
Abstract
A series of azo-ester linked benzothiazole mesogen containing new polymerizable liquid crystalline (LC) methacrylate monomers M1–M4 having different terminal substituents (H, CH3, OCH3, and OC2H5) on the benzothiazole moiety and their side chain liquid crystalline polymers (SCLCPs) P1–P4 were synthesized and characterized. The chemical structures, thermal stability, and LC phase behaviors of monomers and polymers were examined by the usage of different experimental techniques. All the synthesized monomers and polymers exhibited excellent thermal stability. Monomer M1 (without terminal substitution) displayed both nematic and smectic phases while monomers M2–M4 (CH3, OCH3, and OC2H5 substituted) showed only nematic phase. On the other hand, all the prepared polymers exhibited only nematic LC phase. Polymers P1–P4 showed two strong absorption bands in the range of 261–262 and 376–413 nm whereas polymers P1–P4 exhibited strong fluorescence emission in the range of 522–524 nm. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of the polymers were found to be −4.87 to −4.80 eV and −2.36 to −2.14 eV, respectively. The synthesized polymers could be potential candidate as fluorescent materials in the polymer light-emitting diode applications.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Side chain liquid crystalline polymers (SCLCPs) are a class of macromolecular compounds, which show the anisotropic optical, electrical, and mechanical characteristics of the liquid crystals and at the same time, demonstrate many useful and versatile properties of polymers [1, 2]. Due to this unique duality of properties, SCLCPs exhibit some enhanced thermal and mechanical properties. As a result, these polymers find numerous applications as high-tensile strength fibers, self-strengthened materials, optoelectronic materials, reversible data storage devices, elastomeric products, thermal or barometric sensors, chromatographic separations, solid polymer electrolytes, separation membranes, light-emitting diodes, and display materials [3, 4]. The properties of SCLCPs depend on a number of parameters, such as length of the flexible spacer, nature of the polymer backbone and the type of mesogen [5, 6]. The mesogenic unit in SCLCPs is mainly comprised of benzene rings connected by different linking groups. In recent years, single ring (pyridine, thiophene, and thiazole) and fused ring (benzothiazole, benzoxazole) heterocyclic compounds have also been used as mesogenic core in SCLCPs [7, 8].
Benzothiazole-incorporated molecules are very fascinating and promising class of compounds because of their interesting photophysical properties. These derivatives have been exploited in many applications, such as organic light-emitting diodes, chromogenic chemosensor for metal ion detection, potential sensitizers for photodynamic therapy, fluorescent tracer for diseases diagnosis, photovoltaic cell, memory devices, and photoconductive materials [9–15]. In addition, benzothiazole is considered as a good mesogen forming molecule due to the presence of electron rich heteroatoms (S and N) in its moiety [16]. As a result, calamitic LC compounds comprising benzothiazole moiety has gained considerable research attention. The fast hole-transportation properties of a photoconductive calamitic LC compound containing benzothiazole moiety have been reported by Funahashi and Hanna [17]. Dutta et al. have investigated the field-effect transistor performance of LC compounds having benzothiazole moiety, and these materials may be excellent candidates for application in organic electronics [18]. Benzothiazole moiety is usually connected with the mesogen of calamitic LC compounds by different linking groups, e.g., azomethine (–C = N–), azo –N = N–), and ester (–COO–). The choice of linking groups in LC compounds is very crucial because a linking group can increase the overall molecular length and polarizable anisotropy of mesogen and hence may provide favorable geometry of molecule [19]. For example, ester group is considered as a versatile and most commonly used linking unit in LC materials, which increases the polarizability of molecules and provides relatively stable mesophase. On the other hand, the photochromic azo group can be exploited to control phase behavior and optical properties of LC materials [20]. Furthermore, the efficient and reversible photoisomerization makes azo compounds potential in a variety of applications, such as optical data storage devices, photo-switching materials, nonlinear optics (NLO), and liquid crystal display [21–25]. The incorporation of benzothiazole moiety into an azo backbone enhances some optoelectronic properties compared to their benzenoid analogues [26]. For example, benzothiazole moiety containing azo compounds exhibit large molecular hyperpolarizability which is one of the desired criteria for the NLO materials [27]. Thus, a combination of azo and ester linking groups with benzothiazole moiety in the mesogenic core leads to unique mesomorphic properties as well as may provide multifunctional material with improved properties. Although azo-ester bridged benzothiazole moiety containing LC materials have been investigated for many applications, SCLCPs having azo-ester linked benzothiazole moiety in the mesogenic side chain have rarely been reported. In this connection, we have described the synthesis and characterization of azo-ester connected benzothiazole mesogen containing LC monomers and their SCLCPs. We have also evaluated the effect of terminal electron donating/pushing substituent on the thermal stability, LC phase behavior, and optical and electrochemical properties of the synthesized monomers and polymers.
Experimental
Materials and measurements
All reagents were purchased from Aldrich, Merck, and Acros Organics and used as received, unless otherwise stated. Chlorobenzene, toluene, dimethylformamide (DMF), and tetrahydrofuran (THF) were distilled over sodium/benzophenone under an inert atmosphere prior to use. Initiator benzoyl peroxide (BPO) was recrystallized two times from ethanol and dried under vacuum at 40 °C for 24 h before use.
Nuclear magnetic resonance (NMR) spectra (1H and 13C) were recorded with a JEOL spectrometer (400 MHz, JNM-LA400, Japan). Fourier transform infrared (FT-IR) spectra were taken on a PerkinElmer Spotlight 400 spectrometer (Waltham, MA, USA) with 16 scans using attenuated total reflectance (ATR) method and a resolution of 4 cm−1. The thermal decomposition data were obtained on a SDT Q600 thermogravimetric analyzer (TA Instruments, New Castle, DE, USA) under nitrogen atmosphere at a heating rate of 20 °C min−1. Different thermal transitions (melting, liquid crystalline and glass transition) were performed by differential scanning calorimetry (DSC) using a PerkinElmer DSC6 instrument (Waltham, MA, USA) with scan rate of 10 and 20 °C min−1 for monomers and polymers, respectively. The LC phase transitions were investigated using an Olympus BH-2 (Tokyo, Japan) polarized optical microscope (POM) equipped with a Mettler Toledo hot stage FP-82 (Columbus, OH, USA). Molecular weights were determined on a gel permeation chromatography (GPC) instrument (Waters 2414 refractive index detector coupled with a Waters 717 plus Auto sampler and Waters 600 Controller, all from Waters, Milford, MA, USA) with polystyrene standards as reference and THF as the eluent. UV–vis absorption and fluorescence spectra in dilute chloroform (CHCl3) solution (1 × 10−6 M) were recorded on Cary 60 UV–vis and Cary Eclipse Fluorescence spectrophotometer (Santa Clara, CA, USA). Cyclic voltammetric experiments were conducted using a potentiostat/galvanostat AUTOLAB/PGSTAT302N (Ecochemic, Netherlands) which was run by General Purpose Electrochemical System (GPES) software installed in computer. All measurements were performed with a conventional three-electrode configuration, a working glassy carbon, an auxiliary platinum electrode, and an Ag/AgCl reference electrode with tetrabutylammonium perchlorate (n-Bu4NClO4) in chloroform solution as the supporting electrolyte. The potentials were measured against Ag/AgCl reference electrode, and ferrocene/ferrocenium ion (Fc/Fc+) was used as the internal standard. The solutions were purged with nitrogen before measurement in order to remove traces of oxygen.
Synthesis of monomers
The synthetic routes of monomers M1–M4 are illustrated in Scheme 1. Azo-benzothiazole dyes 1a–1d were synthesized according to the similar method reported earlier [28]. Compounds 4-(6-hydroxyhexyloxy)benzoic acid (2) and 4-(6-methacryloxyhexyloxy)benzoic acid (3) were prepared by the similar procedures described in the literature with modifications [29].

Synthetic pathways of monomers M1–M4 and polymers P1–P4
2-[4′-(4″-(6-Methacryloxyhexyloxy)benzoyloxy)phenylazo]benzothiazole (M1)
In a reaction flask, azo-benzothiazole dye 1a (2.55 g, 10 mmol), compound 3 (3.06 g, 10 mmol), and N,N′-dimethylaminopyridine (DMAP) (0.244 g, 2 mmol) were dissolved a mixture of DMF and DCM (1:1) and stirred in an ice bath. To this mixture, N,N′-dicyclohexylcarbodiimide (DCC) (2.1 g, 10 mmol) dissolved in DCM (10 mL) was added dropwise for half an hour and stirring was continued for another hour in the ice bath. After stirring for 24 h at room temperature, the precipitated solid was filtered out and DCM was removed under reduced pressure, and the resulting concentrated mixture was poured into distilled water (100 mL). The obtained colored product was again extracted with DCM, and the organic phase was washed with water several times, separated and dried over anhydrous magnesium sulfate. DCM was removed under reduced pressure and the obtained product was recrystallized two times from 2-propanol to get the orange-colored crystalline product M1. The monomer M1 was purified by column chromatography using chloroform as eluent and silica gel as stationary phase. Yield 40 %, T m = 103–105 °C; FT-IR (cm−1): 3063 (C–H, aromatic), 2938, 2909, 2866 (C–H, aliphatic), 1717 (C = O in benzoate), 1706 (C = O in methacrylate), 1639 (C = C), 1604 (C = N), 1578 (C–C, aromatic), 1491, 1472, 1456 (–N = N–), 1396 (C–N), 1315, 1301, 1259 (C–O), 1220, 1195, 1167, 1122, 1106, 1060 (benzothiazole), 1005, 939, 844, 804, 756, 728, 691, 665 (C–S–C); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.18–8.05 (m, 5H, benzo-H + Ar-H), 7.90–7.83 (d, 1H, benzo-H), 7.56–7.39 (m, 4H, benzo-H + Ar–H), 6.99–6.91 (d, 2H, Ar–H), 6.08 (s, 1H, =CH2), 5.54 (s, 1H, =CH2), 4.18–4.12 (t, 2H, –OCH2–), 4.06–3.99 (t, 2H, –OCH2–), 1.92 (s, 3H, –CH3), 1.89–1.77 (m, 2H, –CH2–), 1.77–1.66 (m, 2H, –CH2–),1.68–1.40 (m, 4H, –CH2–), 13C NMR (400 MHz, CDCl3) δ (ppm): 175.6 (–O–CO–), 167.5 (benzo-C–N = N–), 164.3 (Ar–O–CO–Ar), 163.8 (Ar–C–O–), 155.3 (Ar–C–O–CO–), 152.7 (benzo-C–N), 149.2 (benzo-C–S), 136.5 (–C(CH3) = CH2), 134.4 (Ar–C–N = N–), 132.5, 131.5, 127.6, 126.7, 126.7, 125.7,125.0, 122.9, 122.3, 121.0, 114.4, 114.0 (Ar–C), 125.2 (–C(CH3) = CH2), 68.2 (–OCH2–), 64.7 (–CH2–O–), 30.9, 29.0, 28.7, 28.6, 25.8, 25.7, 18.4 (aliphatic-C).
2-[4′-(4″-(6-Methacryloxyhexyloxy)benzoyloxy)phenylazo]-6-methylbenzothiazole (M2)
Monomer M2 was synthesized by the esterification reaction of azo-benzothiazole dye 1b with compound 3 according to similar synthetic techniques described for M1. Orange-yellow crystal, yield 25 %, T m = 110–112 °C; FT-IR (cm−1): 3060 (C–H, aromatic), 2940, 2910, 2867 (C–H, aliphatic), 1720 (C = O in benzoate), 1705 (C = O in methacrylate), 1640 (C = C), 1608 (C = N), 1579 (C–C, aromatic), 1492, 1475, 1429 (–N = N–), 1396 (C–N), 1315, 1301, 1261 (C–O), 1222, 1182, 1168, 1125, 1107, 1060 (benzothiazole), 1006, 942, 844, 805, 756, 730, 691, 655 (C–S–C); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.22–8.13 (m, 4H, Ar–H), 8.09–8.04 (d, 1H, benzo-H), 7.70 (s, 1H, benzo-H), 7.49–7.42 (d, 2H, Ar-H), 7.39–7.34 (d, 1H, benzo-H), 7.03–6.98 (d, 2H, Ar–H), 6.11(s, 1H, =CH2), 5.56 (s, 1H, =CH2), 4.21–4.14 (t, 2H, –OCH2–), 4.11–4.01 (t, 2H, –OCH2–), 2.53 (s, 3H, –CH3), 1.95 (s, 3H, –CH3), 1.91–1.79 (m, 2H, –CH2–), 1.78–1.68 (m, 2H, –CH2–),1.67–1.43 (m, 4H, –CH2–), 13C NMR (400 MHz, CDCl3) δ (ppm): 174.7 (–O–CO–), 167.5 (benzo-C–N = N–), 164.3 (Ar–O–CO–Ar), 163.8 (Ar–C–O–), 155.3(Ar–C–O–CO–), 150.9 (benzo-C–N), 149.3 (benzo-C–S), 136.5 (–C(CH3) = CH2), 138.2 (CH3-Ar-C), 134.4 (Ar–C–N = N–), 132.4, 128.4, 125.7,124.6, 122.8, 122.0, 121.0, 114.4 (Ar–C), 125.2 (–C(CH3) = CH2), 68.2 (–OCH2–), 64.6 (–CH2–O–), 29.0, 28.6, 25.8, 25.7, 21.9, 18.3 (aliphatic-C).
2-[4′-(4″-(6-Methacryloxyhexyloxy)benzoyloxy)phenylazo]-6-methoxybenzothiazole (M3)
Monomer M3 was synthesized by the esterification reaction of azo-benzothiazole dye 1c with compound 3 according to similar synthetic techniques described for M1. Orange crystal, yield 28 %, T m = 120–122 °C, FT-IR (cm−1): 3062 (C–H, aromatic), 2938, 2867, 2833(C–H, aliphatic), 1719 (C = O in benzoate), 1708 (C = O in methacrylate), 1636 (C = C), 1595 (C = N), 1579 (C–C, aromatic), 1496, 1476, 1436 (–N = N–), 1393 (C–N), 1325, 1310, 1264 (C–O), 1217, 1194, 1164, 1129, 1103, 1062 (benzothiazole), 1008, 988, 937, 876, 843, 760, 725, 691, 652 (C–S–C); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.08–8.01 (m, 4H, Ar–H), 7.98–7.93 (d, 1H, benzo-H), 7.34–730 (d, 2H, Ar–H), 7.22 (s, 1H, benzo-H), 7.05–7.01 (d, 1H, benzo-H), 6.91–6.85 (d, 2H, Ar–H), 6.00(s, 1H, =CH2), 5.55 (s, 1H, =CH2), 4.09–4.03 (t, 2H, –OCH2–), 3.98–3.92 (t, 2H, –OCH2–), 3.81 (s, 3H, –OCH3) 1.84 (s, 3H, –CH3), 1.79–1.69 (m, 2H, –CH2–), 1.67–1.57 (m, 2H, –CH2–), 1.50–1.33 (m, 4H, –CH2–); 13C NMR (400 MHz, CDCl3) δ (ppm): 173.3 (–O–CO–), 167.5 (benzo-C–N = N–), 164.3 (Ar–O–CO–Ar), 163.7 (Ar–C–O–), 159.7 (CH3O–Ar–C), 154.9 (Ar–C–O–CO–), 149.3 (benzo-C–N), 147.5 (benzo-C–S), 136.5 (–C(CH3) = CH2), 136.4 (Ar–C–N = N–), 132.4, 125.8, 125.4, 122.8, 121.1, 116.6, 104.3 (Ar–C), 125.2 (–C(CH3) = CH2), 68.2 (–OCH2–), 64.6 (–CH2–O–), 55.9 (–OCH3) 30.9, 29.0, 28.9, 28.6, 25.8, 25.7, 18.3 (aliphatic-C).
2-[4′-(4″-(6-Methacryloxyhexyloxy)benzoyloxy)phenylazo]-6-ethoxybenzothiazole (M4)
Monomer M4 was synthesized by the esterification reaction of azo-benzothiazole dye 1d with compound 3 according to similar synthetic techniques described for M1. Orange crystal, yield 35 %, T m = 126–128 °C, FT-IR (cm−1): 3087 (C–H, aromatic), 2949, 2896 (C–H, aliphatic), 1726 (C = O in benzoate), 1707 (C = O in methacrylate), 1644 (C = C), 1603 (C = N), 1572 (C–C, aromatic), 1495, 1477, 1460 (–N = N–), 1404 (C–N), 1329, 1310, 1261 (C–O), 1222, 1197, 1165, 1143, 1116, 1060 (benzothiazole), 1000, 940, 897, 839, 760, 732, 688, 655 (C–S–C); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.15–8.05 (m, 4H, Ar–H), 8.2–7.97 (d, 1H, benzo-H), 7.41–7.34 (d, 2H, Ar-H), 7.27–7.23 (d, 1H, benzo-H), 7.09–7.04 (m, 1H, benzo-H), 6.96–6.90 (d, 2H, Ar–H), 6.04(s, 1H, =CH2), 5.49 (s, 1H, =CH2), 4.15–3.98 (m, 6H, –OCH2–), 1.88 (s, 3H, –CH3), 1.83–1.74 (m, 2H, –CH2–), 1.72–1.64 (m, 2H, –CH2–),1.57–1.38 (m, 7H, –CH3 and –CH2–); 13C NMR (400 MHz, CDCl3) δ (ppm): 173.2 (–O–CO–), 167.5 (benzo-C–N = N–), 164.3 (Ar–O–CO–Ar), 163.7 (Ar–C–O–), 159.1 (CH3O–Ar–C), 154.8 (Ar–C–O–CO–), 149.3 (benzo-C–N), 147.5 (benzo-C–S), 136.5 (–C(CH3) = CH2), 136.4 (Ar–C–N = N–), 132.4, 131.5, 125.9, 125.4, 123.0, 122.8, 121.1, 117.0, 114.4, 114.0 104.9 (Ar–C), 125.2 (–C(CH3) = CH2), 68.2 (–OCH2–), 64.6 (–CH2–O–), 64.2 (–OCH2), 29.0, 28.9, 28.7, 28.6, 25.8, 25.7, 18.3, 14.8 (aliphatic-C).
Synthesis of polymers
The synthetic pathways of polymers P1–P4 are outlined in Scheme 1. In a Schlenk tube, monomer (2 mmol) and BPO (5 mol.% with respect to the monomer) were dissolved in anhydrous chlorobenzene (5 mL). The reaction mixture was degassed by several vacuum/nitrogen cycles. The resulting solution was heated at 90 °C in a thermostated oil bath and stirred at this temperature for 24 h. The polymer mixture was then cooled to room temperature and poured into large excess of ethanol under vigorous stirring. The precipitated polymer was filtered off and purification was carried out by dissolving the polymer in chloroform and re-precipitated it in excess hot ethanol until complete removal of any unreacted monomers.
Results and discussion
Synthesis and structural characterization
LC monomers M1–M4 were synthesized via multistepped reactions: azo-coupling, etherification, condensation, and esterification reactions (Scheme 1). The orange-colored crystalline products (M1–M4) were obtained in fairly good yield (25–40 %) and were soluble in common organic solvents including THF, DMSO, CHCl3, CH2Cl2, and chlorobenzene. The chemical structure of the synthesized monomers was confirmed by FT-IR, 1H, and 13C NMR spectroscopic techniques. Azo-ester bridged benzothiazole mesogen containing methacrylate monomers M1–M4 were polymerized via conventional free radical polymerization method at 90 °C using BPO as initiator and chlorobenzene as solvent (Scheme 1). The weight average molecular weights (M w) and polydispersity indices (PDI) of the synthesized polymers were obtained from GPC measurements in the range of 12,300–13,900 and 1.62–1.66, respectively. Although adopting a high initiator concentration (5 mol.% with respect to the monomer) and elevated temperature (90 °C), the conversion rate (30–40 %) and the obtained molecular weights of the prepared polymers are not satisfactory. However, these values are typical for azo chromophore-containing polymers [30, 31]. The lower molecular weights of polymers may be linked to a relatively high concentration of growing chain radicals in the reaction mixture, as a result of the high BPO/monomer ratio adopted, which could favor the termination of reactions [14].The prepared polymers were obtained as orange colored solid, which were soluble in DMSO, THF, CHCl3, and CH2Cl2. The chemical structure and purity of the prepared polymers were investigated by FT-IR and 1H NMR spectroscopies.
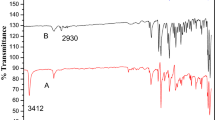
The FT-IR spectra of monomer M1 and its polymer P1 are depicted in Fig. 1. The band at 1639 cm−1 corresponding to the stretching vibration of C = C bond in methacrylate group disappeared completely after polymerization. Furthermore, the stretching vibrational band of C = O in methacrylate functional group shifted from 1706 to 1733 cm−1 due to the reduced electron delocalization in carbonyl group. These observations indicate that C = C bond in terminal methacrylate group was involved in the polymerization.

FT-IR spectra of monomer M1 and polymer P1
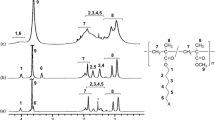
Figure 2 shows 1H NMR spectra of monomer M1 and its corresponding polymer P1. It can be clearly seen that the characteristic vinylidene proton peaks of monomer M1 located at δ = 5.54 ppm and δ = 6.08 ppm have disappeared completely from corresponding polymer (P1) spectrum. Additionally, the chemical shifts of all protons in polymer P1 became quite broad, which are consistent with the expected polymer structure. Moreover, a new chemical shift is observed at 1.04 ppm in P1 spectrum, which is due to the methylene (–CH2–) protons of polymer backbone (main chain). The complete disappearance of C = C stretching vibrational band as well as the chemical shifts of vinylidene protons from FT-IR and 1H NMR spectra respectively confirms the total removal of unreacted monomer (M1) from the purified polymer (P1). Similar spectral behaviors were also observed for other polymers (P2–P4).

1H NMR spectra of monomer M1 and its corresponding polymer P1 in CDCl3
Thermal properties
The TG and DTG traces of polymer P1 are depicted in Fig. 3, and the relevant thermal analysis data of monomers M1–M4 and SCLCPs P1–P4 are summarized in Table 1. All the monomers (M1–M4) and polymers (P1–P4) exhibited two-staged thermal decomposition. The first-staged decomposition of M1–M4 was observed from 223 to 342 °C while polymers P1–P4 were decomposed around 293–359 °C with estimated mass losses of 8–11 %. These cleavages of monomers and polymers may be due to the thermal decomposition of azo-heterocyclic segments located in the mesogen [32, 33]. In the second stage, M1–M4 were degraded around 342–541 °C and their polymers P1–P4 were decomposed from 352 to 537 °C with estimated weight losses of 59–64 %. The second-staged decomposition may be ascribed to the thermal degradation of aliphatic segments and heat resistant aromatic moiety [34, 35].

TG and DTG traces of polymer P1 recorded at a heating rate of 20 °C min−1 under nitrogen atmosphere
The thermal decomposition temperatures corresponding to 5 % weight losses (T d) of monomers and polymers are in the range of 316–324 and 337–343 °C, respectively. These observations reveal that the synthesized monomers and polymers have high thermal stability [36, 37]. However, the thermal stability of polymers P1–P4 was ca. 20 °C higher than those of corresponding monomers M1–M4. This result may be attributed to the fact that the introduction of the mesogenic groups as side chains into the poly-methacrylate backbone has enhanced the thermal stability of the polymers [38]. The char yields of M1–M4 and P1–P4 were estimated around 25–32 % at 600 °C, supporting the oxidative stability of the monomers and polymers [39].
Liquid crystalline phase behaviors
The thermal transition temperatures and LC behaviors of monomers and polymers were investigated by DSC and POM. The DSC traces of monomers M1–M4 are illustrated in Fig. 4, and the thermal transition results are presented in Table 2. Monomer M1 exhibited enantiotropic phase transition during heating and cooling processes. The isotropization temperatures of monomers M2–M4 are equal/greater than their first decomposition temperatures determined from TGA measurements (Table 1). As a result, no distinct LC phase transition peaks of M2–M4 were identified during cooling scan due to the partial decomposition of the monomers. Wei et al. also reported similar observation for their studied compounds [40]. Thus, only first heating data of monomers M2–M4 are considered for further discussion. It can be seen from Fig. 4 that monomer M1 exhibited three thermal transitions during heating scan: (i) a crystal to smectic at 104.8 °C, (ii) smectic to nematic at 143.3 °C, and (iii) nematic to isotropic at 197.2 °C. Similarly, M1 showed three thermal transitions on cooling process: (i) isotropic to nematic at 193.6 °C, (ii) nematic to smectic at 140.7 °C, and (iii) crystallization at 53.3 °C.

DSC curves of monomers M1 (a) and M2–M4 (b) at heating and cooling rates of 10 °C min−1
On the other hand, monomer M2 showed two thermal transitions: (i) a crystal to nematic at 106.6 °C and (ii) nematic to isotropic at 229.6 °C. Similarly, monomer M3 displayed a crystal to nematic transition at 121.0 °C and (ii) nematic to isotropic transition at 236.7 °C. Likewise M2 and M3, monomer M4 showed two thermal transitions: (i) crystal to nematic at 127.9 °C and (ii) nematic to isotropic at 232.5 °C. The observed POM textures of M1–M4 are demonstrated in Fig. 5. The identification of nematic and smectic phases was made by comparison of the detected textures with those reported in the literatures. The optical photomicrographs of monomer M1 were recorded during cooling scan whereas POM images of M2–M4 were taken during heating. Monomer M1 exhibited schlieren texture of nematic phase at 193.2 °C upon cooling from isotropic liquid and further cooling schlieren texture of smectic C phase appeared at 140.5 °C. The identification of SmC phase was made on the basis of the characteristic grey schlieren texture (Fig. 5b) which appeared during nematic to smectic C transition [41]. During heating, monomer M2 melted around 107.0 °C and upon further heating schlieren texture of nematic phase with fourfold brushes (Fig. 5c) started appearing and the image was taken at 225.5 °C. Monomers M3 and M4 also exhibited schlieren texture of nematic phases upon heating scan, and the POM images of M3 and M4 were recorded at 229.4 and 226.5 °C, respectively. Monomer M1 exhibited both nematic and smectic phases whereas M2–M4 showed only nematic phase. The polarization or electron distribution in electron-deficient benzothiazole moiety may be affected by the electron-donating substituent (CH3, OCH3, and OC2H5) which could facilitate the formation of nematic phase.

POM images of monomers M1–M4: M1 displays schlieren texture of nematic phase at 193.2 °C and smectic C phase at 140.5 °C (a, b); M2 shows nematic phase at 225.5 °C (c); M3 exhibits nematic phase at 229.4 °C (d); and M4 reveals nematic phase at 226.5 °C (e) (magnification × 50)
Replacement of hydrogen atom by methyl, methoxy, and ethoxy groups at the sixth position on benzothiazole moiety has substantial influence on mesophase stability. Methyl-substituted compound (M2) exhibited greater mesophase stability (123.0 °C) than those of methoxy-substituted (109.7 °C) and ethoxy-substituted (100.6 °C) compounds (M3 and M4). The reduced nematic phase stability of monomers M3 and M4 may be attributed to the fact that the oxygen being in conjugation with the heteroaromatic core extends the length of the rigid core as well as enhances the polarizability anisotropy [42]. As the isotropization temperature of polymers P1–P4 is almost equal or greater than the first decomposition temperature (Table 1) determined from TGA analysis, the DSC curve of polymers P1–P4 started declining after isotropization point. As a result, no distinct DSC peaks was observed during cooling process. Only glass transition temperature (T g) was clearly identified from DSC curve during first heating cycle (Table 3). The optical photomicrographs of polymers P1–P4 are depicted in Fig. 6. All the POM images were recorded during heating process and each polymer exhibited nematic LC phase. The POM images of P1–P4 were taken at 200.5, 210.8, 220.4, and 215.6 °C, respectively.

POM images of polymers P1–P4: P1 shows nematic phase at 200.5 °C (a); P2 exhibits nematic phase at 210.8 °C (b); P3 displays nematic phase at 220.4 °C (c); P4 reveals nematic phase at 215.6 °C (d) (magnification × 50)
Optical properties of polymers
The UV–vis absorption spectra of SCLCPs P1–P4 in dilute chloroform solutions (1 × 10−6 M) are illustrated in Fig. 7, and their spectral results are presented in Table 4. All the polymers showed two absorption bands in their UV–vis spectra: (i) a high-energy absorption bands around 245–295 nm and (ii) a low-energy absorption bands about 320–520 nm. The absorption maxima (λ max) of short wavelength region are in the range of 260–262 nm, and this high-energy absorption band may be attributed to a π–π* electronic transition of the heterocyclic moiety and phenyl rings [43, 44]. On the other hand, the absorption maxima (λ max) of longer wavelength region are in the range of 376–413 nm and this low-energy absorption band may be regarded as a π–π* transition involving the π-electronic system throughout the whole molecule with a considerable charge transfer (CT) character [45, 46]. The absorption maxima (λ max) of polymers P2–P4 in the longer wavelength region were red-shifted compared to polymer P1 by 12, 35, and 37 nm, respectively, with the influence of electron donating/pushing terminal substituent located on the benzothiazole moiety. This result may be ascribed to the fact that the electron donating/pushing group tends to provide extra electron density toward benzothiazole moiety through resonance effect which could reduce HOMO-LUMO energy gaps of the molecules.

UV–vis absorption spectra of P1–P4 in dilute CHCl3 solutions (1 × 10−6 M)
The normalized fluorescence spectra of SCLCPs P1–P4 in dilute chloroform solutions (1 × 10−6 M) are depicted in Fig. 8, and the relevant data are summarized in Table 4. The emission spectra of polymers P1–P4 are identical pattern because of the structural similarities in the mesogenic side chain. The fluorescence emission maxima of polymers are in the range of 522–524 nm which may be categorized as green emission. Polymer P3 exhibited highest fluorescence intensity among all the studied polymers. The electron donating/pushing terminal substituent situated on benzothiazole moiety may affect the HUMO-LUMO energy gaps which could play crucial role on the fluorescence intensities of the polymers. The highest fluorescence intensities of polymers P3 may be due to the largest electron pushing power of OCH3 group whereby resonance effect on the benzothiazole moiety could probably extend conjugation. The obtained fluorescence intensities of polymers P1–P4 are comparable with standard compound pyrene (see inset in Fig. 8). As a result, SCLCPs P1–P4 may be potential candidate as fluorescent materials in polymer light emitting diode (PLED) applications. Azo chromophore based materials are generally considered useful for their fascinating photochemistry. Specially, π-conjugated system containing azo compounds absorb UV and/or visible portion of the electromagnetic spectrum and the absorption maxima can be shifted with the appropriate ring substitution in the benzene ring. The most interesting property of azo compounds is the reversible trans-cis photoisomerism upon absorption of a photon within the absorption band. The lifetime of the cis state may be elongated by bulky ring substitution and it is the crucial factor for the efficient photoisomerism azo derivatives [47]. In addition, conformational strain or steric hindrance also plays a vital role to lock the cis state, which may delay the isomerism process [48]. In case of SCLCPs P1–P4, benzothiazole molecule is directly attached to the azo (–N = N–) linking group and benzothiazole moiety is relatively bulkier than benzene ring. As a result, photoisomerism of polymers P1–P4 could be quenched or suppressed due to the conformational strain and/or steric hindrance. Although limited number of azo compounds exhibit fluorescence property, SCLCPs P1–P4 showed green fluorescence emission in solution due to the presence of benzothiazole moiety. Moreover, suppression of photoisomerism could also facilitate strong fluorescence emission of the polymers.

Fluorescence spectra of P1–P4 in dilute CHCl3 solutions (1 × 10−6 M). Inset shows fluorescence spectrum of standard compound pyrene
Electrochemical properties of polymers
The redox properties of the newly synthesized SCLCPs P1–P4 were investigated by cyclic voltammetry to estimate the HOMO and LUMO energy levels. These energy levels are crucial for determining the band gaps as well as the selection of cathode and anode materials for OLED devices. The measurements were carried out in anhydrous CHCl3 solution containing 0.1 M tetrabutylammonium perchlorate (n-Bu4NClO4) as supporting electrolyte. The conventional three electrodes system was employed to carry out the measurements: Ag/AgCl, platinum wire, and glassy carbon served as reference, counter, and working electrodes, respectively. All the measurements were performed at room temperature in the potential range from −1.0 to +2.0 V with a scan rate of 50 mV s−1. The external standard potential of the ferrocene/ferricenium ion couple (E Fc/Fc+) was estimated under the same experimental condition and the value was located at 0.30 V to the Ag/AgCl electrode. The onset oxidation potentials of polymers P1–P4 were calculated from the intersection of two tangents drawn at the rising and background currents of the cyclic voltammogram. The typical voltammogram of polymer P1 is shown in Fig. 9. The oxidation potentials of polymers P1–P4 are characterized by an irreversible wave with onset oxidation potentials at 0.37, 0.35, 0.32, and 0.30 V respectively. The HOMO energy levels of polymers can be calculated by using the empirical equation E HOMO = −[E ox (onset) − E Fc/Fc+ + 4.8] eV, where E ox represents the onset oxidation potential [49]. The HOMO energy levels of the polymers P1–P4 were estimated to be −4.87, −4.85, −4.82, and −4.80 eV, respectively. The LUMO energy levels of the polymers can be calculated by equation E LUMO = [E HOMO + E g (opt.)] eV, where optical band gap (E g) is estimated from the onset of the absorption spectra for polymers. The LUMO energy levels of polymers P1–P4 were found to be −2.14, −2.22, −2.25, and −2.36 eV, respectively. Table 4 lists the HOMO-LUMO energy levels of polymers P1–P4 and their corresponding band gap values. The HOMO energy levels of the polymers increase whereas the LUMO energy levels of the polymers decrease by the electronic effect of electron pushing terminal substituents situated on the mesogenic side chain.

Cyclic voltammogram (CV) of polymer P1 in dilute CHCl3 solution (1 × 10−3 M) with 0.1 M tetrabutylammonium perchlorate (Bu4NClO4) as supporting electrolyte. Inset and dotted line show CV of ferrocene and blank respectively run under identical condition as polymer
Conclusions
Azo-ester linked benzothiazole moiety containing new polymerizable LC monomers M1–M4 and their SCLCPs P1–P4 having different terminal substituents were successfully synthesized and characterized. Polymers P1–P4 exhibited improved thermal stability of ca. 20 °C compared to their corresponding monomers M1–M4. All the synthesized monomers and polymers exhibited LC properties. The LC phase transitions and stability of mesophase were influenced by the terminal substituents located on the benzothiazole moiety. The absorption maxima of polymers in the longer wavelength region were red-shifted with the incorporation of electron donating/ pushing terminal substituents situated on the benzothiazole moiety of the mesogenic side chain. All the polymers exhibited strong green fluorescence emission. The HOMO and LUMO energy levels of polymers were also affected by the electron donating/pushing terminal substituents. The newly synthesized SCLCPs could be potential candidate as fluorescent materials in the polymer light emitting diode (PLED) applications.
References
Cui Z, Zhang Y, He S (2008) Synthesis of a side chain liquid crystalline polymer containing the cholesteryl moiety via ROP and “click” chemistry. Colloid Polym Sci 286:1553–1559
Wang JW, Zhang BY (2013) Effect of fluorinated nematic mesogens on phase behaviorsand optical properties of chiral liquid crystalline polysiloxanes. Colloid Polym Sci 291:2917–2925
WewerkaA VK, VlassopoulosD SF (2001) Structure and rheology of model side-chain liquid crystalline polymers with varying mesogen length. Rheol Acta 40:416–425
Xiao W, Zhang B, Cong Y (2008) Preparation and characterization of side-chain liquidcrystalline polymers containing chenodeoxycholicacid residue. Colloid Polym Sci 286:267–274
Watanabe M, Tsuchiya K, Shinnai T, Kijima M (2012) Liquid crystalline polythiophene bearing phenylnaphthalene side-chain. Macromolecules 45:1825–1832
Misra G, Srivastava AK (2008) Liquid crystalline side chain polymer with a poly(Geraniol-co-MMA) backbone and phenylbenzoatemesogenic group: synthesis and characterization. Colloid Polym Sci 286:445–451
Cui L, Zhao Y (2004) Azopyridine side chain polymers: an efficient way to prepare photoactive liquid crystalline materials through self-assembly. Chem Mater 16:2076–2082
KarimMR SMRK, YahyaR SNM, AzzahariAD HA, Sarih NM (2013) Thermal, optical and electrochemical study of side chain liquid crystalline polymers bearing azo-benzothiazole chromophore in the mesogen. J Polym Res 20:1–7
FuHY YXT, ZhongGY ZZY, Xiao F (2010) White organic light-emitting diodes based on benzothiazole derivative. Curr Appl Phys 10:1326–1330
BingolH KE, ZorE CA (2010) A novel benzothiazole based azocalix[4]arene as a highly selective chromogenic chemosensor for Hg2+ ion: a rapid test application in aqueous environment. Talanta 82:1538–1542
FaustinoH SRME, ReisLV SPF, Almeida P (2008) 2-Nitrosobenzothiazoles: useful synthons for new azobenzothiazole dyes. Tetrahedron Lett 49:6907–6909
Ono M, Hayashi S, Kimura H, Kawashima H, Nakayama M, Saji H (2009) Push–pull benzothiazole derivatives as probes for detecting β-amyloid plaques in Alzheimer’s brains. Bioorg Med Chem 17:7002–7007
Iwan A, Palewicz M, Krompiec M, Grucela-Zajac M, Schab-Balcerzak E, Sikora A (2012) Synthesis, materials characterization and opto(electrical) properties of unsymmetrical azomethines with benzothiazole core. Spectrochim Acta A 97:546–555
Cojocariu C, Rochon P (2004) Synthesis and optical storage properties of a novel polymethacrylate with benzothiazole azo chromophore in the side chain. J Mater Chem 14:2909–29016
Tokunaga K, Takayashiki Y, Iino H, Hanna J (2009) Electronic conduction in nematic phase of smallmolecules. Phys Rev B 79:1–4
Thaker BT, Patel BS, Dhimmar YT, Chothani NJ, Solanki DB, Patel N, Patel KB, Makawana U (2013) Mesomorphic studies of novel azomesogens having a benzothiazole core: synthesis and characterisation. Liq Cryst 40:237–248
FunahashiM HJ (1997) Fast hole transport in a newcalamitic liquid crystal of 2-(4-heptyloxyphenyl)-6-dodecylthiobenzothiazole. Phys Rev Lett 78:2184–2187
DuttaGK GS, Patil S (2010) Synthesis of liquid crystalline benzothiazole based derivatives: a study of their optical and electrical properties. Org Electron 11:1–9
Dunmur DA (2002) Liquid crystals: fundamentals. World Scientific, Singapore
PrasadSK NGG, Rao DSS (2009) Photoinduced phase transitions. Liq Cryst 36:705–716
Åstrand PO, Ramanujam PS, Hvilsted S, Bak KL, Sauer SPA (2000) Ab initio calculation of the electronic spectrum of azobenzene dyes and its impact on the design of optical data storage materials. J Am Chem Soc 122:3482–3487
Kang X, Zhao J, Li H, He S (2013) Synthesis of a main-chain liquid crystalline azo-polymer via “click” chemistry. Colloid Polym Sci 291:2245–2251
Gibbons WM, Shannon PJ, Sun ST, Swetlin BJ (1991) Surface-mediated alignment of nematic liquid crystals with polarized laser light. Nature 351:49–50
Li CE, Zhong SA, Li XJ, Guo M (2013) Silica particles coated with azobenzene-containing photoresponsive molecule-imprinted skin layer. Colloid Polym Sci 291:2049–2059
Luk YY, Abbott NL (2003) Surface-driven switching of liquid crystals using redox-active groups on electrodes. Science 301:623–626
Yu G, Yin S, Liu Y, Shuai Z, Zhu D (2003) Structures, electronic states, and electroluminescent properties of a Zinc(II) 2-(2-hydroxyphenyl)benzothiazolate complex. J Am Chem Soc 125:14816–14824
Wang X, Yang K, Kumar J, Tripathy SK, Chittibabu KG, Li L, Lindsay G (1998) Heteroaromatic chromophore functionalized epoxy-based nonlinear optical polymers. Macromolecules 31:4126–4134
Karim MR, Sheikh MRK, Salleh NM, Yahya R, Hassan A, Hoque MA (2013) Synthesis and characterization of azo benzothiazole chromophore based liquid crystal monomers: effects of substituents on benzothiazole ring and terminal group on mesomorphic, thermal and optical properties. Mater Chem Phys 140:543–552
Portugall M, Ringsdorf H, Zentel R (1982) Synthesis and phase behaviour of liquid crystalline polyacrylates. Makromol Chem 183:2311–2321
TourasanidisEV KGP (1999) Synthesis and characterization of new polymethacrylates bearing an azo-dye in the side chain. J Macromol Sci A36:1241–1258
Tsutsumi O, Kitsunai T, Kanazawa A, Shiono T, Ikeda T (1998) Photochemical phase transition behavior of polymer azobenzene liquid crystals with electron-donating and -accepting substituents at the 4,4‘-positions. Macromolecules 31:355–359
Karipcin F, Kabalcilar E, Ilican S, Caglar Y, Caglar M (2009) Synthesized some 4-(2-thiazolylazo)resorcinol complexes: Characterization, thermal and optical properties. Spectrochim Acta A 73:174–180
Rameshbabu K, Kannan P (2004) Synthesis and characterization of thermotropic liquid crystalline polyphosphates containing photoreactive moieties. Liq Cryst 31:843–851
Chen BK, Tsay YS, Chen JY (2005) Synthesis and properties of liquid crystalline polymers with low Tm and broad mesophase temperature ranges. Polymer 46:8624–8633
Cui Y, Qian G, Chen L, Wang Z, Wang M (2008) Synthesis and nonlinear optical properties of a series of azo chromophore functionalized alkoxysilanes. Dyes Pigments 77:217–222
Wang Y, Zhang BY, He XZ, Wang JW (2007) Side-chain cholesteric liquid crystalline polymers containingmenthol and cholesterol—synthesis and characterization. Colloid Polym Sci 285:1077–1084
Lam JWY, Kong X, Dong Y, Cheuk KKL, Xu K, Tang BZ (2000) Synthesis and properties of liquid crystalline polyacetylenes with different spacer lengths and bridge orientations. Macromolecules 33:5027–5040
Zhang XA, Zhao H, Gao Y, Tong J, Shan L, Chen Y, Zhang S, Qin A, Sun JZ, Tang BZ (2011) Functional poly(phenylacetylene)s carrying azobenzene pendants: polymer synthesis, photoisomerization behaviors, and liquid-crystalline property. Polymer 52:5290–5301
Kumaresan S, Kannan P (2003) Substituent effect on azobenzene-based liquid-crystalline organophosphorus polymers. J Polym Sci Pol Chem 41:3188–3196
Wei Q, Shi L, Cao H, Wang L, Yang H, Wang Y (2008) Synthesis and mesomorphic properties of two series of new azine‐type liquid crystals. Liq Cryst 35:581–585
Guillevic MA, Light ME, Coles SJ, Gelbrich T, Hursthouse MB, Bruce DW (2000) Synthesis of dinuclear complexes of rhenium(I) as potential metallomesogens. Dalton T 9:1437–1445
Collings PJ, Hird M (1997) Introduction to liquid crystals: chemistry and physics. Taylor & Francis, USA
Issa RM, Khedr AM, Rizk HF (2005) UV–vis, IR and 1H NMR spectroscopic studies of some Schiff bases derivatives of 4-aminoantipyrine. Spectrochim Acta A 62:621–629
Gup R, Giziroglu E, Kırkan B (2007) Synthesis and spectroscopic properties of new azo-dyes and azo-metal complexes derived from barbituric acid and aminoquinoline. Dyes Pigments 73:40–46
Mabrouk A, Azazi A, Alimi K (2010) On the properties of new benzothiazole derivatives for organic light emitting diodes (OLEDs): a comprehensive theoretical study. J Phys Chem Solids 71:1225–1235
Shi HP, Shi LW, Dai JX, Xu L, Wang MH, Wu XH, Fang L, Dong C, Choi MMF (2012) Synthesis, photophysical and electrochemical properties and theoretical studies on three novel indolo[3,2-b]carbazole derivatives containing benzothiazole units. Tetrahedron 68:9788–9794
Barrett CJ, Mamiya J-I, Yager KG, Ikeda T (2007) Photo-mechanical effects in azobenzene-containing soft materials. Soft Matter 3:1249–1261
Han M, Ishikawa D, Muto E, Hara M (2009) Isomerization and fluorescence characteristics of sterically hindered azo benzene derivatives. J Lumin 129:1163–1168
Pommerehe J, Vestweber H, Guss W, Mahrt RF, Bassler H, Porsch M, Daub J (1995) Efficient two layer leds on a polymer blend basis. Adv Mater 7:551–554
Acknowledgments
The authors would like to thank University of Malaya for the financial support through the research grant PV041-2012A and BK0662014.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Karim, M.R., Sheikh, M.R.K., Yahya, R. et al. Synthesis of polymerizable liquid crystalline monomers and their side chain liquid crystalline polymers bearing azo-ester linked benzothiazole mesogen. Colloid Polym Sci 293, 1923–1935 (2015). https://doi.org/10.1007/s00396-015-3578-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-015-3578-8