
Abstract
Lissencephaly with agenesis of the corpus callosum and rudimentary dysplastic cerebellum may represent a subset of lissencephaly with cerebellar hypoplasia (LCH) of unknown etiology, one that is distinct from other types of LCH. We present a detailed neuropathological description of an autopsy brain from a 7-day-old neonate born at 38-gestational weeks, presenting with this malformation. The brain was severely hydrocephalic and totally agyric. The corpus callosum was absent and deep gray matter structures indistinct. A rudimentary dysplastic cerebellum, dysplastic olivary nuclei and nearly complete absence of corticospinal tracts were also noted. Microscopic examination revealed various types of dysplastic and malformative features throughout the brain in addition to the classic four-layered neocortical structure characteristic of type I lissencephaly. Unique features in the present case were (1) bilateral periventricular undulating cortical ribbon-like structures mimicking fused gyri and sulci, associated with aberrant reelin expression, (2) large dysplastic neocortical neurons positive for phosphorylated neurofilament, calbindin-D28K, tuberin, hamartin, doublecortin, LIS1, reelin and Dab1, (3) derangement of radial glial fibers, and (4) disorganized cerebellar cortex and heterotopic gray matter composed exclusively of granule cells in the cerebellar deep white matter. The clinicopathological features in the present case are suggestive of a distinct category of lissencephaly with cerebellar involvement. We suggest a possible classification of this unique case among the LCH syndromes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lissencephaly is a severe developmental brain malformation characterized by loss of the normal gyral pattern (agyria and/or pachygyria) in the cerebrum, marked disorganization of the cerebral cortical cytoarchitecture, and a strong association with profound neurological deficits and epilepsy. This type of malformation comprises a heterogeneous group of disorders sharing similar pathological features that are caused by alterations in distinct genes. Over the past decade, several genetic mutations responsible for lissencephalic phenotypes have been identified (for review see [7]): these include: (1) LIS1 on chromosome 17p13 for Miller-Dieker type lissencephaly [41], (2) doublecortin (DCX) on chromosome Xq22 for X-linked lissencephaly [9, 15, 16], (3) FCMD on chromosome 9q31 for Fukuyama congenital muscular dystrophy [26], (4) POMGnT1 (protein O-mannose β1,2-N-acetylglucosaminyltransferase) on chromosome 1p32 for muscle-eye-brain disease (MEB) [49], and (5) reelin (RELN) on chromosome 7q22 for lissencephaly with cerebellar hypoplasia (LCH) [19]. We describe the anatomical, histological and immunohistochemical features observed in an autopsy case of lissencephaly with agenesis of the corpus callosum and rudimentary dysplastic cerebellum as a distinct subset of LCH, and discuss the pathogenesis of this disorder.
Materials and methods
Case history
This male infant was vaginally delivered after induction, following 38 weeks of gestation (by dates), to a 27-year-old, gravida 3, para 1 (2 spontaneous abortions), mother. There is no known history of congenital abnormalities in the family. At 27 weeks of gestation, an ultrasound examination of the fetus revealed dilatation of the fourth ventricle and absence of the cerebellum. Subsequent ultrasound examinations at 30 and 36 weeks of gestation revealed intrauterine growth retardation consistent with 29 weeks gestation, without further fetal growth, and a Dandy-Walker abnormality as well as hydrocephalus. Prenatal laboratory testing of the mother was unremarkable. At birth, the baby was intubated due to respiratory insufficiency. Birth weight was 2,100 g. The head was microcephalic (circumference 27 cm) with small anterior and posterior fontanelles and closed sutures. The baby moved all extremities with appropriate muscle tone but had diminished reflexes in his upper and lower extremities. On the second day of life, magnetic resonance imaging (MRI) of the head (Fig. 1) confirmed severe brain malformation. The infant died 7 days after birth. Chromosomal analysis on the patient’s blood revealed a normal male karyotype (46, XY). Fluorescence in situ hybridization (FISH) chromosomal analysis did not detect deletions in the DiGeorge and Miller-Dieker critical regions. Gene analysis of reelin (RELN) was not available.

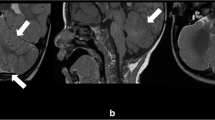
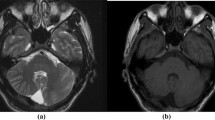
MRI of the head confirming near total absence of the cerebellum with prominent cystic dilatation of the fourth ventricle (A, B), consistent with Dandy-Walker syndrome. The cerebrum shows complete lack of gyral formation (lissencephaly), with a thin continuous zone of T2 hypointensity in what seems to be the subcortical area (arrowheads in B, arrows in D), bilaterally. Note a thickened calvarium (asterisk in A), hypoplastic cerebellum (arrows in B), agenesis of the corpus callosum (C), hypoplastic brainstem (arrow in A), basal ganglia and thalami (A, C), and undulating ventricular surface (arrowheads in C, D). A: T1-weighted image, sagittal, B,C: T2-weighted image, coronal, D: T2-weighted image, axial
Histological and immunohistochemical procedures
Formalin-fixed paraffin-embedded tissue blocks were cut at 5-µm thickness, and subjected to hematoxylin and eosin (HE) staining, Klüver-Barrera, and modified Bielschowsky methods as routine procedures. Consecutive serial sections were used for immunohistochemical studies. Immunostaining was performed with the avidin-biotin-peroxidase complex (ABC) (Vector, Burlingame, CA) and/or the Labeled Polymer (goat anti-rabbit or anti-mouse immunoglobulins conjugated to peroxidase labeled-dextran polymer, EnVision+™, DAKO, Carpinteria, CA) method using 3,3’-diaminobenzidine tetrahydrochloride (DAB) as chromogen. Antibodies and/or antisera (Table 1) were used according to the commercial product specification and previously published protocols [22, 23] with appropriate positive and negative controls. Specimens were subjected to microwave boiling, if needed, in a 0.015 M sodium citrate buffer solution (pH 6.0) for 12 min as an antigen retrieval procedure before blocking with appropriate normal animal serum. Sections were counterstained with hematoxylin. A combination of the ABC method with DAB and alkaline phosphatase with 5-bromo-4-chloro-3-indoxyl phosphate and nitro blue tetrazolium chloride (BCIP/NBT; DAKO) was also employed for double-label immunohistochemistry.
Control cases
Formalin-fixed, paraffin-embedded cerebral hemispheres from 11 normal human fetuses (12, 16, 19, 20, 24, 26, 28, 30, 34, 39 and 41 weeks of gestation), and cerebella from 6 normal human fetuses (24, 29, 30, 36, 39 and 41 weeks of gestation), all obtained at autopsy, were used for controls. All cases were stillborn infants without any recorded abnormalities in the central nervous system. Brains of 12, 16, 19 and 20 weeks of gestation were a generous gift from Professor Eisaku Ohama (Department of Neuropathology, Institute of Neurological Sciences, Faculty of Medicine, Tottori University, Japan).
Neuropathological findings
Gross appearance
External examination of the head showed microcephaly, mild hypertelorism and low-set ears. The palate was intact. The external genitalia were normally formed for a male. A closed or fused anterior fontanelle and a small posterior fontanelle (1-cm diameter) were seen along with fused cranial sutures. The skull showed a thickened calvarium surrounding normal-appearing dura mater. The brain was of extremely small size for its gestational age [14, 17], weighing 80 g after formalin fixation. It showed total absence of gyri and sulci without a gradient of severity (Fig. 2). Coronal sections of the cerebral hemispheres revealed hypoplastic pallium with markedly enlarged lateral and third ventricles (Fig. 3A–C), and an unusual undulation of the ependymal surface of the lateral ventricles bilaterally (Fig. 3B). Corpus callosum was absent and Probst bundles were not observed. Ammon’s horns were indistinct. The basal ganglia consisted of 1.1×1.0×1.0-cm masses of tissue without distinctions between the lentiform nuclei and the diencephalon. The cerebellum was extremely hypoplastic (Fig. 2A) with complete absence of the vermis. There was marked cystic dilatation of the fourth ventricle consistent with Dandy-Walker malformation. The pons and medulla oblongata were markedly flattened (Fig. 2A). There were no discrete abnormalities in the spinal cord.

Gross appearance of base of the brain (A) and convexity (B) showing total absence of gyri and sulci. Note hypoplastic brainstem and rudimentary cerebellum (arrow). Asterisk indicates tentorium cerebelli

Coronal sections of the cerebral hemispheres (A–C) reveal hypoplastic pallium with markedly enlarged lateral ventricles, and an unusual undulation of the ependymal surface of the lateral ventricles bilaterally (B). D demonstrates a four-layered structure (1–4) of neocortex observed in the right parietal convexity (section is from the slice shown in C). Note a hypocellular lamina (arrowhead) splitting the outer neuronal layer, a thick cord of neurons running across the pallium (arrow), and focal polymicrogyria (double arrowheads). The superficial and deep neuronal layers are often associated with acellular islands or glomeruli (asterisks in E) that are accentuated by synaptophysin immunoreactivity (asterisks in F). Note some pyramidal neurons strongly positive for cytoplasmic synaptophysin (arrows and inset). G A heterotopic cluster of germinal matrix cells (arrow). H Abnormally directed fiber bundle formations within the subpial marginal layer (arrowheads). I Focal heterotopic clusters of granule type neurons in the subpial marginal layer of the frontal lobe (arrows). Bars A–C 1 cm. D Klüver-Barrera, ×6; E HE, ×100; F synaptophysin immunostaining, ×100; G Klüver-Barrera, ×3; H, I HE, ×40
Microscopic observation
Cerebrum
Consistent with type I lissencephaly, a four-layered neocortex was observed predominantly in the cerebral convexities; this was composed of (1) a marginal layer, (2) superficial cellular layer or outer neuronal layer, (3) sparsely cellular or paucicellular layer with a few unmyelinated fiber bundles, and (4) deep cellular layer or inner gray matter (Fig. 3D). The gray-white matter ratio in this region varied from 5:1 to 10:1. In addition, however, the greater part of the pallium showed more complex malformative features (Fig. 3D–I). The thickness of the inner gray matter gradually tapered and eventually disappeared at the medial aspect of the cerebral hemisphere, whereas the cortical ribbon extended down from the outer neuronal layer and showed undulations without accompanying sulcal or gyral formation, i.e., focal polymicrogyria (Fig. 3D). Large dysplastic neurons (LDNs) were observed predominantly in the posterior part of the cerebrum (Fig. 4A).

LDNs (arrows in A) are strongly positive for pNF (B). Many of them express CB (C) and are scattered throughout the cortex with a slight accentuation in the superficial marginal (1 in C) and superficial cellular layers (2 in C). In age-matched control brain (39 weeks), CB-immunoreactive cells predominate around layer IV of the neocortex (D). E High-power view of CB-positive LDN. F High-power view of CB-positive multipolar interneuron in age-matched control brain (39 weeks) (LDN large dysplastic neuron, pNF phosphorylated neurofilament, CB calbindin-D28K). A HE, ×100; B 2F11 immunostaining, ×200; C, D CB immunostaining, ×40; E, F CB immunostaining, ×400
LDNs were strongly positive for phosphorylated neurofilament (pNF) (Fig. 4B), and were observed throughout the thickness of the cortex; 50–60% of them were also positive for calbindin-D28K (CB) (Fig. 4C, E). These CB-positive cells were scattered throughout the cortex with a slight accentuation in the superficial marginal and superficial cellular layers (Fig. 4C), as opposed to those in a normal age-matched control brain, where they predominate in deeper layers, around layer IV of the neocortex (Fig. 4D, F). Most LDNs were also positive for tuberin (Fig. 5A) and hamartin (Fig. 5B). Some were positive for doublecortin (DCX) (Fig. 5C), LIS1 (Fig. 5D), reelin (Fig. 5E) and Dab1 (Fig. 5F) in their cytoplasm. In addition to abundant expression of DCX in germinal matrix, some pyramidal neurons in the superficial cellular layer were also positive for DCX (Fig. 5C). DCX expression in normal developing neocortex was observed only in younger individuals (12, 16, 19 weeks), but not in age-matched controls as previously described [40]. LIS1 immunohistochemistry failed to detect cells that would normally be positive in normal developing brain, including Cajal-Retzius (C-R) cells, subplate neurons and others [6, 42].

Most LDNs are immunoreactive for tuberin (arrowhead in A) and hamartin (B). Some of them are positive for DCX (arrow in C), LIS1 (D), reelin (E) and Dab1 (F). Note tuberin immunoreactivity in LDN and relatively normal pyramidal neuron (arrow in A), DCX-positive LDN (arrow in C) and DCX-negative LDN (arrowhead in C). A thin band consisting of pNF-positive fibers and/or neurites is observed in the superficial marginal layer beneath the pial surface (arrowheads in G). H By contrast, in control fetal brains, neocortical layer I is mostly occupied by sparse pNF-positive fibers. I pNF-positive band in G is immunoreactive for reelin. Strong immunoreaction is observed in subpial Cajal-Retzius cells (arrow) (DCX doublecortin). A Tuberin immunostaining, ×200; B hamartin immunostaining, ×200; C DCX immunostaining, ×260; D LIS1 immunostaining, ×400; E reelin immunostaining, ×400; F Dab1 immunostaining, ×400; G, H 2F11 immunostaining, ×100; I reelin immunostaining, ×260
There was a thin horizontal band consisting of pNF-positive fibers and/or neurites beneath the pial surface (Fig. 5G) in the present case but not in any of the control brains (Fig. 5H). Strong reelin expression was observed in what appeared to be C-R cells (Fig. 5I) in both the present and control cases. Diffuse neuropil staining of reelin was observed in layer I of cerebral cortex in 12–39 week control fetal brains, whereas this pattern of reelin-labeling was less prominent in the present case. However, a thin horizontal band consisting of reelin-positive C-R cells and their neurites and/or diffuse neuropil staining of reelin (Fig. 5I) was observed in the subpial area anatomically corresponding to the subpial pNF-positive band. This structure predominated in dorso- and ventro-medial aspects of the cerebral hemisphere.
There were periventricular undulating cortical ribbon-like structures (PUC) mimicking fused gyri and sulci observed bilaterally and symmetrically in the area adjacent to the germinal matrix (Fig. 6A). PUC consisted of the extension of single streaks of gliovascular tissue from the subventricular germinal matrix (SGM), surrounded by a thin band of molecular layer-like paucicellular structure and an underlying single band of disorganized neuronal cell layer (Fig. 6B) that was continuous with the inner gray matter of the four-layered cortex. Neither an arachnoidal nor ependymal covering was observed. Profound derangement of radial glial fibers was also noted (Fig. 6C–E). There were increased numbers of GFAP-positive astrocytes throughout the cerebrum, and most of them had morphological features of Alzheimer type II astrocytes. SGM contained a substantial number of these astrocytes and some foci of ependymal differentiation forming ependymal rosette, canals and tubules (Fig. 6F). Ki-67 immunohistochemistry revealed a high labeling index within SGM and scattered positive nuclei throughout the pallium. Double-label immunohistochemistry confirmed the co-localization of Ki-67 and GFAP in only a few cells within the pallium (Fig. 6G). Patchy islands of reelin immunoreaction were observed mostly in a disorganized neuronal cell layer within PUC (Fig. 6H), consisting of diffuse neuropil staining and cytoplasmic reelin-expressing small, round neurons with scant cytoplasm that are morphologically distinct from subpial C-R cells (Fig. 6I). A small number of reelin-expressing small neurons were also scattered throughout the pallium, whereas no reelin-immunoreactive cells were observed in the area deeper than layer I of developing normal cerebral cortex. DCX was abundantly expressed in both the subventricular and heterotopic germinal matrix (Fig. 6J). Many apparently migrating neurons appeared positive for DCX (Fig. 6K) and were observed predominantly in the depths of the PUC subjacent to SGM and more superficial area of the pallium.

A A PUC is demonstrated in a whole mount section of the right cerebral hemisphere shown in Fig. 3B. It gradually tapers off in the ventro-medial portions (arrow) or merges with the outer neuronal layer in the dorso-medial portions of the pallium (left upper corner). Note the indistinct Ammon’s horn (asterisk). B Microscopic feature of PUC consisting of the extension of single streaks of gliovascular tissue (arrow) from the SGM, surrounded by a molecular layer-like paucicellular structure (asterisk) and an underlying single band of disorganized cells. Vimentin-positive radial glial fibers appear to originate also from the gliovascular streaks and run through the surrounding neuronal layer (C), but do not appear to properly reach the overlying superficial cortex where there were only a few neurons observed (D). Note radial glial fibers originating from a heterotopic cluster of germinal matrix cells (asterisk in E) and approaching, in oblique or asymptotic fashion, the pial surface where there was no well-formed cortical plate (E). SGM contains areas of both astrocytic and ependymal differentiation (asterisk in F). Co-localization of MIB-1 (brown by DAB) and GFAP (purplish blue by BCIP/NBT) is observed in a few cells (arrow in G) in the pallium. H, I Unique reelin immunoreactivity within the PUC (arrowhead in H) and reelin-expressing small granule type neurons (arrows in I). Note diffuse neuropil staining of reelin (arrow in H). DCX is abundantly expressed in SGM cells sparing the area of astrocytic and ependymal differentiation (asterisk in J). DCX is also expressed in what appear to be migrating neurons (arrows in K) (PUC periventricular undulating cortical ribbon-like structures, SGM subventricular germinal matrix). A Klüver-Barrera method, ×3; B HE, ×100; C–E vimentin (V9) immunostaining, C ×133, D ×13, E ×26; F HE, ×200; G double-label immunohistochemistry for MIB-1 and GFAP, ×400; H reelin immunostaining, ×3; I reelin immunostaining, ×400; J DCX immunostaining, ×260; K DCX immunostaining, ×370
Ammon’s horns could not be clearly identified at the microscopic level (Fig. 6A). Neither the pyramidal cell layer nor fascia dentata appeared to have formed properly, with only a mixture of disoriented pyramidal neurons and patchy islands of granule cells randomly scattered about.
Brainstem and cerebellum
The cerebral peduncles were almost completely absent. The pontine base was extremely thin and gliotic with nearly complete lack of pontine nuclei, corticospinal and pontocerebellar tracts (Fig. 7A). Medullary pyramids were almost totally absent (Fig. 7B). The cerebellum was severely hypoplastic and dysplastic with only rudimentary hemispheres and complete aplasia of the vermis (Fig. 7A). The external granular layer was hypoplastic (Fig. 7C). The cellular arrangement of the cortex was severely disorganized (Fig. 7C, D). Large clusters of heterotopic gray matter composed exclusively of granule cells (Fig. 7E, F), and scattered single heterotopic LDNs immunoreactive for pNF and CB (Fig. 7G) were also observed in the cerebellar deep white matter. The dentate nucleus was dysplastic and identified as a collection of gray matter islands lacking the normal undulating contour (Fig. 7H). Most of the astrocytes observed in the cerebellar white matter were multinucleated (Fig. 7I), consistent with Alzheimer type I astrocytes.

A Whole mount transverse section of pons and cerebellum showing hypoplastic cerebellar hemispheres. Note severely hypoplastic pontine base. Asterisks indicate trigeminal nerves. B Inferior olivary nuclei (arrow) are hypoplastic and dysplastic, lacking hilus directed medially. Asterisk indicates hypoplastic medullary pyramids. C, D Microscopically clusters of disoriented Purkinje cells (arrow in C) and granule cells are randomly intermingled with each other without normal laminar cytoarchitecture, better demonstrated with CB immunostaining (D). The cellularity of external and internal granular layers appears less than normal. Note a Purkinje cell just beneath the external granular layer (arrow in D). E, F There is also a relatively well-organized Purkinje cell layer, although situated in the middle of the internal granular layer (asterisks in E). Note a large cluster of heterotopic gray matter composed exclusively of granule cells (arrow in E) positive for synaptophysin (F). G There are also scattered single heterotopic LDNs immunoreactive for pNF and CB observed in the deep cerebellar white matter. H The dentate nucleus is dysplastic and identified beneath the dysplastic cerebellar cortex (asterisk) as a collection of gray matter islands lacking the normal undulating ribbon-like contour (arrowheads). I Most of the astrocytes observed in the white matter are multinucleated consistent with Alzheimer type I astrocytes. A Klüver-Barrera, ×4.5; B Klüver-Barrera, ×40; C HE, ×200; D CB immunostaining, ×100; E HE, ×42; F synaptophysin immunostaining, ×66; G CB immunostaining, ×400; H synaptophysin immunostaining, ×20; I HE, ×400
Discussion
Agyria/pachgyria associated with cerebellar hypoplasia has recently been recognized as a heterogeneous group of brain malformations [2, 11, 19, 27, 44] described as lissencephaly with cerebellar hypoplasia (LCH). The pathological features in the present case are characterized by type I lissencephaly (complete agyria) associated with agenesis of corpus callosum, rudimentary dysplastic cerebellum, and hypoplastic brainstem.
The significance of the present case with relevance to current concepts of LCH
A small number of familial cases that may overlap with the pathological features in the present case have been described (Table 2). Kerner et al. [24] described the clinicopathological features observed in two siblings as a distinct phenotype of familial lissencephaly with cleft palate and severe cerebellar hypoplasia. These cases seem to be distinct from the present case on the basis of clinicopathological features, including a diffuse single band of disorganized neurons and glia rather than a four-layered cortex. In cases with lissencephaly associated with skin and skeletal abnormalities [1], severe cerebellar and brainstem hypoplasia may be observed. However, histological features of diffuse neuronal degeneration resulting in severe micrencephalic brain in those cases implicate a pathogenesis similar to that suggested in Neu-Laxova syndrome (NLS) [37]. Although the brain in our case was also extremely small, weighing only 80 g [14, 17], histological features of a putative neuroblast migration disorder associated with hypoplastic pallium and severe hydrocephalus appear more likely to account for this brain weight rather than the degenerative process of NLS. Lack of specific anomalies such as ichthyosis, edema and a characteristic facial abnormality in the present case is also against the diagnosis of NLS [25]. Recently, mutations in the RELN gene have been reported to be associated with autosomal recessive LCH [19]. In mice carrying mutations in RELN (reeler mice) and in disabled-1 (Dab1) as well as in mice carrying double mutations of both very low density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2), normal neuroblast migration with an “inside-out” fashion is inverted [46]. This suggests a role for these genes in the control of cell positioning in the developing central nervous system and predicts a pattern of cytoarchitectural alteration in patients carrying alterations in reelin/lipoprotein receptor/Dab1 pathway as well as RELN mutations.
Recently, six categories of LCH (LCHa–f) have been proposed mainly based on the neuroradiographic findings [44]. Of note, two pediatric cases are listed as a heterogeneous group of agyria/pachygyria associated with absent corpus callosum, moderate to severe cerebellar hypoplasia and brainstem hypoplasia of unknown inheritance (LCHf in the series). One presented with complete agyria and severe hypoplasia of cerebellum and brainstem, and had a thin continuous band of high signal intensity on T2-weighted sequences in what seems to be the subcortical area, as well as focal undulation of the ventricular surface—almost identical to that observed in MR images of the present case (Fig. 1). Mutations in LIS1 can also cause a LCH phenotype (LCHa) that displays mild vermian hypoplasia, lissencephaly with either an anterior-to-posterior or posterior-to-anterior gradient of severity, and presence of the corpus callosum, even if it is hypoplastic—the features do not match those in the present case. Furthermore, in the recent classification of cerebellar malformations, a heterogeneous group of lissencephaly with cerebellar dysplasia has been proposed, listing three cases in a series of 70 MRI studies obtained in patients with cerebellar anomalies [39]. Apart from one case with a RELN mutation, the other two cases without known gene mutations were associated with agenesis of the corpus callosum, small-sized brainstem and tiny cerebellar hemispheres with no visible vermis, implying similar anatomical features to those in the present case. However, there has been neither a detailed histopathological verification nor an immunohistochemical characterization of LCH reported.
A four-layered neocortical structure is, together with preserved superficial marginal layer, a characteristic histological abnormality that distinguishes type I from type II lissencephaly [18, 35]. MRI-neuropathological correlations in the present case revealed that the band of high T2 signal represents the MRI counterpart of a sparsely cellular layer, as has been previously suggested [28]. Hence, the presence of this T2 high intensity band is predictive of a histological four-layered cortex typical of type I lissencephaly, at least in the developing brain.
Other phenotypes of lissencephaly associated with agenesis of corpus callosum
Agenesis of corpus callosum (ACC) per se is often considered as nonspecific since it is found with variable frequency in various syndromes (for review see [34]). Among lissencephaly phenotypes, however, association with ACC seems to be relatively rare and may even comprise a distinct group of disorders. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia [3, 10, 36] has been described as another entity of X-linked lissencephaly that is distinct from that caused by the DCX mutation, although it is not a case of LCH (Table 2).
The unique features of PUC, LDNs and disorganization of the radial glial fibers
The PUC in this case was continuous with the deep cellular layer of the four-layered cortex and was associated with streaks of gliovascular tissue extending from the subventricular germinal matrix. Therefore, the possibility that the PUC represents “band heterotopia” or “periventricular nodular heterotopia” appears to be excluded. The fact that the majority of neurons existed within the PUC but not in the superficial layer, and that the germinal matrix cells were also observed within the gliovascular streaks, suggests that the PUC is associated with abnormalities from the earlier stage of neuroblast migration to the later stage of gliogenesis within the subventricular germinal matrix. Furthermore, the presence of reelin-expressing cells and diffuse neuropil staining of reelin within the PUC are even suggestive of an abnormal formation of preplate or abnormal generation and migration of reelin-expressing cells. These reelin-expressing cells may represent aberrant C-R cells playing a role in the formation of an aberrant radial glial scaffold [12] and the formation of PUC in this case, although reelin-positive cells in the PUC are of round, small-sized and granular type, morphologically distinct from subpial C-R cells [31]. The PUC seems to be an unique feature in the present case.
Studies of macaque monkey brain have suggested that calcium binding protein-containing interneurons make up 90% of all GABAergic neurons in the cerebral cortex [30]. Many LDNs in our case were strongly immunoreactive for CB, suggesting that they have the features of GABAergic interneurons. These cells were scattered throughout the cortex with a slight accentuation in the superficial marginal and superficial cellular layers (Fig. 4C). This observation is in contrast to that of age-matched controls, in which CB-immunoreactive cells predominated in deeper layers, around layer IV of the neocortex (Fig. 4D). It remains unclear whether the laminar distribution of CB-positive LDNs represents their final distribution or a temporary profile in the present case. The number and distribution of calcium binding protein-containing neurons, including CB-immunoreactive cells, in the neocortex can be reorganized in early postnatal life [43]; by 28 weeks after birth, the laminar distribution of CB-immunoreactive cells shifts from layer IV to layer II.
Abnormalities of radial glia may occur with various molecular mechanisms either focally or diffusely in malformations of cortical development including Fukuyama congenital muscular dystrophy [45] and tuberous sclerosis [38]. Furthermore, alterations of signaling pathways that regulate microtubule dynamics either directly or indirectly may result in a derangement of radial glial fibers, as demonstrated in reeler mice [21] and Lis1 mutant mice [4]. The latter may also result in abnormal neuronal morphology [4]. Interestingly, single heterotopic large dysplastic neurons observed in the cerebellum were also strongly positive for pNF and CB. Therefore, it is plausible that CB-expressing GABAergic interneurons in the cerebrum and the CB-expressing heterotopic Purkinje cells in the cerebellum are specifically affected by a yet to be determined alteration of intracellular signaling, which would result in a disorder of generation and migration of inhibitory neurons at either ventricular and subventricular zones of the dorsal forebrain and hindbrain or medial ganglionic eminence of the ventral forebrain or both [29], eventually presenting with dysplastic morphology. Overexpression of proteins that are known to play important roles in regulating cell size, shape, cell cycle and neuroblast migration during brain development (for review see [7]) among the population of LDNs may support this idea. Alternatively, since lissencephaly is strongly associated with epilepsy [7], these abnormal cells may account in part for the epileptogenicity of lissencephaly.
Study of morphological and functional correlation has demonstrated abnormal electrophysiological properties of cytomegalic neurons in surgically resected focal cortical dysplasia from patients with intractable epilepsy [5].
Rare type of heterotopic granule cells in the cerebellum
One of the striking histological features observed in the cerebellum of this patient is the presence of heterotopias composed exclusively of granule cells without accompanying Purkinje cells (Fig. 7D, E). Heterotopias in the developing cerebellum [13, 20, 32] often coexist with other cerebellar malformations [14]. Heterotopic neurons in developing cerebellum, particularly the white matter, are often incidentally observed, but not rare, even during normal development, possibly as a temporary phenomenon during cerebellar development [32]. However, the fact that those heterotopias have been reported to be constantly associated with a Purkinje cell component [32] and that there has been no report of cerebellar white matter heterotopias composed exclusively of granule cells, suggests another yet to be defined role for Purkinje cells in the migration of external granule cells not only in normal cerebellar corticogenesis, but also in the formation of heterotopia. In fact, the Purkinje cell-derived sonic hedgehog has a mitogenic effect on granule cell precursors [8, 47, 48], and the external granule cells secrete reelin [33]. Interestingly, the granule cell heterotopias observed in the present case were morphologically well differentiated, containing synaptophysin-immunoreactive glomeruli (Fig. 7D, E) but not spindle-shaped cells described as “matrix cell heterotopia” or “residual matrix cells” [14]. Further detailed characterization is necessary to determine the precise origin of these malpositioned granular neurons.
In conclusion, the overall neuropathological features along with other external manifestations in the present case are quite unique, and do not fit with any previously reported cases in which histological details have been presented (Table 2). We believe that this is the first neuropathological description of a brain malformation that may fit with the categories that have been proposed, as a distinct subset of LCH syndrome, e.g., LCHf [44], LCH with ACC, brainstem and cerebellar hypoplasia [2], and lissencephaly with cerebellar dysplasia [39]. We propose the term “lissencephaly with agenesis of corpus callosum and rudimentary dysplastic cerebellum” to describe the brain malformation in the present case based on its neuropathological features. Histological verification appears to be an important procedure for a better understanding of the pathogenesis of each LCH subset and for the establishment of more precise classification incorporated with future clinicopathological, genetic, and mechanistic considerations [7].
References
Attia-Sobol J, Encha-Razavi F, Hermier M, Vitrey D, Verloes A, Plauchu H (2001) Lissencephaly type III, stippled epiphyses and loose, thick skin: a new recessively inherited syndrome. Am J Med Genet 99:14–20
Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB (2001) Classification system for malformations of cortical development: update 2001. Neurology 57:2168–2178
Bonneau D, Toutain A, Laquerriere A, Marret S, Saugier-Veber P, Barthez MA, Radi S, Biran-Mucignat V, Rodriguez D, Gelot A (2002) X-linked lissencephaly with absent corpus callosum and ambiguous genitalia (XLAG): clinical, magnetic resonance imaging, and neuropathological findings. Ann Neurol 51:340–349
Cahana A, Escamez T, Nowakowski RS, Hayes NL, Giacobini M, Holst A von, Shmueli O, Sapir T, McConnell SK, Wurst W, Martinez S, Reiner O (2001) Targeted mutagenesis of Lis1 disrupts cortical development and LIS1 homodimerization. Proc Natl Acad Sci USA 98:6429–6434
Cepeda C, Hurst RS, Flores-Hernández J, Hernández-Echeagaray E, Klapstein GJ, Boylan MK, Calvert CR, Jocoy EL, Nguyen OK, André VM, Vinters HV, Ariano MA, Levine MS, Mathern GW (2003) Morphological and electrophysiological characterization of abnormal cell types in pediatric cortical dysplasia. J Neurosci Res 72:472–486
Clark GD, Mizuguchi M, Antalffy B, Barnes J, Armstrong D (1997) Predominant localization of the LIS family of gene products to Cajal-Retzius cells and ventricular neuroepithelium in the developing human cortex. J Neuropathol Exp Neurol 56:1044–1052
Crino PB, Miyata H, Vinters HV (2002) Neurodevelopmental disorders as a cause of seizures: Neuropathologic, genetic, and mechanistic considerations. Brain Pathol 12:212–33
Dahmane N, Ruiz-i-Altaba A (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126:3089–3100
Des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J (1998) A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92:51–61
Dobyns WB, Berry-Kravis E, Havernick NJ, Holden KR, Viskochil D (1999) X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet 86:331–337
Farah S, Sabry MA, Khuraibet A, Khaffagi S, Rudwan M, Hassan M, Haseeb N, Abulhassan S, Abdel-Rasool MA, Elgamal S, Qasrawi B, Al-Busairi W, Farag TI (1997) Lissencephaly associated with cerebellar hypoplasia and myoclonic epilepsy in a Bedouin kindred: a new syndrome? Clin Genet 51:326–330
Förster E, Tielsch A, Saum B, Weiss KH, Johanssen C, Graus-Porta D, Müller U, Frotscher M (2002) Reelin, Disabled 1, and beta 1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proc Natl Acad Sci USA 99:13178–13183
Friede RL (1973) Dating the development of human cerebellum. Acta Neuropathol (Berl) 23:48–58
Friede RL (1989) Developmental neuropathology, 2nd rev. and expanded edn. Springer, Berlin Heidelberg
Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA (1998) Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92:63–72
Gleeson JG, Minnerath SR, Fox JW, Allen KM, Luo RF, Hong SE, Berg MJ, Kuzniecky R, Reitnauer PJ, Borgatti R, Mira AP, Guerrini R, Holmes GL, Rooney CM, Berkovic S, Scheffer I, Cooper EC, Ricci S, Cusmai R, Crawford TO, Leroy R, Andermann E, Wheless JW, Dobyns WB, Ross ME, Walsh CA (1999) Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Ann Neurol 45:146–153
Guihard-Costa AM, Ménez F, Delezoide AL (2002) Organ weights in human fetuses after formalin fixation: standards by gestational age and body weight. Pediatr Dev Pathol 5:559–578
Harding BN, Copp AJ (2002) Malformations. In: Graham DI, Lantos PL (eds) Greenfield’s neuropathology, 7th edn, Vol I. Arnold, London, pp 357–483
Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA (2000) Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet 26:93–96
Hori A, Matsushita M, Murofushi K, Iizuka R (1974) Heterotopien im Kleinhirnmark. No To Hattatsu 6:404–408
Hunter-Schaedle KE (1997) Radial glial cell development and transformation are disturbed in reeler forebrain. J Neurobiol 33:459–472
Johnson MW, Emelin JK, Park SH, Vinters HV (1999) Co-localization of TSC1 and TSC2 gene products in tubers of patients with tuberous sclerosis. Brain Pathol 9:45–54
Johnson MW, Kerfoot C, Bushnell T, Li M, Vinters HV (2001) Hamartin and tuberin expression in human tissues. Mod Pathol 14:202–210
Kerner B, Graham JM Jr, Golden JA, Pepkowitz SH, Dobyns WB (1999) Familial lissencephaly with cleft palate and severe cerebellar hypoplasia. Am J Med Genet 87:440–445
King JAC, Gardner V, Chen H, Blackburn W (1995) Neu-Laxova syndrome: pathological evaluation of a fetus and review of the literature. Pediatr Pathol Lab Med 15:57–79
Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T (1998) An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 394:388–392
Kroon AA, Smit BJ, Barth PG, Hennekam RC (1996) Lissencephaly with extreme cerebral and cerebellar hypoplasia. A magnetic resonance imaging study. Neuropediatrics 27:273–276
Landrieu P, Husson B, Pariente D, Lacroix C (1998) MRI-neuropathological correlations in type 1 lissencephaly. Neuroradiology 40:173–176
Letinic K, Zoncu R, Rakic P (2002) Origin of GABAergic neurons in the human neocortex. Nature 417:645–649
Lund JS, Lewis DA (1993) Local circuit neurons of developing and mature macaque prefrontal cortex: Golgi and immunocytochemical characteristics. J Comp Neurol 328:282–312
Marín-Padilla M (1998) Cajal-Retzius cells and the development of the neocortex. Trends Neurosci 21:64–71
Miyata M, Miyata H, Mikoshiba K, Ohama E (1999) Development of Purkinje cells in humans: an immunohistochemical study using a monoclonal antibody against the inositol 1,4,5-triphosphate type 1 receptor (IP3R1). Acta Neuropathol 98:226–232
Miyata T, Nakajima K, Aruga J, Takahashi S, Ikenaka K, Mikoshiba K, Ogawa M (1996) Distribution of a reeler gene-related antigen in the developing cerebellum: an immunohistochemical study with an allogeneic antibody CR-50 on normal and reeler mice. J Comp Neurol 372:215–228
Norman MG (1996) Malformations of the brain. J Neuropathol Exp Neurol 55:133–143
Norman MG, McGillivray BC, Kalousek DK, Hill A, Poskitt KJ (1995) Congenital malformations of the brain: pathological, embryological, clinical, radiological and genetic aspects. Oxford University Press, New York
Ogata T, Matsuo N, Hiraoka N, Hata JI (2000) X-linked lissencephaly with ambiguous genitalia: delineation of further case. Am J Med Genet 94:174–176
Ostrovskaya TI, Lazjuk GI (1988) Cerebral abnormalities in the Neu-Laxova syndrome. Am J Med Genet 30:747–756
Park SH, Pepkowitz SH, Kerfoot C, De Rosa MJ, Poukens V, Wienecke R, DeClue JE, Vinters HV (1997) Tuberous sclerosis in a 20-week gestation fetus: immunohistochemical study. Acta Neuropathol 94:180–186
Patel S, Barkovich AJ (2002) Analysis and classification of cerebellar malformations. AJNR Am J Neuroradiol 23:1074–1087
Qin J, Mizuguchi M, Itoh M, Takashima S (2000) Immunohistochemical expression of doublecortin in the human cerebrum: comparison of normal development and neuronal migration disorders. Brain Res 863:225–232
Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH (1993) Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 364:717–721
Reiner O, Albrecht U, Gordon M, Chianese KA, Wong C, Gal-Gerber O, Sapir T, Siracusa LD, Buchberg AM, Caskey CT, Eichele G (1995) Lissencephaly gene (LIS1) expression in the CNS suggests a role in neuronal migration. J Neurosci 15:3730–3738
Reynolds GP, Beasley CL (2001) GABAergic neuronal subtypes in the human frontal cortex—development and deficits in schizophrenia. J Chem Neuroanat 22:95–100
Ross ME, Swanson K, Dobyns WB (2001) Lissencephaly with cerebellar hypoplasia (LCH): a heterogeneous group of cortical malformations. Neuropediatrics 32:256–263
Takada K, Nakamura H, Suzumori K, Ishikawa T, Sugiyama N (1987) Cortical dysplasia in a 23-week fetus with Fukuyama congenital muscular dystrophy (FCMD). Acta Neuropathol (Berl) 74:300–306
Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J (1999) Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97:689–701
Wallace VA (1999) Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 9:445–448
Wechsler-Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22:103–114
Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, Herrmann R, Straub V, Talim B, Voit T, Topaloglu H, Toda T, Endo T (2001) Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 1:717–724
Acknowledgement
The authors wish to thank Alexander Blooks and Beth Johnson (Section of Neuropathology, UCLA Medical Center) for invaluable technical assistance. H.M. is supported in part by a grant from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Miyata, H., Chute, D.J., Fink, J. et al. Lissencephaly with agenesis of corpus callosum and rudimentary dysplastic cerebellum: a subtype of lissencephaly with cerebellar hypoplasia. Acta Neuropathol 107, 69–81 (2004). https://doi.org/10.1007/s00401-003-0776-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-003-0776-0