
Abstract
A synthesis strategy for low molecular weight organogelators using the ureidopyrimidinone (UPy) group is reported. The prepared gelators showed robust thermal reversible gelation abilities in various solvents, including dimethyl sulfoxide. The morphology of the dried gels was determined using scanning electron microscopy, revealing a macroscopic porous structure of the gels. Rheology was performed to determine storage (G′) and loss modulus (G″) confirming network gel structures.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Low molecular weight organogelators (LMOGs) are organic molecules that possess applications in a large number of areas including food, medicine, materials science, cosmetics, and pharmacology [1]. The molecular weights of LMOGs typically range from 300 to 1,000 Da, and LMOGs exhibit striking self-assembly properties through non-covalent interactions such as hydrogen-bonding, π–π stacking, donor-acceptor interactions, metal coordination, solvation forces, and van der Waals interactions [2, 3]. Gels made using LMOGs are usually produced by heating the gelator in an appropriate organic solvent to afford a homogenous solution and then cooling the solution. Broadly speaking, as the hot solution is cooled the gelators self-assemble, forming a matrix spanning the solvent [4]. Different classes of LMOGs exist including dendritic systems [5], nucleobases [6], metallogels [7], amides/peptides [8], and ureas [9]. LMOGs containing quadruple hydrogen bonding systems are an interesting class of organogelators that form gels by strong complimentary hydrogen bonding recognition. For example, Simeone et al. [10] reported that amphiphilic sugar-modified guanosine derivatives gelled in polar organic solvents. The lipophilic guanosine derivative also provided excellent gelation properties in chloroform and some hydrocarbon solvents [11]. Li’s group [12] developed hydrazide derivatives as a quadruple hydrogen bonding system forming organogels in hydrocarbon solvents. It is interesting to note that despite the fact the ureidopyrimidinone (UPy) group is well-known as an excellent quadruple hydrogen bonding group [13], UPy-based LMOGs have not been widely reported. In one of the few related examples, Cho and coworkers [14] recently reported using a UPy-modified low molecular weight organosilane in ethyl acetate. In contrast to low molecular weight gels, UPy-based macromolecular gels have been more studied, for example, Bon and coworkers reported their studies of gels using UPy functioned polymer latex particles [15], and UPy-modified poly(N-isopropylacrylamide) nanogels [16].
Recently, we reported that an N-alkyl-N,N-linked urea oligomer containing six urea functional groups formed fiber-like aggregates in different solvents [17]. We called the N-alkyl-N,N-linked urea oligomers “N-alkyl urea peptoids” as they share some structural similarities with N-acyl glycine oligomers that are commonly referred to as “peptoids” [18–20]. Our N-alkyl urea linked oligomers are further analogous to peptoids in that a large number of N-alkyl side chains are readily incorporated into the molecules, making N-alkyl urea peptoids ideal candidates for structure/property relationship studies and the synthesis of novel soft materials. Considering our observations of the fiber-like aggregates in the N-alkyl urea peptoid oligomers and inspired by examples of using the UPy group in polymeric gels, we rationalized that combining UPy-moieties with N-alkyl urea oligomers would be a promising route to new organogelators with diverse and functional N-alkyl groups, thus creating a new set of materials. Herein, we report a facile approach to low molecular weight organogels using UPy functionalized N-alkyl urea peptoid dimers. The potential synthetic diversity of N-alkyl urea peptoid oligomers is highly applicable to this type of investigation, where we have compared two different N-alkyl functional groups in the afforded organogelators—namely benzyl and pyrene groups.
Experimental
Materials and methods
All starting reagents were purchased from Aldrich at the highest purity available and used as received unless stated otherwise. 2(6-isocyanatohexylaminocarbonylamino)-6-methyl-4[1H]pyrimidinone (UPy) was synthesized according to previous literature proceedures [21]. 1H and 13C{1H} NMR measurements were recorded in CDCl3 and DMSO(d 6 ) with Si(CH3)4 as an internal standard using a Bruker Ultrashield 400 MHz (100 MHz for 13C); NMR spectra were processed using UXNMR version 2.5 and MestRe-C. Fourier transform infrared (FT-IR) spectra were collected on a Nicolet 6700 spectrometer and analyzed with OMNIC 32 software. Mass spectrometry was performed using a Micromass Q-TOF-2™ spectrometer. Scanning electron microscopy (SEM) investigations were carried out on an XL30-ESEM instrument operating at an energy of 20 and 30 kEV. Gels were carefully placed on an SEM plate and allowed to air dry for a week and further dried in a vacuum oven for another 24 h. The critical gelation concentration values were determined in the following process: Solvent was added to the gelator and then the mixture was heated up to make homogenous ca. 1.0 ml solution in a 3.0-ml vial. A gel was formed once the solution was cooled down to room temperature. The vial was inverted each time to ensure the absence of any observed flow in the solutions. This process was repeated until the gel was not formed upon adding another small volume of solvent. Gel transition temperatures were determined by placing a screw-cap glass vial containing the gel in a temperature controlled water bath and visually observing the temperature at which flow upon tilt occurred on heating with one tenth degree temperature increments. All gel to sol transition temperature experiments were repeated twice. Rheological measurements were made with a TA Instruments ARES-G2 rheometer with a Couette cell consisting of 30 mm diameter cup with a 27.7-mm diameter recessed bob (length, 41.49 mm; minimum sample volume, 5.8 ml). The temperature was controlled using an Advanced Peltier System. An oscillatory time sweep was performed to monitor the modulus change of the gels at first. To prepare the sample, a gel was reheated at 80 °C for 30 min in a 20-ml vial in a temperature controlled stirplate to obtain a homogeneous solution. Then ca. 6 ml of solution was poured into the Couette cell preheated to 80 °C, followed by cooling the solution to 25 °C. The gelation was monitored by oscillatory shear at a strain amplitude of 1 % and a frequency of 1 rad/s. At the initiation of the experiment, the set temperature was changed from 80 °C to 25 °C. The sample was monitored for 24 h and a data point was acquired every 10 min. An oscillatory frequency sweep was conducted immediately after annealing the gel for 24 hours at 25 °C. The test was performed over a frequency range of 100–0.01 rad/s at 25 °C, using a strain amplitude of 1 %. A seperate oscillatory strain sweep was performed over a strain range of 0.01–100 % at a frequency of 1 rad/s at 25 °C. The gel was annealed for 72 h prior to this measurement.
Gelator 1
2(6-isocyanatohexylaminocarbonylamino)-6-methyl-4[1H]pyrimidinone (400 mg, 1.36 mmol) was added to a solution of N,N′-dibenzylethylenediamine (166 μl, 0.68 mmol) in 6 ml of dimethylformamide (DMF). The reaction flask was sealed with a rubber septum, purged with N2 for 30 min, and then stirred at room temperature overnight. After this time, the solvent was removed using a rotary evaporator, the residue dissolved in CH2Cl2 and purified by column chromatography using silica gel (silica gel 60 Å, 70–230 mesh) with CH2Cl2: methanol (20:1 v/v) as the mobile phase. The product was dried in a vacuum oven to afford 560 mg of white solid. Yield: 99 %. 1H NMR (d 6−DMSO): δ(ppm) 1.25–1.41 (m, 16 H, 2 NCH2 (CH 2 )4CH2N), 2.09 (s, 6 H 2 CH 3 ), 3.04–3.12 (m, 12 H, 6 CH 2 N), 4.37 (s, 4 H, 2 NCH 2 Ar), 5.76 (s, 2 H, 2 CH=C); 6.63 (bs, 2 H, 2 PhCH2NCONH), 7.12–7.73 (m, 10 H, 10 CH on phenyl ring), 7.49 (2 CH3CNH), 9.72 (bs, 4 H, 2 NHCONHCH2); 13C NMR (d 6−DMSO, 100 MHz): δ (ppm) 23.62 (2C), 26.51 (2C), 26.56 (2C), 29.63 (2C), 30.17 (2C), 45.42 (2C), 47.53 (2C), 50.39 (2C), 52.70 (2C), 104.94 (2C), 127.29 (2C), 127.58 (4C), 128.77 (4C), 139.47 (2C), 152.17 (2C), 155.28 (2C), 158.11 (4C), 158.52 (2C); FT-IR: (cm−1) ν(NH) = 3,429, ν(alkanes) = 2,997, ν(CO) = 1,667, ν(Phenyl) = 1,436; MS (TOF MS ES+): 827.4633 M + 1
Gelator 2
Methanesulfonyl chloride (225.8 mg, 2.16 mmol) was added to a solution of 1-pyrenemethanol (500 mg, 2.16 mmol) and triethylamine (436.3 mg, 4.32 mmol) in 10 ml of CHCl3. The reaction flask was chilled using an ice bath and the solution stirred for 2 h. After this time, ethylene diamine (64.8 mg, 1.08 mmol) was added and the reaction mixture stirred at room temperature overnight. CHCl3 was removed using a rotary evaporator, the residue dissolved in CH2Cl2 and purified by column chromatography using silica gel (silica gel 60 Å, 70–230 mesh) with CH2Cl2: methanol (20:1 v/v) as the mobile phase. The product N,N′-dipyrene-yl-ethylenediamine was dried in a vacuum oven to afford 312 mg of pale yellow powder. Yield: 59 %. 1H NMR (CDCl3): δ (ppm) 2.10 (bs, 2 H, 2 NH), 2.94 (s, 4 H, NCH 2 CH 2 N), 4.37 (s, 4 H, 2 NCH 2 Ar), 7.83–8.16 (m 18 H, 18 CH on aromatic ring); 13C NMR (CDCl3, 100 MHz): δ (ppm) 47.58 (2C), 50.77 (2C), 122.66 (2C), 124.62 (2C), 124.84 (2C), 125.13 (2C), 125.17 (2C), 125.89 (2C), 126.94 (2C), 127.10 (2C), 127.27 (2C), 127.33 (2C), 127.89 (2C), 127.97 (2C), 130.88 (4C), 131.18 (2C), 131.64 (2C); FT-IR: (cm−1) ν(NH) = 3289, ν(alkenes) = 3,040, ν(alkenes) = 2,831, ν(pyrene-yl) = 1,448; MS (TOF MS ES+): 489.2335 M + 1.
2(6-isocyanatohexylaminocarbonylamino)-6-methyl-4[1H]pyrimidinone (360 mg, 1.23 mmol) was added to a solution of N,N′-dipyrene-yl-ethylenediamine (300 mg, 0.61 mmol) in 6 ml of DMF. The reaction flask was sealed with a rubber septum, purged with N2 for 30 min, and then stirred at room temperature overnight. After this time, the solvent was removed using a rotary evaporator, the residue dissolved in CH2Cl2 and purified by column chromatography using silica gel (silica gel 60 Å, 70–230 mesh) with CH2Cl2: methanol (20:1 v/v) as the mobile phase. The product was dried in a vacuum oven to afford 600 mg of white solid. Yield: 91 %. 1H NMR (d 6−DMSO): δ (ppm) 1.23–1.43 (m, 16 H, 2 NCH2 (CH 2 )4CH2N), 2.07 (s, 6 H 2 CH 3 ), 3.05–3.13 (m, 12 H, 6 CH 2 N), 5.05 (s, 4 H, 2 NCH 2 Ar), 5.75 (s, 2 H, 2 CH = C); 6.85 (bs, 2 H, 2 ArCH2NCONH), 7.37 (2 CH3CNH), 7.58 (d, J = 7.2 Hz, 2 H, 2 CH = CCH2N), 7.95-8.27 (m, 16 H, 16 CH on pyrene-yl), 9.67 (bs, 2 H, 2 NHCONHCH2), 11.56 (bs, 2 H, 2 NHCONHCH2); 13C NMR (d 6−DMSO, 100 MHz): δ (ppm) 23.70 (2C), 26.51 (4C), 29.57 (2C), 30.18 (2C), 41.29 (2C), 44.88 (2C), 48.37 (2C), 56.50 (2C), 104.97 (2C), 123.41 (2C), 124.29 (2C), 124.46 (2C), 124.98 (2C), 125.56 (2C), 125.59 (2C), 126.11 (2C), 126.60 (2C), 127.31 (2C), 127.68 (2C), 127.77 (2C), 128.63 (2C), 130.38 (2C), 130.66 (2C), 131.19 (2C), 132.49 (2C), 151.86 (2C), 155.12 (2C), 158.15 (2C), 162.74 (2C), 165.09 (2C); FT-IR: (cm−1) ν(NH) = 3,435, ν(alkanes) = 2,996, ν(CO) = 1,665, ν(Phenyl) = 1,436; MS (TOF MS ES+): 1,075.5247 M + 1
Compound A
Butyl isocyanate (198 mg, 2.0 mmol) was added to a solution of N,N′-dipyrene-yl-ethylenediamine (240 mg, 1.0 mmol) in 6 ml of DMF. The reaction was performed in the same manner as for gelator 2 and purified by column chromatography using silica gel (silica gel 60 Å, 70–230 mesh) with ethyl acetate as the mobile phase. The product was dried in a vacuum oven to afford 430 mg of white solid. Yield: 98 %. 1H NMR (CDCl3): δ (ppm) 0.88 (t, J = 7.2 Hz, 6 H, 2 CH2CH2 CH 3 ), 1.25–1.45 (m, 8 H, 2 CH 2 CH 2 CH3), 3.20–3.30 (m, 8 H, 4 NCH 2 ), 4.38 (s, 4 H, 2 NCH 2 Ar), 5.22 (bs, 2 H, 2 CONH), 7.17–7.32 (m, 10 H, phenyl ring protons); 13C NMR (CDCl3, 100 MHz): δ (ppm) 13.80 (2C), 20.02 (2C), 32.17 (2C), 40.69 (2C), 46.49 (2C), 51.64 (2C), 127.04 (4C), 127.51 (2C), 128.79 (4C), 137.97 (2C), 158.52 (2C); FT-IR: (cm−1) ν(NH) = 3,287, ν(alkanes) = 2,953, ν(CO) = 1,614, ν(Phenyl) = 1,536; MS (TOF MS ES+): 439.30006 M + 1.
Compound B
A 50-ml round bottom flask was charged with hexyl diisocyanate (0.5 ml, 2.98 mmol), 2-amino-4-hydroxy-6-methylpyrimidine (0.71 g, 5.96 mmol), and 5 ml DMF. The reaction flask was sealed with a rubber septum and heated at 80 °C overnight. DMF was removed by vacuum and the residue was washed with ether and dichloromethane to give 1.2 g of white solid at a yield of 99 %. One milligram of the white solid was mixed with 600 μl of d 6 -DMSO in an NMR tube and heated at 100 °C until completely dissolved. The d 6 -DMSO solution was kept warm and examined by NMR instrument immediately at 60 °C. 13C NMR of the white solid was not obtained due to solubility difficulty. 1H NMR (CDCl3): δ (ppm) 1.30–1.57 (m, 8H, 4 CH 2 ), 2.10 (s, 6 H, 2 CH 3 ), 3.08–3.14 (m, 2 CH 2 N), 5.77 (s, 2 H, 2 CH=C), 7.40 (bs, 2 H, 2 CH3CNH), 9.72 (bs, 2 H, 2 NHCONHCH2), 11.51 (bs, 2 H, 2 NHCONHCH2); FT-IR: (cm−1) ν(alkanes) = 2,936, ν(CO) = 1,699, ν(Phenyl) = 1,579; MS (TOF MS ES+): 419.2090 M + 1.
Compound C
Compound C was synthesized according to previously reported procedures [22]. A Schlenk tube was charged with 2,7-dichloro[1,8]naphthyridine (50 mg, 0.25 mmol), hexanoamide (57.6 mg, 0.5 mmol), xantphos (86.7 mg, 0.15 mmol), Pd (OAc)2 (11.2 mg, 0.05 mmol), K2CO3 (69.0 mg, 0.5 mmol), and 15 ml of dioxane. The reaction flask was sealed with a rubber septum and purged with N2 for 30 min and then heated at 80 °C for 24 h. The resulting dark solution was filtered through a thin layer of celite to remove the palladium complex. Dioxane was removed by vacuum and the residue was purified by silica gel column using ethyl acetate/hexane (1:1 v/v) as eluent. The obtained solid was recrystallized from ethanol to give 61.9 mg off-white flakes. Yield: 70 %. 1H NMR (CDCl3): δ (ppm) 0.92 (t, J = 6.2 Hz, 6 H, 2 CH 3 ), 1.34–1.39 (m, 8 H, 4 CH 2 ), 1.73–1.80 (m, 4 H, 2 CH 2 CH2CO), 2.46 (t, J = 7.2 Hz, 2 CH 2 CO), 8.09 (s, CONH), 8.13 (d, J = 8.8 Hz, 2 H, 2 CH=CH–C–NH), 8.43 (d, J = 8.8 Hz, 2 H, 2 CH–C–NH); 13C NMR (CDCl3, 100 MHz): δ (ppm) 13.90, 22.39, 24.95, 31.29, 37.95, 113.53 (2 C), 118.31, 139.02 (2 C), 153.65, 154.00, 172.48; FT-IR: (cm−1) ν(NH) = 3,324, ν(alkanes) = 2,952, ν(CO) = 1,671, ν(Phenyl) = 1,539; MS (TOF MS ES+): 357.2236 M + 1.
Results and discussions
We chose to prepare gelator 1 as a proof-of-principle and gelator 2 to demonstrate that this chemistry can easily be extended into more practical directions, for example potential energy transfer and photochemistry applications [23]. We decided to use pyrene specifically for gelator 2 based on previous reports that bipyrene organogelators exhibit thermal-driven fluorescence through gel to sol transitions as a potential fluorescence switch [24]. Maitra and coworkers have pioneered pyrene containing LMOGs, especially pyrene substituted bile acid–base molecular tweezers. Pyrene is a frequently used fluorescent compound due to its properties including long singlet lifetime, readily formed excimers, in addition to being an environment sensitive energy acceptor [25]. For example, pyrene-modified oligonucleotides and locked nuleic acids have been proven as useful tools for fundamental research, diagnostics, and nanotechnologies [26]. Gelators 1 and 2 were synthesized via reactions of a symmetrical diamine and a UPy-functionalized isocyanate in near quantitative yields (Scheme 1) and their afforded structures were confirmed using 1H nuclear magnetic resonance (NMR) spectroscopy. Gelator 1 afforded a single peak in the NMR spectrum at 2.09 ppm arising from the pyrimidone methyl groups, and the phenyl ring protons resulted in a multiplet peak at 7.12–7.33 ppm. The spectrum of gelator 2 showed a peak at 2.07 ppm due to the methyl groups of the pyrimidone ring, and the pyrene-yl protons were present at 7.58–8.27 ppm. Mass spectrometry further confirmed the structures, and molecular ion peaks were observed m/z = 827.4633 (calculated m/z = 827.4681 M + 1) for 1 and m/z = 1,075.5247 (calculated m/z = 1,075.5297 M + 1) for 2, respectively.

Synthesis of gelator 1 and 2
The gelation abilities of both gelators in various organic solvents were investigated using the “stable to inversion in a test tube” method [27]. Figure 1a shows the stable gels formed from solutions of gelators 1 and 2 in dimethyl sulfoxide (DMSO). As summarized in Table 1, both gelators formed gels in selected polar aprotic solvents.

Inverted vials showing gels formed from DMSO solutions of gelators 1 and 2 at their critical gelation concentration
The critical gelation concentration (CGC) of compound 1 was determined in DMSO (CGC = 4.3 wt%), DMF (CGC = 6.5 wt%), and pyridine (CGC = 7.5 wt%). In contrast, compound 1 was observed to precipitate from chloroform solutions. We rationalized this precipitation as resulting from the well-known supramolecular dimerization of the UPy groups, which does not occur in the polar aprotic solvents tested. Gelator 2 was observed to form gels in DMSO (CGC = 6.0 wt%), DMF (CGC = 6.5 wt%), and pyridine (CGC = 5.8 wt%). However, gelator 2 is completely soluble in chloroform; this is contradictory to the observations of the precipitation of 1 in chloroform at low gelator concentrations. We hypothesize that the larger, bulky, pyrene groups prevent organization and assembly mediated by UPy dimerization with gelator 2.
We first examined the stability of the DMSO gels by adding excess DMSO after the gels had formed and observing that the gels remained intact without noticeable changes. The morphologies of dried organogels were examined using scanning electron microscopy (SEM). SEM images (provided in Supporting Information) of dried DMSO gels of gelator 1 showed disordered porous networks. We observed that the dried gel from gelator 2 formed a denser porous material in DMSO compared with that from gelator 1 consistent with the experimental data of gelator 2 requiring a higher critical gelation concentration than gelator 1. However, caution should always be used in the interpretation of the xerogel morphology as collapse can occur due to the strong capillary forces during drying [28].
The gel transition temperature (T gel) is the temperature at which a gel undergoes a gel–sol transition and the value of T gel reflects the thermodynamic stability of a gel [29]. It is usually determined by placing a sealed glass vial containing the organogel in a temperature controlled water bath and visually monitoring if the gel flows upon tilting the vial as the temperature is increased. We studied organogels formed by gelators 1 and 2 in DMSO, as this was the highest boiling point solvent that formed a gel in our study. The results from this experiment are shown in Fig. 2. All measurements of T gel were repeated twice to ensure reproducibility and reversibility. As shown in Fig. 2, T gel increased as the weight percentage of gelators was increased until a plateau region was reached, denoted by a concentration-independent gel–sol transition temperature [30]. The organogels from gelator 1 show higher T gel values than those from gelator 2. The differences in T gel values between the two gelators presumably reflect differences in network organization in the gelled state due to the differences in solubility and behavior of the gelators. This argument is supported by the SEM images of dried networks of gelator 1 and 2 from DMSO (Fig. 1c, d). The lower CGC values are reflected in the low concentration range of Fig. 2, where 2 does not form a gel below 6 wt % in DMSO.

Gel–sol transition temperatures of gelator 1 and 2 for different concentrations of the gelator in DMSO. Data were collected twice and both runs are plotted
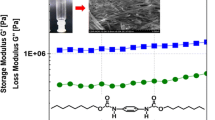
Rheological measurements provide determinative evidence for gel formation and so we further characterized the gels using rheology. We focused on gelator 1 for the rheology measurements as it affords more robust gels than gelator 2. A weight ratio of 5 wt% in DMSO, slightly above the critical gelation concentration of gelator 1, was used to make a gel in the rheometer fixture. The storage modulus, G′, and the loss modulus, G″, of the 5 wt% gelator solution were plotted against time upon quenching from 80 °C to 25 °C (Fig. 3a). The moduli increased continuously over the time span of the experiment, and eventually reached equilibrium values of approximately 103 and 40 Pa for G′ and G″ respectively after 5,000 s, meaning formation of a stable gel. It should be noted that G′ > G″ was observed for the first data point in the time sweep, which is not expected for a liquid. However, we believe this is an experimental artifact due to the sensitivity of the instrument and not more complex viscoelastic behavior as the data at the first time point was acquired below the torque limit of the instrument (0.05 μN).

a Time sweep of the storage (G′) and loss (G″) modulus at 25 °C after a quench from 80 °C at frequency of 1 rad/s and strain of 1 %. b Frequency sweep at 25 °C at a strain of 1 %. c Strain sweep at 25 °C at a frequency of 1 rad/s. Symbols: black squares G′, red circles G″
The gel was then subjected to a frequency sweep over a frequency range of 100 to 0.01 rad/s. As shown in Fig. 3b, G′ was invariant with frequency. The fact that G′ was much greater than G″ over the observed frequency range (0.04 ≤ tan δ ≤ 0.15) confirms the dominant elastic (i.e., gel) character of the material. A strain sweep of the gel at 25 °C is shown in Fig. 3c. The critical strain of the network was estimated to be 3 % from the intersection of extrapolated lines from the linear sections of the curve at low and high strain [31].
Two dominant pathways of gelation in LMOGs are microphase separation (e.g., wormlike micelles) and macrophase separation (e.g., fibrillar crystals) [32]. We believe that macrophase separation is driving gelation in these systems. It has been established in a number of systems that the solubility of the LMOG is a key parameter driving gelation [29, 33–36]. Solubility, in turn, will depend on the chemical structure of the LMOG. We have previously reported that an N-alkyl urea peptoid oligomer containing six urea groups was observed to self-assemble into uniform fibers in DMF, while shorter oligomers did not self-assemble. The driving force for fiber formation was determined to be intermolecular hydrogen bonding. In the current work, gelator 1 has only two N-alkyl urea functional groups, and is much shorter than the oligomer previously studied. This suggests the driving force to form the observed gels was not merely based on the N-alkyl urea groups. In order to test this hypothesis we prepared the analogous molecule to gelator 1 without UPy groups (compound A) to demonstrate that the observed gelation was dependent upon the presence of UPy, and therefore, not driven primarily by the N-alkyl urea groups. The synthesis of compound A is shown in Scheme 2a.

Synthesis of compounds a, b, and c
Significantly, we did not observe any organogels formed by compound A in the organic solvents listed in Table 1. Furthermore, gels were observed to form when A was mixed with either gelator 1 or 2, and gelator 1 or 2 was at a concentration above its CGC. Conversely, if gelator 1 or 2 was mixed with compound A and the total concentration of both molecules was equal to the CGC but A was present in any ratio to gelator 1 or 2, gels were not observed to form. Therefore, compound A shows little effect on the ability of gelators 1 and 2 to form gels in organic solvents. These results show that the presence of UPy in the molecules is required to form an organogel. Similarly, compound B (Scheme 2b) was prepared to demonstrate the importance of the alkyl urea groups for gelation with compounds 1 and 2. Compound B was synthesized through a single step reaction between hexyl diisocyanate and 2-amino-4-hydroxy-6-methylpyrimidine in near quantitative yield and does not contain any alkyl urea groups, but is of a similar length to gelator 1 and 2. Compound B precipitated as a white powder from the reaction mixture and did not dissolve in a range of solvents, including DMSO, CHCl3, pyridine, DMF and water. Compound B was observed to dissolve slightly in DMSO at elevated temperature, but precipitated out of solution upon cooling to room temperature (image of B in DMSO is available in the ESI). Therefore, in addition to the N-alkyl urea groups acting as a synthetic “handle” to incorporate a desired chemical group into the gelator, it appears that the combination of the alkyl urea (high solubility) and UPy groups (low solubility) tune the solubility of gelators 1 and 2 to drive gelation in a number of organic solvents. Indeed, the use of UPy in supramolecular chemistry can be limited by low solubility of the UPy moieties in some solvents. For instance, Long’s group incorporated UPy groups into high molecular weight polymers as side chains via random copolymerization of a UPy-functional alkene or methacrylate monomer. The UPy methacrylate was limited to 10 mol% or less incorporation in the copolymers due to the low solubility of the monomer [37, 38]. Collectively, the observations that the gelators must possess both N-alkyl urea groups and UPy groups raise the possibility that hydrogen bonding is occurring between these groups, and this hydrogen bonding is promoting the formation of a solid gel.
While the solubility of compounds 1 and 2 is tuned by their chemical structure it also depends on the choice of solvent. Due to its high dimerization constant (>106 M−1 in CHCl3)12 and synthetic accessibility, UPy has attracted considerable attention as a building block for various supramolecular architectures and materials [39, 40]. Meijer et al. [13]. demonstrated that the UPy quadruple hydrogen bonding group exists as three different reversible tautomeric forms, a mixture of dimers formed from two tautomeric 4-[1H] pyrimidinone and pyrimidin-4-ol forms, and a monomeric 6-[1H]-pyrimidinone form. However, the UPy moieties were shown by Meijer to be in the 6-[1H]-pyrimidinone monomeric form in DMSO. The UPy groups in both gelator 1 and 2 exist in the monomeric forms in deuterated DMSO solutions just below the CGC, as confirmed by the peak positions of the urea and amide protons in the 1H NMR spectra (spectra available in ESI). Moreover, 1H NMR spectra of gelator 1 and 2 did not show any shift of the urea protons and aromatic protons at different concentrations, indicating no intermolecular hydrogen bonding interactions. We also observed no shift of the urea protons and aromatic protons of 1 and 2 at the concentration below critical gelation concentration (i.e., 20 mg/ml) at different temperatures ranging from 283 K to 325 K (data not shown). Fourier transform infrared (FTIR) spectra of 1 and 2 in DMSO solutions below the CGC further supported existence of the monomeric form of UPy, showing broad signature absorption bands arising from free urea and amide N-H stretch around 3,300 cm−1 (Figs. S2 and S3). Cho and co-workers [14] observed UPy-modified organosilanes showed an N-H stretching absorbance at 3,343 cm−1 in CHCl3, while the same molecules showed a N-H stretching absorbance moved to 3,150 cm−1 in the gel state. However, our data combined with these literature examples imply that the UPy groups in our gelators exist in the monomeric form rather than as hydrogen bonded dimers in DMSO. This led us to speculate that the opaque gels formed by gelators 1 and 2 are not caused by formation of supramolecular self-recognition and hydrogen bonding of UPy, but rather through a phase-separation driven by the poor solubility of the UPy groups. We tested this hypothesis using a di-2,7-diamido-1,8-naphthyridine (Napy) functionalized molecule, shown as compound C in Scheme 2c. The Napy group has been shown to form strong and selective complementary complexation with UPy via four hydrogen bonds between acceptor–donor–donor–acceptor (ADDA) and donor–acceptor–acceptor–donor (DAAD) groups on the respective molecules [41]. We rationalized that compound C would disrupt the organogel if the gel formation did in fact depend on UPy dimerization instead of phase separation of the UPy groups. Indeed, addition of compound C to a solution of compound 1 in chloroform resulted in the partial solubilization of compound 1, likely as a result of this competitive dimerization of Napy and UPy groups. Upon adding an equal amount of compound C (on a molar ratio to gelator 1 or 2) to gels formed by gelator 1 or 2, the gels did not break or rupture. The mixtures containing the gelator and compound C were reheated to 80 °C to form homogenous solutions followed by cooling to room temperature. Gels still formed from the resulting solutions (Fig. S4 in ESI). In other words, the presence of compound C had little effect on the ability of gelator 1 or 2 to form organogels. These observations are consistent with our hypothesis that the interactions between the gelators are not only dimerization of the UPy moieties, but a more complex interplay between the N-alkyl urea and UPy functional groups.
Conclusion
We have successfully prepared UPy-functionalized N-alkyl urea dimers, and shown that these gelators form gels in various aprotic polar organic solvents including DMSO and DMF. Rheological characterization gave determinative evidence for the gelled nature of the solutions. The driving force for gel formation is the relationship of solubility and interactions of UPy and N-alkyl groups in the gelators, rather than only supramolecular hydrogen bonding interactions between UPy groups as might be expected. To the best of our knowledge, this is the first example of UPy functionalized low molecular weight organogelators. Considering the inherent potential synthetic diversity of N-alkyl urea oligomers, this strategy opens new opportunities to create gelled materials with a range of functional groups and potential applications.
References
Abdallah DJ, Weiss RG (2000) Organogels and low molecular mass organic gelators. Adv Mater 12:1237–1247
Terech P, Weiss RG (1997) Low molecular mass gelators of organic liquids and the properties of their gels. Chem Rev 97:3133–3159
Sangeetha NM, Maitra U (2005) Supramolecular gels: functions and uses. Chem Soc Rev 34:821–836
Buerkle LE, Rowan SJ (2012) Supramolecular gels formed from multi-component low molecular weight species. Chem Soc Rev 41:6089–6102
Smith DK (2006) Dendritic supermolecules – towards controllable nanomaterials. Chem Commun 7:34–44
Sivakova S, Rowan SJ (2005) Nucleobases as supramolecular motifs. Chem Soc Rev 34:9–21
Piepenbrock MOM, Lloyd GO, Clarke N, Steed JW (2010) Metal-and anion-binding supramolecular gels. Chem Rev 110:1960–2004
Suzuki M, Hanabusa K (2009) L-Lysine-based low-molecular-weight gelators. Chem Soc Rev 38:967–975
Rubio J, Martí-Centelles V, Burguete MI, Luis SV (2013) Synthesis and organogelating ability of bis-urea pseudopeptidic compounds. Tetrahedron 69:2302–2308
Simeone L, Milano D, De Napoli L, Irace C, Di Pascale A, Boccalon M, Tecilla P, Montesarchio D (2011) Design, synthesis and characterisation of guanosine-based amphiphiles. Chem Eur J 17:13854–13865
Wang X, Zhou L, Wang H, Luo Q, Xu J, Liu J (2011) Reversible organogels triggered by dynamic K + binding and release. J Colloid Interface Sci 353:412–419
Du P, Wang G, Zhao X, Li G, Jiang X, Li Z (2010) Two novel quadruple hydrogen-bonding motifs: the formation of supramolecular polymers, vesicles, and organogels. Tetrahedron Lett 51:188–191
Beijer FH, Sijbesma RP, Kooijman H, Spek AL, Meijer EW (1998) Strong dimerization of ureidopyrimidones via quadruple hydrogen bonding. J Am Chem Soc 120:6761–6769
Han JT, Lee DH, Ryu CY, Cho K (2004) Fabrication of superhydrophobic surface from a supramolecular organosilane with quadruple hydrogen bonding. J Am Chem Soc 126:4796–4797
Chen Y, Ballard N, Gayet F, Bon SAF (2012) High internal phase emulsion gels (HIPE-gels) from polymer dispersions reinforced with quadruple hydrogen bond functionality. Chem Commun 48:1117–1119
Chen Y, Ballard N, Bon SAF (2013) Moldable high internal phase emulsion hydrogel objects from non-covalently crosslinked poly(N-isopropylacrylamide) nanogel dispersions. Chem Commun 49:1524–1526
Chen X, Ayres N (2010) Synthesis of novel polymer/urea peptoid conjugates using RAFT polymerization. Macromolecules 43:1341–1348
Chen X, Ayres N (2011) Synthesis of low grafting density molecular brush from a poly(N-alkyl urea peptoid) backbone. J Polym Sci Polym Chem 49:3030–3037
Chen X, Ding K, Ayres N (2011) Investigation into fiber formation in N-alkyl urea peptoid oligomers and the synthesis of a water-soluble PEG/N-alkyl urea peptoid oligomer conjugate. Polym Chem 2:2635–2642
Taylor L, Chen X, Ding K, Ayres N (2013) Synthesis of a glycosaminoglycan polymer mimetic using an N-alkyl-N, N-linked urea oligomer containing glucose pendant groups. Polym Int. doi:10.1002/pi.4567
Folmer BJB, Sijbesma RP, Versteegen RM, van der Rijt JAJ, Meijer EW (2000) Supramolecular polymer materials: chain extension of telechelic polymers using a reactive hydrogen-bonding synthon. Adv Mater 12:874–878
Ligthart GBWL, Ohkawa H, Sijbesma RP, Meijer EW (2006) Pd-catalyzed amidation of 2-chloro-and 2,7-dichloro-1,8-naphthyridines. J Org Chem 71:375–378
Ajayaghosh A, Praveen VK, Vijayakumar C (2008) Organogels as scaffolds for excitation energy transfer and light harvesting. Chem Soc Rev 37:109–122
Wang C, Wang Z, Zhang D, Zhu D (2006) Thermal modulation of the monomer/excimer fluorescence for bispyrene molecules through the gel-solution transition of an organogel: a thermo-driven molecular fluorescence switch. Chem Phys Lett 428:130–133
Winnik FM (1993) Photophysics of preassociated pyrenes in aqueous polymer solutions and in other organized media. Chem Rev 93:587–614
Østergaard ME, Hrdlicka PJ (2011) Pyrene-functionalized oligonucleotides and locked nucleic acids (LNAs): tools for fundamental research, diagnostics, and nanotechnology. Chem Soc Rev 40:5771–5788
Hardy JG, Hirst AR, Smith DK, Brennan C, Ashworth I (2005) Controlling the materials properties and nanostructure of a single-component dendritic gel by adding a second component. Chem Commun 3:385–387
Brinker C, Scherer G (1990) Sol–gel Science: The Physics and Chemistry of Sol–gel Processing. Academic Press, San Diego, CA
Hirst AR, Coates IA, Boucheteau TR, Miravet JF, Escuder B, Castelletto V, Hamley IW, Smith DK (2008) Low-molecular-weight gelators: elucidating the principles of gelation based on gelator solubility and a cooperative self-assembly model. J Am Chem Soc 130:9113–9121
Lescanne M, Colin A, Mondain-Monval O, Fages F, Pozzo JL (2003) Structural aspects of the gelation process observed with low molecular mass organogelators. Langmuir 19:2013–2020
Li JL, Yuan B, Liu XY, Xu HY (2010) Cryst Growth Design 10:2699–2706
Raghavan SR (2009) Distinct character of surfactant gels: a smooth progression from micelles to fibrillar networks. Langmuir 25:8382–8385
Zhu G, Dordick JS (2006) Solvent effect on organogel formation by low molecular weight molecules. Chem Mater 18:5988–5995
Feng L, Cavicchi KA (2012) Investigation of the relationships between the thermodynamic phase behavior and gelation behavior of a series of tripodal trisamide compounds. Soft Matter 8:6483–6492
Gao J, Wu S, Rogers MA (2012) Harnessing Hansen solubility parameters to predict organogel formation. J Mater Chem 22:12651–12658
Raynal M, Bouteiller L (2011) Organogel formation rationalized by Hansen solubility parameters. Chem Commun 47:8271–8273
Elkins CL, Park T, McKee MG, Long TE (2005) Synthesis and characterization of poly(2-ethylhexyl methacrylate) copolymers containing pendant, self-complementary multiple-hydrogen-bonding sites. J Polym Sci Part A Polym Chem 43:4618–4631
McKee MG, Elkins CL, Park T, Long TE (2005) Influence of random branching on multiple hydrogen bonding in poly(alkyl methacrylate)s. Macromolecules 38:6015–6023
Brunsveld L, Folmer BJB, Meijer EW, Sijbesma RP (2001) Supramolecular polymers. Chem Rev 101:4071–4098
Sijbesma RP, Meijer EW (2003) Quadruple hydrogen bonded systems. Chem Commun 1:5–16
Wang XW, Li XQ, Shao XB, Zhao X, Deng P, Jiang XK, Li ZT, Chen GJ (2003) Selective rearrangements of quadruply hydrogen-bonded dimer driven by donor–acceptor interaction. Chem Eur J 9:2904–2913
Acknowledgments
Acknowledgment is made to the donors of the American Chemical Society Petroleum Research Fund (51850-DN17) and The University of Cincinnati for support of this research for N.A. and X.C. P.F. and K.A.C. thank The National Science Foundation (NSF-CHE 1012237).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 9783 kb)
Rights and permissions
About this article
Cite this article
Chen, X., Fei, P., Cavicchi, K.A. et al. The poor solubility of ureidopyrimidone can be used to form gels of low molecular weight N-alkyl urea oligomers in organic solvents. Colloid Polym Sci 292, 477–484 (2014). https://doi.org/10.1007/s00396-013-3087-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-013-3087-6