incubation of sections with anti-Prx2 antibody that had been pretreated with an excess of the synthetic Prx2 produced no staining (Fig. 3D).

Serial transverse sections through the lumbar segments of the normal human spinal cords. A Light microscopic preparation stained with HE. B, C Immunostaining for GPx1 (B) and Prx2 (C). GPx1 and Prx2 immunoreactivities are found diffusely in the neuropil with considerably less intensity (arrowheads). No counterstaining (HE hematoxylin and eosin, GPx1 glutathione peroxidase1, Prx2 peroxiredoxin2). Bar A (also for B, C) 2 mm
Detection of Prx2 and GPx1 in the normal motor neurons of the human spinal cord. A–D Serial sections. A Staining with HE. B Immunostaining with the antibody against GPx1, showing GPx1-positive neurons. C Immunostaining with the antibody to Prx2. Immunoreactivity is identified in most of the neurons. Thus, most of the normal motor neurons in the spinal cord co-express both GPx1 (B) and Prx2 (C), although their staining intensities in neurons vary. D Immunostaining with anti-Prx2 antibody pretreated with an excess of the synthetic Prx2 peptide. No immunoreaction products are observed in the motor neurons and neuropil. E GPx1 immunostaining of the neuronal cytoplasm and proximal dendrites is observed, but no intranuclear localization is seen. F Prx2 immunostaining of the neuronal cytoplasm and proximal dendrites is observed, and a nucleus of the neuron is also immunostained by the anti-Prx2 antibody. B–F No counterstaining (HE hematoxylin and eosin, GPx1 glutathione peroxidase1, Prx2 peroxiredoxin2). Bars A (also for B–D) 100 µm; E, F 50 µm
A neuropil staining pattern similar to that for Prx2 was observed with GPx1; weak GPx1 immunoreactivity was diffusely seen in the neuropil in transverse sections of the spinal cords (Fig. 2A, B). GPx1 immunostaining was observed in the cytoplasm with cell bodies and proximal dendrites being essentially identified (Fig. 3A, B, E), but no intranuclear staining was observed (Fig. 3B, E). The stainability and intensity of Prx2 and GPx1 in the normal anterior horn cells of the spinal cords in humans were identical to those in rats. Therefore, almost all of the normal motor neurons in the spinal cords co-expressed both Prx2 and GPx1 (Fig. 3A–C), although the staining intensities of positively stained neurons varied.
Corroborating recent findings [12, 13, 16, 19, 27, 30], almost all of the neuronal LBHIs in both the FALS patients from two different families and races (Japanese Oki family and American C family) and the transgenic rats expressing two different human SOD1 mutations (H46R and G93A) were intensely immunostained by the antibody against human SOD1 (Figs. 4A, D; 5A, D; 6A; 7A). Most neuronal LBHIs were also immunoreactive for Prx2, although the intensity of Prx2 immunoreactivity in the LBHIs varied (Figs. 4C, F; 5C, F; 6B). The LBHIs in the neurons of the FALS patients and transgenic rats (H46R and G93A) showed a similar immunoreactivity for Prx2. The Prx2 immunolocalization in many intracytoplasmic and intraneuritic LBHIs was similar to that of SOD1 in both diseases. In core and halo-type LBHIs, the reaction product deposits of the antibody against Prx2 were generally restricted to the periphery (Fig. 4C, F), and were sometimes localized in the cores alone (Fig. 5C). In ill-defined LBHIs, Prx2 immunostaining was distributed throughout each inclusion. In some inclusions, however, expression of Prx2 was observed in only part of the inclusion (Fig. 5F). With respect to the GPx1 immunostaining in the neuronal LBHIs, similar stainability and immunolocalization to Prx2 were confirmed in the core and halo types as well as the ill-defined forms; most LBHIs in neurons were immunostained by the anti-GPx1 antibody with various intensities (Figs. 4B, E; 5B, E; 7B). The immunoreactivity for GPx1 in the FALS patients was similar to that in the transgenic rats (H46R and G93A). Like Prx2, the immunolocalization of GPx1 was similar to that of SOD1 in both diseases. GPx1-immunoreactive products in many core and halo-type inclusions were mainly localized in the periphery portions (Fig. 4B, E), but sometimes in the core portions alone (Fig. 5B). In some inclusions, the reaction products were confined to certain regions of each inclusion (Fig. 5E).

Serial sections of a typical LBHI with a core and halo in neurons from the spinal cord of an FALS patient with a two-base pair deletion in the SOD1 gene. A Immunostaining for SOD1: immunoreactivity is mostly restricted to the halo. B Immunostaining for GPx1: immunoreactivity is located in the SOD1-positive portion of the LBHI. C Immunoreactivity for Prx2. Co-localization of the three proteins SOD1, GPx1 and Prx2 in the LBHI is evident. D–F Serial sections of a core and halo-type LBHI in a transgenic rat expressing human SOD1 with an H46R mutation. Immunostaining for SOD1 (D), GPx1 (E) and Prx2 (F). Similar stainability and immunolocalization of SOD1, GPx1 and Prx2 in the LBHI are observed (LBHI Lewy body-like hyaline inclusion, FALS familial amyotrophic lateral sclerosis, SOD1 superoxide dismutase 1, GPx1 glutathione peroxidase1, Prx2 peroxiredoxin2). A–F No counterstaining. Bars A (also for B, C), D (also for E, F) 25 µm

Serial sections of an LBHI in an FALS patient with an A4V mutation in SOD1 gene. Immunostaining for SOD1 (A), GPx1 (B) and Prx2 (C). Co-localization of the three proteins in the LBHI is mainly observed in the core (A–C). D–F Serial sections of an LBHI in an FALS patient with a two-base pair deletion in the SOD1 gene. Immunostaining for SOD1 (D), GPx1 (E) and Prx2 (F). Immunostaining GPx1 (E) and Prx2 (F) are observed in only part of the SOD1-positive LBHI. The precise intra-inclusional immunolocalizations of these three proteins differ from each other in this LBHI (LBHI Lewy body-like hyaline inclusion, FALS familial amyotrophic lateral sclerosis, SOD1 superoxide dismutase 1, GPx1 glutathione peroxidase1, Prx2 peroxiredoxin2). Bars A (also for B, C), D (also for E, F) 25 µm

Serial sections of the spinal anterior horn in a transgenic rat expressing human SOD1 with an H46R mutation immunostained with antibodies against SOD1 (A) and Prx2 (B). Round and sausage-like LBHIs in the neuropil are positive for both SOD1 and Prx2 (arrows) (SOD1 superoxide dismutase1, LBHI Lewy body-like hyaline inclusion, Prx2 peroxiredoxin2). Bar A (also for B) 50 µm

Serial sections of the spinal anterior horn in a transgenic rat expressing human SOD1 with an H46R mutation immunostained with antibodies against SOD1 (A) and GPx1 (B). Round LBHIs in the neuropil are positive for both SOD1 and GPx1 (arrows) (SOD1 superoxide dismutase1, LBHI Lewy body-like hyaline inclusion, GPx1 glutathione peroxidase1). Bar A (also for B) 50 µm
Noticeably, the co-localization of the three proteins SOD1, Prx2 and GPx1 in neuronal LBHIs in SOD1-mutated FALS patients and transgenic rats (H46R and G93A) was evident (Figs. 4, 5, 6, 7), although all three immunoreactive intensities varied. With respect to the intra-inclusional localization, many inclusions showed similar co-localizations of these three proteins (Figs. 4, 5A–C). In some LBHIs, the precise intra-inclusional immunolocalizations of the three proteins differed: Prx2 (Fig. 5D, F) and GPx1 (Fig. 5D, E) immunostaining was observed in only some areas of the SOD1-positive LBHIs.
Discussion
Under normal physiological conditions, Prx2 and GPx1 immunoreactivities in the spinal cord anterior horns in humans and rats are primarily identified in the neurons: cytoplasmic staining with both antibodies is observed in almost all of the anterior horn cells. Like Prx1 [26, 33], intranuclear localization in some neurons is also observed in Prx2 immunostaining. Considering that endogenous Prx2 and GPx1 within the neuronal cytoplasm are extremely effective regulators of the redox system, our immunohistochemical finding that almost all of the normal spinal motor neurons co-expressed both Prx2 and GPx1 confirms that these motor neurons maintain themselves using the intracellular Prx2/GPx1 system, that is, the redox system.
As expected [12, 13, 16, 27, 30], SOD1 protein (probably the mutant form) was found to aggregate in the anterior horn cells as neuronal LBHIs in FALS patients with SOD1 gene mutations and transgenic rats expressing human SOD1 with H46R and G93A mutations. Intense co-expression of SOD1, Prx2, and GPx1 in neuronal LBHIs in both diseases was evident. To eliminate ROSs, SOD1-mutated motor neurons in mutant SOD1-linked FALS and transgenic rats (G46R and G93A) induce mutant/wild-type SOD1 as an antioxidant system and Prx2/GPx1 as a redox system. In this in vivo milieu where mutant SOD1 exists, Prx2 and GPx1 would aberrantly interact with the mutant SOD1, which is assumed to aggregate easily by itself [8]. Among the multiple theories of how mutant SOD1 contributes to motor neuron death in mutant SOD1-related FALS and transgenic animal models expressing human mutant SOD1, the aggregation of mutant SOD1 in neurons leads to part of the mutant SOD1-mediated toxicity through the formation of advanced glycation endproduct-modified SOD1 that is insoluble and cytotoxic [16]. Our recent study of FALS patients with a two-base pair deletion at codon 126 of the SOD1 gene (Oki family) and G85R transgenic mice has revealed that not only does mutant SOD1 provoke inclusion formation, but that normal SOD1 also co-aggregates in these inclusions [3]. Together with the facts that there are neuronal LBHIs positive for SOD1, Prx2, and GPx1 in the milieu where mutant SOD1 exists but no LBHIs (no aggregations) exist under physiological conditions, our study demonstrates an aberrant interaction of Prx2/GPx1 with mutant SOD1, the aggregation of which results in neuronal LBHIs. In addition, intra-inclusional co-aggregation of Prx2/GPx1 with mutant SOD1 causes the intracytoplasmic reduction of Prx2/GPx1, thereby reducing the availability of the redox system. A similar aberrant interaction of the copper chaperone for SOD (CCS) and SOD1 (probably CCS-mutant SOD1) also occurs in the formation of the neuronal LBHIs in mutant SOD1-linked FALS [19] and the mutant SOD1 transgenic mouse model [32]. Such sequestration into LBHIs has also been observed for normal cytosolic constitutive proteins including tubulin and tau protein, as well as neuron-specific constitutive proteins containing phosphorylated neurofilament proteins (NFP), nonphosphorylated NFP, synaptophysin, and neuron-specific enolase [13, 17, 18]; this results in partial impairment of the maintenance of cell metabolism [13, 17, 18]. Although we cannot readily compare the sequestration of normal constitutive proteins with the aberrant interaction of cytotoxic mutant SOD1 with Prx2/GPx1 directly regulating a redox system, our finding leads us to speculate that not only co-aggregation of Prx2/GPx1 and SOD1 into LBHIs, but also intracytoplasmic reduction of Prx2/GPx1 in both diseases may partly contribute to the breakdown of the redox system itself in these SOD1-mutated neurons, and this may be one of the endogenous mechanisms that accelerate neuronal death. This hypothesis would appear to be compatible with the aggregation toxicity theory. It remains to be determined whether this aberrant interaction of Prx2/GPx1 with mutant SOD1 is a direct or an indirect effect based on the pathogenesis of SOD1-mutated FALS disease itself or whether Prx2 and GPx1 play a primary or a secondary role to mutant SOD1. Consequently, we would like to emphasize that the aberrant interaction and co-aggregation of Prx2/GPx1 and SOD1 (probably Prx2/GPx1 and mutant SOD1) in FALS patients with SOD1 gene mutations and transgenic rats expressing human SOD1 mutations may amplify a more marked mutant SOD1-mediated toxicity.
References
Asayama K, Burr IM (1984) Joint purification of mangano and cuprozinc superoxide dismutases from a single source: a simplified method. Anal Biochem 136:336–339
Asayama K, Yokota S, Dobashi K, Hayashibe H, Kawaoi A, Nakazawa S (1994) Purification and immunoelectron microscopic localization of cellular glutathion peroxidase in rat hepatocytes: quantitative analysis by postembedding method. Histochemistry 102:213–219
Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281:1851–1854
Chae HZ, Kim IH, Kim K, Rhee SG (1993) Cloning, sequencing, and mutation of thiol-specific antioxidant gene of Saccharomyces cerevisiae. J Biol Chem 268:16815–16821
Chae HZ, Chung SJ, Rhee SG (1994) Thioredoxin-dependent peroxide reductase from yeast. J Biol Chem 269:27670–27678
Chae HZ, Robison K, Poole LB, Church G, Storz G, Rhee SG (1994) Cloning and sequencing of thiol-specific antioxidant from mammalian brain: alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci USA 91:7017–7021
De Haan JB, Bladier C, Griffiths P, Kelner M, O’Shea RD, Cheung NS, Bronson RT, Silvestro MJ, Wild S, Zheng SS, Beart PM, Hertzog PJ, Kola I (1998) Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J Biol Chem 273:22528–22536
Durham HD, Roy J, Dong L, Figlewicz DA (1997) Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol 56:523–530
Fridovich I (1986) Superoxide dismutases. Adv Enzymol Relat Area Mol Biol 58:61–97
Jin D-Y, Chae HZ, Rhee SG, Jeang K-T (1997) Regulatory role for a novel human thioredoxin peroxidase in NF-kappaB activation. J Biol Chem 272:30952–30961
Kato S, Shimoda M, Morita T, Watanabe Y, Nakashima K, Takahashi K, Ohama E (1996) Neuropathology of familial ALS with a mutation of the superoxide dismutase 1 gene. In: Nakano I, Hirano A (eds) Amyotrophic lateral sclerosis: progress and perspectives in basic research and clinical application. Elsevier Science, Amsterdam, pp 117–122
Kato S, Shimoda M, Watanabe Y, Nakashima K, Takahashi K, Ohama E (1996) Familial amyotrophic lateral sclerosis with a two base pair deletion in superoxide dismutase 1 gene: multisystem degeneration with intracytoplasmic hyaline inclusions in astrocytes. J Neuropathol Exp Neurol 55:1089–1101
Kato S, Hayashi H, Nakashima K, Nanba E, Kato M, Hirano A, Nakano I, Asayama K, Ohama E (1997) Pathological characterization of astrocytic hyaline inclusions in familial amyotrophic lateral sclerosis. Am J Pathol 151:611–620
Kato S, Horiuchi S, Nakashima K, Hirano A, Shibata N, Nakano I, Saito M, Kato M, Asayama K, Ohama E (1999) Astrocytic hyaline inclusions contain advanced glycation endproducts in familial amyotrophic lateral sclerosis with superoxide dismutase 1 gene mutation: immunohistochemical and immunoelectron microscopical analyses. Acta Neuropathol 97:260–266
Kato S, Saito M, Hirano A, Ohama E (1999) Recent advances in research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 gene mutations: neuronal Lewy body-like hyaline inclusions and astrocytic hyaline inclusions. Histol Histopathol 14:973–989
Kato S, Horiuchi S, Liu J, Cleveland DW, Shibata N, Nakashima K, Nagai R, Hirano A, Takikawa M, Kato M, Nakano I, Ohama E (2000) Advanced glycation endproduct-modified superoxide dismutase-1 (SOD1)-positive inclusions are common to familial amyotrophic lateral sclerosis patients with SOD1 gene mutations and transgenic mice expressing human SOD1 with G85R mutation. Acta Neuropathol 100:490–505
Kato S, Takikawa M, Nakashima K, Hirano A, Cleveland DW, Kusaka H, Shibata N, Kato M, Nakano I, Ohama E (2000) New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: inclusions containing SOD1 in neurons and astrocytes. Amyotroph Lateral Scler Other Motor Neuron Disord 1:163–184
Kato S, Nakashima K, Horiuchi S, Nagai R, Cleveland DW, Liu J, Hirano A, Takikawa M, Kato M, Nakano I, Sakoda S, Asayama K, Ohama E (2001) Formation of advanced glycation end-product-modified superoxide dismutase-1 (SOD1) is one of the mechanisms responsible for inclusions common to familial amyotrophic lateral sclerosis patients with SOD1 gene mutation, and transgenic mice expressing human SOD1 gene mutation. Neuropathology 21:67–81
Kato S, Sumi-Akamaru H, Fujimura H, Sakoda S, Kato M, Hirano A, Takikawa M, Ohama E (2001) Copper chaperone for superoxide dismutase co-aggregates with superoxide dismutase 1 (SOD1) in neuronal Lewy body-like hyaline inclusions: an immunohistochemical study on familial amyotrophic lateral sclerosis with SOD1 gene mutation. Acta Neuropathol 102:233–238
Kato T, Hirano A, Kurland LT (1987) Asymmetric involvement of the spinal cord involving both large and small anterior horn cells in a case of familial amyotrophic lateral sclerosis. Clin Neuropathol 6:67–70
Kosower NS, Kosower EM (1978) The glutathione status of cells. Int Rev Cytol 54:109–160
Kurland LT, Mulder DW (1955) Epidemiologic investigations of amyotrophic lateral sclerosis. II. Familial aggregations indicative of dominant inheritance. Neurology 5:249–268
Matsumoto A, Okado A, Fujii T, Fujii J, Egashira M, Niikawa N, Taniguchi N (1999) Cloning of the peroxiredoxin gene family in rats and characterization of the fourth member. FEBS Lett 443:246–250
Meister A, Anderson ME (1983) Glutathione. Annu Rev Biochem 52:711–760
Mills GC (1957) Hemoglobin catabolism. 1. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J Biol Chem 229:189–197
Mu ZM, Yin XY, Prochownik EV (2002) Pag, a putative tumor suppressor, interacts with the Myc Box II domain of c-Myc and selectively alters its biological function and target gene expression. J Biol Chem 277:43175–43184
Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, Brown RH Jr, Itoyama Y (2001) Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci 21:9246–9254
Nakano I, Hirano A, Kurland LT, Mulder DW, Holley PW, Saccomanno G (1984) Familial amyotrophic lateral sclerosis. Neuropathology of two brothers in American “C” family. Neurol Med (Tokyo) 20:458–471
Sen CK, Packer L (1996) Antioxidant and redox regulation of gene transcription. FASEB J 10:709–720
Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, Hung WY, Kato T, Asayama K (1996) Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol 55:481–490
Takahashi K, Nakamura H, Okada E (1972) Hereditary amyotrophic lateral sclerosis. Histochemical and electron microscopic study of hyaline inclusions in motor neurons. Arch Neurol 27:292–299
Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD (2001) Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis 8:933–941
Wen ST, Van Etten RA (1997) The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev 11:2456–2467
Acknowledgements
This study was supported in part by a Grant-in-Aid for Scientific Research (c) (2) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (S.K.: 13680821) and by a Grant from the Ministry of Health, Labour and Welfare of Japan (S.K. and Y.I.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kato, S., Saeki, Y., Aoki, M. et al. Histological evidence of redox system breakdown caused by superoxide dismutase 1 (SOD1) aggregation is common to SOD1-mutated motor neurons in humans and animal models. Acta Neuropathol 107, 149–158 (2004). https://doi.org/10.1007/s00401-003-0791-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-003-0791-1