
Abstract
We report a familial disorder occurring in three patients that presented as frontotemporal dementia (FTD). A neuropathological study was performed in a 58-year-old patient, who developed FTD 2 years prior to the onset of motor neuron disease (MND), and died at age 62. Lesions indicative of associated MND were observed: neuronal loss in the anterior horns of the spinal cord, Bunina bodies, axonal spheroids, degeneration of the pyramidal tracts, and of FTD: decreased neuronal density and laminar microvacuolation of layers II and III in the frontal and temporal cortex. Ubiquitin-only-immunoreactive changes were found in the spinal cord and medulla, but were absent from the temporal and frontal cortex. There were also widespread deposits of various neuronal and glial inclusions containing abnormally phosphorylated tau protein, the Western blotting pattern of which was characterized by two major bands of 64 and 69 kDa. There were no abnormalities of the entire coding sequences of microtubule-associated protein tau (MAPT) and copper-zinc superoxide dismutase (SOD 1 ) genes. Our results suggest that FTD associated with MND can be caused by a larger spectrum of neuropathological lesions than commonly accepted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Association of frontotemporal dementia (FTD) and motor neuron disease (MND) is not an exceptional occurrence, and may be observed in 5% of patients with MND. The neuropathological examination reveals frontotemporal lobar degeneration (FTLD) associated with the lesions of MND, including ubiquitin-positive, tau-negative and silver-negative neuronal inclusions (ubiquitin-only-immunoreactive inclusion bodies) in spinal cord and medulla motor neurons, and in the frontal and temporal cortices. The histopathological diagnosis is FTLD with MND (FTLD-MND). Other cases of FTD have been characterized by the presence of similar inclusions in the absence of clinical and pathological evidence of MND. These cases have been classified as FTLD with ubiquitin-only-immunoreactive changes (FTLD-U) [18]. In FTLD-MND and FTLD-U, no abnormal deposits of tau protein are observed in the central nervous system. In contrast, in other cases of FTD without MND and lacking ubiquitin-only-immunoreactive inclusion bodies, abnormal deposits of tau protein have been found in the brain, and these cases are classified among tauopathies. In the majority of cases, they are linked to microtubule-associated protein tau (MAPT) gene mutations, and are classified together into the group of FTD and parkinsonism linked to chromosome 17 (FTDP-17). We report a familial disorder that presented as FTD. In one case clinically associated with MND, the patient died at age 62. The neuropathological examination showed lesions indicative of associated MND and FTDP. Ubiquitin-only-immunoreactive changes were observed in the spinal cord and medulla, but were absent from the temporal and frontal cortex. Immunohistochemistry also revealed widespread deposits of abnormally phosphorylated tau protein, where the Western blotting pattern was characterized by two major bands of 64 and 68 kDa. There were no abnormalities of the entire coding sequences of MAPT and copper-zinc superoxide dismutase (SOD 1 ) genes.
Material and methods
Case history
The patient, a right-handed male electrician from a French family, was in good health until the age of 58, when he developed apathy and irritability. He lost interest in daily household affairs and gradually became antisocial. Dysarthria appeared 2 years later and the initial neurological examination at 60 years of age showed dysphagia and fasciculations of the tongue. The patient appeared calm and lacked spontaneity. Neurophysiological studies revealed neurogenic alterations in the muscles of the face, the upper and lower limbs, with normal motor nerve conduction velocities and sensory action potentials, which confirmed the diagnosis of definite amyotrophic lateral sclerosis (ALS) [2]. Neuropsychological assessment was performed at age 62 and was limited by comprehension deficit and language reduction. Clinical presentation and neuropsychological characteristics were consistent with the diagnosis of severe FTD [24]. Magnetic resonance imaging (MRI) showed nonspecific high signal intensities in the white matter on T2-weighted images (Fig. 1a). Discrete symmetrical high signal intensities in the corticospinal tracts were also observed (Fig. 1b), as described in ALS [1, 32]. Single photon emission tomography using 99mTc-ECD revealed severe reduction of the uptake of tracer in the right frontotemporal cortex (Fig. 1c). One month later, the patient presented with an aggravation of difficulties in swallowing, and was mute. Physical examination revealed bulbar palsy and pyramidal signs of the four limbs with amyotrophy particularly severe in the left upper limb. There were no signs of parkinsonism, postural instability or ocular impairment. The patient died from aspiration pneumonia 4 years after the onset of disease. The final diagnosis was ALS with FTD.

T2-weighted magnetic resonance imaging showing nonspecific high signal intensity in the white matter. b High signal intensity of the corticospinal tracts. c Brain SPECT study showing severe reduced uptake of tracer in the right frontotemporal cortex
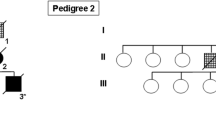
Additional familial history is illustrated in Fig. 2. The patient’s father had been frequently hospitalized in a psychiatric department for behavioral disorders and multiple suicide attempts from the age of 45 years. His daughter reported wandering, change of personality with irritability and violence, lack of personal hygiene and language reduction at the end of his life. He died at age of 53 years. The patient’s sister, previously a farmer, had been in good health until the age of 44 when she presented increasing behavioral disturbances, with a loss of interest and major inertia. At age 50, she became irritable and lost interest in household affairs and personal hygiene. She became rapidly unable to travel alone and to manage her financial affairs. Neuropsychological assessment demonstrated cognitive deficits, predominantly in executive functions. MRI showed a diffuse cerebral cortical atrophy. Single photon emission tomography using 99mTc-ECD revealed a severe bilateral reduced uptake of tracer in the frontotemporal cortex, particularly in the right side of the brain. She was clinically diagnosed as having FTD.

Reduced pedigree
Neuropathological study
Autopsy procedure and postmortem studies
An autopsy restricted to the brain and the spinal cord was performed on the proband (patient III-4). Coronal slices from the left hemisphere and three sections from the dorsal spinal cord were frozen and stored at −70°C until use. The right hemisphere, the brainstem, the cerebellum and the remaining spinal cord were fixed in a 10% formaldehyde buffer solution.
Paraffin-embedded samples were obtained from the middle frontal gyrus, superior temporal gyrus, temporal pole, inferior parietal gyrus, anterior cingulate gyrus, insular and motor cortex, occipital lobe adjacent to the calcarin fissure, hippocampus, nucleus basalis of Meynert, basal ganglia, mesencephalon, pons, medulla oblongata, cerebellum (vermis, right hemisphere, dentate nucleus), and the cervical, thoracic and lumbar spinal cord.
Sections, 7 μm thick, were cut from paraffin-embedded blocks and stained with hematoxylin-eosin, and modified Bielschowsky method. Immunohistochemical evaluation was carried out on all paraffin-embedded blocks using antibodies directed against β-amyloid protein (diluted 1:100), ubiquitin (1:200), glial fibrillary acidic protein (GFAP, 1:300), phosphorylated neurofilaments (1:200) (Dakopatts, Trappes, France), and α-synuclein (1:200), (Zymed Laboratories, Le Perray en Yvelines, France). Immunohistochemistry of PHF tau was performed using monoclonal antibodies AT8 (1:20) and AT100 (1:40) (Innogenetics, Gent, Belgium).
Biochemical analyses
Pathological tau proteins were studied by Western blotting on total brain homogenates as previously described [4].
Molecular analyses
Genomic DNA was extracted from peripheral leukocytes, after informed consent was obtained from the patient’s family. The SOD1 and MAPT genes were studied by sequence analysis, using previously described methods [7]. Genomic DNA sequencing spanned intron–exon boundaries for all MAPT exons, except for those not found in human adult brain (exons 4a, 6 and 8).
Results
Neuropathology
The unfixed brain weighed 1,430 g. Macroscopic examination revealed moderate gyral atrophy of the frontal and temporal lobes (Fig. 3a). Cerebral vessels were normal. On coronal sections, there was neither marked atrophy nor ventricular dilation (Fig. 3b). Transverse sections of the brainstem did not reveal any discoloration of substantia nigra or locus coeruleus. The cerebellum was macroscopically normal. The spinal cord appeared atrophic, mainly at the cervical level.

Macroscopic views of the brain. a Midline sagittal section showing mild atrophy of frontal and temporal cortex. b Absence of atrophy of the caudate nucleus and ventricular dilation on coronal section through the mamillary bodies. c Macroscopic view of the spinal cord showing severe loss of motor neurons, and atrophy and pallor of non-crossed pyramidal tracts. Luxol-phloxin staining
Histopathological examination showed major neuronal loss in the anterior horns of the cervical level of the spinal cord (Fig. 3c), in association with scarce Bunina bodies (Fig. 4a). Degeneration of the pyramidal tracts was evident at the same level, with the presence of axonal spheroids as revealed by neurofilament immunohistochemistry (data not shown). At the lumbar level, motor neurons were more numerous, but some were chromatolytic, without neuronophagia. In the medulla, neuronal loss was found in the nucleus nervi hypoglossi. No lesion was observed in the substantia nigra; in particular, no Lewy bodies were observed in the pigmented nuclei of the brainstem, and there was no noticeable lesion in the cerebellum. In the cerebral hemispheres, histological alterations consisted of decreased neuronal cortical density and of laminar microvacuolation of layers II and III (Fig. 5a) mainly observed in the frontal and temporal areas. Neither senile plaques nor ballooned cells were observed. Myelin pallor was never observed, whatever the structure evaluated.

Histopathology of the spinal cord. a Typical Bunina bodies. b Ubiquitin spherical intraneuronal inclusions in the anterior horn of the cervical spinal cord. c Skein-like inclusions in a motor neuron in the anterior horn of the dorsal spinal cord. d Ubiquitin-positive chromatolytic motoneurons in the anterior horn of the lumbar spinal cord. Bars 10 μm

Histopathology of the temporal cortex. a Decreased neuronal cortical density, associated with superficial microspongiosis. b Tau-positive oligodendroglial cells observed in the superficial white matter. Bar b 15 μm
Scarce ubiquitin-immunoreactive rounded or skein-like intraneuronal inclusions were found in the anterior horns of the cervical and thoracic spinal cord (Fig. 4b, c). Chromatolytic motor neurons also displayed ubiquitin immunoreactivity (Fig. 4d). In contrast, no ubiquitin-positive inclusions were seen in the brainstem and the supratentorial areas, particularly in the dentate gyrus. No intranuclear inclusions were observed in the basal ganglia. Conversely, tau-positive neuronal cytoplasm and adjacent processes, and various tau-positive intraneuronal inclusions, some of them of the globose type (Fig. 6d), others of the Lewy-body-like variety, and tufted astrocytes were abundant in many areas of the brain. They occurred mainly in the dentate gyrus (Fig. 6a, b), the pyramidal cell layer of Ammon’s horn and the entorhinal cortex; they were scarce in the insular, cingular, frontal and temporal isocortices, the nucleus basalis of Meynert (Fig. 6c), the internal part of the putamen, the amygdaloid complex and the thalamus. In the mesencephalon and pons, they were mainly observed in the substantia nigra and the locus coeruleus (Fig. 7a–d). In the other nuclei and cerebellum, inclusions were scanty or absent. In the medulla oblongata, they were observed in the nucleus dorsalis motorius nervi vagi and nucleus nervi hypoglossi. Some coiled bodies and threads were seen in the temporal white matter (Fig. 5b). Astrocytic plaques, thorn astrocytes and grains were not observed. GFAP immunochemistry revealed gliosis in the cortex, in the vicinity of microvacuolar changes. Mild astrocytosis was also present in the basal ganglia. β-Amyloid and α-synuclein immunolabeling were negative.

Histopathology of the dentate gyrus. a Ubiquitin-negative granular cell layer. b Tufted astrocytes (arrow), adjacent to tau positive intraneuronal inclusions (asterisk) in the granular cell layer. c Diffuse tau-immunoreactivity in the nucleus basalis of Meynert: presence of neuronal inclusions, threads, coiled bodies and tufted astrocytes. d Pick-like inclusion in the raphe nuclei

Immunohistochemistry of the pigmented nuclei of the brainstem. a Tau-positive neurons in the substantia nigra. b Tau-positive astrocytes in the vicinity of neurons. c Tau immunoreactivity in the locus coeruleus. d Ubiquitin negativity in the locus coeruleus. Bar a–c 50 μm
Biochemical analysis of tau protein
Western blotting detected a major doublet of pathological tau at 64 and 69 kDa in brain tissue homogenates (Fig. 8) as reported in progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) or some FTDP-17 [4].

Brain distribution of pathological tau doublets by Western blot analysis
Genetic analysis
Sequencing of the SOD 1 and MAPT entire coding sequences failed to detect any mutation.
Discussion
The first prominent clinical signs in the proband were apathy and decline in social conduct, rapidly followed by a major change in speech and reduction of verbal fluency. Two years after the onset of the disease, neurological examination revealed a global cognitive decline. Neuropsychological tests were compatible with FTD. This case was thus consistent with FTD clinical criteria [24], occurring 2 years prior to the onset of MND. Dementia associated with ALS is usually of frontotemporal lobe type and MND generally occurs within a mean delay of 3 years [23, 25]. The reverse pattern, i.e., MND complicated with dementia, has rarely been reported, mainly in sporadic cases [14]. It should be underlined that initial dementia is most often described as overactive, with increasing apathy during course of the disease [21, 26].
The association of FTD with MND is generally sporadic, but a few familial cases have been reported [9, 28]. In the present family (Fig. 2), two affected adult individuals were found in the second generation and the father’s proband presented with an evolutive psychiatric disorder suggestive of FTD, but neuropsychological and neuropathological data were not available. Thus, the mode of inheritance is compatible with a dominant condition. No motor difficulties were noted in the father and the sister. This intra-familial phenotype diversity has been reported in autosomal dominant FTLD-MND [14]. In a single family, patients may be affected by MND, FTLD or FTLD-MND [9]. Even though the precise genetic alteration remains unknown in 80% of familial MND cases [12] and in 60–75% FTLD-MND cases [11], several genes or loci have been reported. Approximately 20% of autosomal dominant familial ALS are associated with mutations in SOD 1 gene [29]. However, to our knowledge, SOD 1 mutations have not been reported in familial FTLD-MND. Hosler et al. [12] have demonstrated a link to chromosome 9q21-q22 for familial FTLD-MND. It has been shown that 10–13% of autosomal dominant FTD are associated with mutations of the MAPT gene on chromosome 17 [13]. In one family previously described as “disinhibition-dementia-parkinsonism-amyotrophic complex” [19, 30], there was a point mutation in intronic region affecting alternative splicing of exon 10 of the MAPT gene [15]. Four cases had clinical features suggestive of FTLD-MND, but they did not appear to fulfill the neurophysiological criteria for MND. Therefore, to our knowledge, no cases of familial FLTD-MND have to date been associated with mutations of the MAPT gene [25] or linked to the second FTD locus, identified on chromosome 3 in a single family [3]. Due to the limited number of cases in our family, genetic linkage analysis was not available.
The neuropathological diagnosis in the proband case does not entirely meet the usual criteria for degenerative dementias [5]. The spinal cord and medullary lesions indicate MND, and explain the motor signs and electrophysiological findings. In addition, some of the hemispheric lesions, namely decreased neuronal density and laminar microvacuolation of layers II and III in the frontal and temporal cortex, indicate FTLD. Therefore, FTLD-MND appears the optimal hypothesis. Isocortical atrophy and neuronal loss are less prominent features in FTLD-MND than in FTLD alone [23]. However, the characteristic ubiquitin-only-immunoreactive inclusion bodies were not found in the dentate gyrus, and no tauopathy has been reported in this disorder [22]. Furthermore, severe nigral damage with heavy loss of pigmented neurons and intense reactive astrocytosis, often observed in MND-FTLD [23, 24, 26] were lacking in the present case. In FTLD-MND, however, nor neurofibrillary tangles, senile plaques [21, 24], or Pick bodies [21, 23, 26] have been described.
Further hypotheses could be a new FTDP-17 phenotype or a chance association of FTLD-MND with Alzheimer’s disease (AD), CBD or PSP that would explain the diffuse tau associated inclusions of the 64- to 69-kDa type observed in our case. The neuropathological findings reported in FTDP-17 associated with MAPT gene mutations are highly variable [8]. In the present case, the lesions may be suggestive of those described in disinhibition-dementia-parkinsonism-amyotrophic complex, although neuronal loss in the hippocampal and parahippocampal gyri were more severe in our case, and the characteristic cortical ubiquitinated neurons seen in this disorder were lacking. Lastly, the phenotype is different from that of the San Francisco family A presenting with three cases of clinical FTLD-MND associated with tau- and α-synuclein-positive inclusions in the temporal cortex, limbic structures and brainstem. This disease is linked to the chromosome 17 without MAPT mutation [31].
The Western blot characteristics of abnormal tau in this case are similar to those reported in PSP, CBD and argyrophilic grain disease, consisting of a characteristic major doublet (tau 64 and 69 kDa) [4]. This would eliminate a chance association with AD and the Guam variants of ALS and parkinsonism dementia complex [20], where the abnormal tau protein is composed of a major tau triplet of 69, 64 and 55 kDa. The clinical symptoms of CBD are more heterogeneous than previously expected, but the characteristic astrocytic plaques [6, 16] were lacking in this case. Even though clinical PSP with MND has been described [27], clinical signs in our case were not suggestive of PSP [17], and the distribution of the lesions (i.e., the absence of involvement of nucleus pallidus and basis pontis) were not distinctive for this disease [10]. Of course, this association of ALS and atypical tauopathy can be fortuitous.
In conclusion, the clinical, neuropathological, biochemical characteristics and genetic results of this case from a family affected by FTLD associated with MND are not consistent with the previously reported entities. Our results suggest that FTLD associated with MND can be caused by a larger spectrum of neuropathological lesions than commonly accepted.
References
Basak M, Erturk M, Oflazoglu B, Ozel A, Yildiz GB, Forta H (2002) Magnetic resonance imaging in amyotrophic lateral sclerosis. Acta Neurol Scand 105:395–399
Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases (2000) El Escorial revisited. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293–299
Brown J, Ashworth A, Gydesen S, Sorensen A, Hardy J, Collinge J (1995) Familial non-specific dementias maps to chromosome 3. Hum Mol Genet 45:1625–1628
Buée L, Delacourte A (1999) Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol 9:681–693
Dickson DW (ed) (2003) Neurodegeneration: the molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, p 414
Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I (2002) Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61:935–946
Dumanchin C, Camuzat A, Campion D, Verpillat P, Hannequin D, Dubois B, Saugier-Veber P, Martin C, Penet C, Charbonnier F, Agid Y, Frebourg T, Brice A (1998) Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet 7:1825–1829
Ghetti B, Hutton M, Wszolek ZK (2003) Frontotemporal dementia and parkinsonism linked to chromosome 17 associated with tau gene mutations (FTDP-17) In: Dickson DW (ed) Neurodegeneration: the molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, pp 86–102
Gunnarsson LG, Dahlbom K, Strandman E (1991) Motor neuron disease and dementia reported among 13 members of a single family. Acta Neurol Scand 84:429–433
Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I (1994) Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy) Neurology 44:2015–2019
Heutink P (2000) Untangling tau-related dementia. Hum Mol Genet 9:979–986
Hosler BA, Siddique T, Sapp PC, Sailor W, Huang MC, Hossain A, Daube JR, Nance M, Fan C, Kaplan J, Hung WY, McKenna-Yasek D, Haines JL, Pericak-Vance MA, Horvitz RH, Horvitz RHB Jr (2000) Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. JAMA 284:1664–1669
Houlden H, Baker M, Adamson J, Grover A, Waring S, Dickson D, Lynch T, Boeve B, Petersen RC, Pickering-Brown S, Owen F, Neary D, Craufurd D, Snowden J, Mann D, Hutton M (1999) Frequency of tau mutations in three series of non-Alzheimer’s degenerative dementia. Ann Neurol 46:243–248
Hudson AJ (1981) Amyotrophic lateral sclerosis and its association with dementia, parkinsonism, and other neurological disorders : a review. Brain 104:217–247
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al (1998) Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705
Komori T, Arai N, Oda M, Nakayama H, Mori H, Yagishita S, Takahashi T, Amano N, Murayama S, Murakami S, Shibata N, Kobayashi M, Sasaki S, Iwata M (1998) Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 96:401–408
Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, McKee A, Dickson D, Bancher C, Tabaton M, Jellinger K, Anderson DW (1996) Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 55:97–105
Lowe J, Rossor M (2003) Frontotemporal lobar degeneration: In: Dickson DW (ed) Neurodegeneration: the molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, pp 342–348
Lynch T, Sano M, Marder KS, Bell KL, Foster NL, Defendini RF, Sima AAF, Keohane C, Nygaard TG, Fahn S, Mayeux R, Rowland LP, Wilhelmsen KC (1994) Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology 44:1878–1884
Mawal-Dewan M, Schmidt ML, Balin B, Perl DP, Lee VM, Trojanowski JQ (1996) Identification of phosphorylation sites in PHF-TAU from patients with Guam amyotrophic lateral sclerosis/parkinsonism-dementia complex. J Neuropathol Exp Neurol 55:1051–1059
Mitsuyama Y (1993) Presenile dementia with motor neuron disease. Dementia 4:137–142
Nakano I (2000) Frontotemporal dementia with motor neuron disease (amyotrophic lateral sclerosis with dementia) Neuropathology 20:68–75
Neary D (1995) Neuropsychological aspects of frontotemporal degeneration. Ann NY Acad Sci 769:15–22
Neary D (1998) Frontotemporal lobar degeneration. A consensus on clinical diagnosis criteria. Neurology 51:1546–1554
Neary D, Snowden JS, Mann DM (2000) Cognitive change in motor neuron disease/amyotrophic lateral sclerosis (MND/ALS) J Neurol Sci 180:15–20
Niizato K, Tsuchiya K, Tominaga I, Kato Y, Ikeda K (1997) Pick’s disease with amyotrophic lateral sclerosis (ALS): report of two autopsy cases and literature review. J Neurol Sci 148:107–112
Paviour DC, Lees AJ, Josephs KA, Ozawa T, Ganguly M, Strand C, Godbolt A, Howard RS, Revesz T, Holton JL (2004) Frontotemporal lobar degeneration with ubiquitin-only-immunoreactive neuronal changes: broadening the clinical picture to include progressive supranuclear palsy. Brain 127:2441–2451
Pinsky L, Finlayson MH, Libman I, Scott BH (1975) Familial amyotrophic lateral sclerosis with dementia: a second Canadian family. Clin Gen 7:186–191
Siddique T, Nijhawan D, Hentati A (1996) Molecular genetic basis of familial ALS. Neurology 47 (Suppl 2):S27–S35
Wilhelmsen KC (1998) Chromosome 17-linked dementias. Cell Mol Life Sci 54:920–924
Wilhelmsen KC, Forman MS, Rosen HJ, Alving LI, Goldman J, Feiger J, Lee JV, Segall SK, Kramer JH, Lomen-Hoerth C, Rankin KP, Johnson J, Feiler HS, Weiner MW, Lee VM, Trojanowski JQ, Miller BL (2004) 17q-linked frontotemporal dementia-amyotrophic lateral sclerosis without tau mutations with tau and alpha-synuclein inclusions. Arch Neurol 61:398–406
Zhang L, Ulug AM, Zimmerman RD, Lin MT, Rubin M, Beal MF (2003) The diagnostic utility of FLAIR imaging in clinically verified amyotrophic lateral sclerosis. J Magn Reson Imaging 17:521–527
Acknowledgements
The authors are grateful to Richard Medeiros, Rouen University Hospital Medical Editor, for his valuable advice in editing the manuscript, and to Siegfried Le Roy for the iconography.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Martinaud, O., Laquerrière, A., Guyant-Maréchal, L. et al. Frontotemporal dementia, motor neuron disease and tauopathy: clinical and neuropathological study in a family. Acta Neuropathol 110, 84–92 (2005). https://doi.org/10.1007/s00401-005-1028-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-005-1028-2