
Abstract
Current concepts of vascular permeability are largely still based on the Starling principle of 1896. Starling’s contribution to understanding vascular fluid homeostasis comes from realising that the transport of fluid to and from the interstitial space of peripheral tissues follows the balance between opposing oncotic and hydrostatic pressures. It is presumed that in peripheral tissues fluid is readily filtered from blood to tissues at the arterial/arteriolar side of the circulation and largely reabsorbed at the venular/venous aspect, excess fluid being removed from the tissue by the lymphatic system. This balance is determined particularly by the properties of the vascular barrier. Recent studies have shown that the endothelial glycocalyx, located with a thickness of at least 200 nm on the luminal side of healthy vasculature, plays a vital role in vascular permeability by constituting the vascular barrier together with the endothelial cells themselves. While water and electrolytes can freely pass through the glycocalyx, plasma proteins, especially albumin, interact strongly. Binding and intercalating plasma constituents with the structural elements of the glycocalyx creates the so-called endothelial surface layer. This is the actual interface between flowing blood and the endothelial cell membrane in vivo. The oncotic pressure difference pertinent to fluid homeostasis is not built up between the intravascular and the interstitial tissue spaces, but within a small protein-free zone beneath the glycocalyx surface layer. This explains why perturbation of the glycocalyx leads to a breakdown of both fluid and protein handling in the coronary vascular bed. Preventing damage to the glycocalyx seems to be a promising goal in cardioprotection in many clinical scenarios, including acute ischaemia, hypoxia and inflammation, and chronic vascular disease as in atherosclerosis, diabetes and hypertension.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Basics
About 60% of the body mass of adults in good cardiopulmonary health consists of water, which is distributed to two-thirds intracellularly and to one-thirds extracellularly. Most of the latter is allocated in the interstitial space (80%), while 20% contribute as the plasma volume to cardiac preload. Intra- and extracellular spaces are separated by the cell membranes. This lipid double layer is not resistant against hydrostatic pressure, but is largely impermeable for electrolytes and proteins. In contrast, the vascular barrier, in total, permits hydrostatic pressure gradients to be established, but does not retain electrolytes, though it is supposedly largely impermeable for proteins. Thus, water distributes passively over the compartments, following the distribution of osmotically and oncotically active substances.
This review was aimed to provide an integrative update on current physiological knowledge of the properties of the vascular barrier, focusing on the coronary system and the role played by the endothelial glycocalyx. Other physiological functions of the glycocalyx such as the mediation of shear stress from flowing blood to the endothelial cells, the regulation of inflammatory interactions and the prevention of firm adhesion of blood platelets and leucocytes to the intact vessel wall will only be mentioned in passing. The readers are referred to recent reviews and papers covering these aspects [5, 14, 16, 26, 40, 54, 63, 79, 80, 85, 93, 102, 114].
Though the endothelial glycocalyx represents the primary, endothelially generated structure at the vessel wall/blood interface, in vivo we are dealing with the endothelial surface layer, formed when plasma constituents interact with the molecules expressed at the cell membrane [86]. The two concepts are often not differentiated in the following text, but there are graded differences between them in regard to hydraulic resistance and permeability.
Background
In 1896, the British physiologist Ernest Starling [100] introduced his famous concept of vascular fluid homeostasis, in which he schematically opposed hydrostatically driven fluid losses towards the tissues against reabsorption on the basis of an inwardly directed oncotic pressure gradient. This model is still favoured in most text books of physiology today and can be mathematically described by the equation
where F/A is filtration rate per area, C H hydraulic conductivity, P HV − P HI = hydrostatic pressure (P H) gradient between the vascular lumen (V) and the interstitial space (I), σ reflection coefficient for the respective colloids at the vascular barrier and πOV − πOI is oncotic pressure (πO) gradient between the vascular (V) lumen and the interstitial space (I).
Starling [100] suggested a high filtration–high resorption scenario even for non-fenestrated microvessels where fluid loss out of the vasculature on the arteriolar side caused by the higher blood pressure in comparison to the oncotic pressure is countered on the venular side by reabsorption due to the overbearing effect of intravascular oncotic pressure (Fig. 1). If these forces are in near balance when integrated along the microvascular length, the lymphatic system should be able to cope with any resulting, relatively low excess of fluid left in the interstitial space. The introduction of the term “sigma”—the reflection coefficient of colloids at the vascular barrier—within the equation results from an uncertainty within the concept concerning possible differences in behaviour of distinct colloidal molecular species and variability of permeability between different organs. In the special case of the blood–brain barrier, σ can be set as 1. However, also in all other vascular beds, one must expect σ to become totally unnecessary under steady-state conditions, once the actual net difference in oncotic pressure (ΔπO) is known.

The classical principle of Ernest Starling for fluid homeostasis in peripheral microvascular beds with continuous endothelium: a high filtration–high resorption concept relying on very low permeability of plasma proteins and low interstitial protein concentration
Most clinical and experimental observations detailing tissue oedema have been based on this historical principle. It was also, apparently successfully, adopted for describing myocardial fluid homeostasis [69]. This appears to be inadequate in the face of several recent physiological findings, but theoretical physiological considerations clearly challenge the traditional view. For instance, Levick [62] first noticed the “low lymph flow paradox”, i.e., the phenomenon that the vascular barrier is still competent even when the interstitial oncotic pressure equals that of the vascular lumen. In rat mesenteric microvessels, the effective inwardly directed oncotic force opposing the hydrostatic gradient was still 70% of the intravascular colloid osmotic pressure even if the non-fenestrated microvessels examined in this model were surrounded by fluid containing colloid at the same concentration [2]. The Starling hypothesis would have predicted the absence of any inwardly directed force in this situation [100], hydrostatically driven outflow of fluid merely being mechanically hindered by physical properties of the vessel wall. As illustrated schematically in Fig. 2, the steepness of the graphs represents the hydraulic conductivity, i.e., the filtration rate per unit area per unit pressure gradient, which is the higher the steeper the line [2]. The intersection with the abscissa represents the effective inwardly directed oncotic force, which should lie at the point of origin if the oncotic pressure gradient across the vessel wall is nullified. Any resorptive force towards the vascular lumen should shift the curve rightwards. Obviously, a non-fenestrated isolated microvessel does not follow Starling’s suggestions concerning a significant role of the interstitial oncotic pressure for vascular barrier competence. Experimental research using the isolated perfused heart, a whole organ model providing a complete vascular bed with large vessels, arterioles, capillaries and venules, plus the naturally surrounding tissue, confirmed these findings. Consistently, the interstitial and intravascular concentrations of any colloid applied into the coronaries came to within about 90–95% of each other inside of 20 min of onset of infusion [17, 48, 91]. Moreover, immunohistochemistry showed an even distribution of albumin throughout the whole interstitial compartment. The vascular barrier, however, was functionally intact in these isolated organs [47].

The effective oncotic gradient across the vessel wall is not determined by the concentration gradient of colloid between intravascular and interstitial compartment (schematic summary of results of studies on isolated microvessels; for detailed description see [2])
A further, clinically important contradiction to Starling’s concept was revealed in the perfused whole organ, namely the “COP paradox” [48]. It is obviously not primarily the intravascular colloid osmotic pressure (COP) which determines the power limiting transvascular fluid loss. Rather, the pivotal issue might be the type of colloid active at the endothelial surface. Similar to the isolated microvessel model, the coronary system of isolated guinea pig hearts was pressurised with artificial perfusate at different levels. Using a Langendorff setup, hearts were perfused with Krebs-Henseleit buffer alone, buffer diluted with isotonic saline, or buffer containing one of two different kinds of colloid at various concentrations [48]. The resulting transvascular filtration rates gave rise to transudate fluid appearing on the epicardial surface, comparable to the lymph flow in vivo, and were related to the respective perfusion pressures (Fig. 3). Just as in the isolated vessel model (Fig. 2) [2], a strictly linear dependency was observed. Adding a colloid to the artificial perfusate significantly lowered pressure-dependent transvascular filtration. The natural protein human albumin, however, appeared to be much more effective in sealing the vasculature than the artificial alternative hydroxyethyl starch, frequently used in clinically relevant bleeding to maintain the plasma COP [48]. The sealing effect of albumin was practically independent of the intracoronary COP it provided, down to a value only one quarter of the physiological level. The special ability of albumin to prevent coronary vascular extravasation of fluid has been noted repeatedly before [42–44]. If such behaviour were to be considered mathematically, then the reflection coefficient σ of the Starling formula would need to become greater than unity for low concentrations of albumin.

The pressure dependency of net fluid filtration in the intact coronary system of guinea pig hearts perfused with Krebs-Henseleit buffer containing no colloid or 2 g% hydroxyethyl starch (HES) or human albumin (1.7 or 0.85 g%). Lines represent mean values of five hearts per condition, with the two albumin concentrations being practically equipotent. COP colloid osmotic pressure; data taken from [47, 48]
Neither the “low lymph flow paradox” [62] nor the “COP paradox” [48] is really compatible with the traditional concept of Ernest Starling [100]. Furthermore, it surprises that physiologists, including Starling, have ignored the fact that lymphatic fluid is potentially able to coagulate. This ability, recognised already in 1653 (see [61]), implies that the extravascular fluid must contain significant amounts of coagulation factors derived from plasma and also all kinds of other plasmatic proteins. Indeed, the interstitial space of human heart tissue is rich in albumin, seen to be coating all cardiomyocyte cell surfaces (Fig. 4) [52].

Albumin (green stain) in samples of human heart tissue. Left panel immunohistology of a healthy human heart; right panel explanted human heart with dilated cardiomyopathy; for details see [52]. Albumin coats all extravascular cell surfaces and fills the interstitial matrix
Introducing the endothelial glycocalyx
The endothelial glycocalyx is an intravascular fringe of astounding functional significance. It participates in numerous physiological processes, foremost in regulating vascular permeability, preventing firm adhesion of leucocytes and blood platelets to the vessel wall, transmission of shear stress, and in modulation of inflammatory and haemostatic processes [5, 16, 85, 86, 93, 102, 114]. Pathophysiological sequelae of glycocalyx failure or perturbation include generation of tissue oedema, systemic inflammatory response syndrome, diabetic angiopathy and, possibly, atherogenesis. Situations in which damage to the glycocalyx has been reported include ischaemia/reperfusion, hypoxia, sepsis, volume overload, diabetes and atherosclerosis [74, 76, 88, 108, 112]. As already stated, this review shall focus on the role played in fluid homeostasis and colloid permeability in the heart.
Hydraulic conductance
First suspicions that there was something on the surface of the vessel wall helping plasma proteins to attenuate the development of oedema were published in 1940, but without any supportive optical evidence [29]. On the basis of chemical analysis, showing high content of sugars, this lining was soon termed the “endothelial glycocalyx”; the physiological properties and true dimensions, however, remained unclear for a long time [67]. There were first substantial considerations about a significant contribution of the endothelial glycocalyx to vascular barrier competence in the early 1980s [24]. Furthermore, the ability of plasma albumin to significantly lower hydraulic conductivity in conjunction with the endothelial glycocalyx was established [24, 42]. However, despite the formal attribution of a role as a “second barrier” [91], in addition to that posed by the endothelial cell bodies themselves, the exact contribution of the endothelial glycocalyx to vascular barrier competence remained unclear until very recently. Meanwhile, improved electron microscopical fixation techniques [106], in particular one based on stabilisation of the negatively charged structure (see below) by lanthanum ions during fixation reaction with glutaraldehyde [111], revealed that the first descriptions had gravely underestimated its actual dimension [67, 112]. It could be demonstrated repeatedly to be at least thicker than 200 nm in situ, both in the coronary system and the human umbilical vein (Fig. 5) [14, 15, 17, 19, 21, 47, 48, 54, 55, 91].

Transmission electron microscopy of the endothelial glycocalyx fixed with glutaraldehyde in the presence of lanthanum nitrate. a, b Coronary capillary of guinea pig heart, in total and at high magnification, respectively, perfused ex vivo after isolation (taken from Chappell et al. [15]); c human umbilical vein, perfused ex vivo within minutes of birth (for details, see Chappell et al. [21])
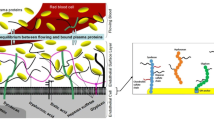
In its strictest sense, the endothelial glycocalyx consists mainly of membrane-bound glycoproteins and proteoglycans, chiefly syndecans and glypicans, carrying negatively charged heparan sulphate (about 70%) and chondroitin and dermatan sulphate side chains (30%), accounting for much of the biophysical properties [5, 85, 86, 114]. In addition, the glycocalyx harbours the outer aspects of membrane receptors, channels and adhesion molecules (within the closest 10–20 nm). The non-sulphated, receptor-attached, long-chain molecule hyaluronan is also often considered to be an essential part of the surface lining (Fig. 6) [86, 103]. The endothelial glycocalyx by itself, however, is relatively functionless in a model vasculature perfused with a pure crystalloid buffer since it poses no great resistance against passage of water, electrolytes and small uncharged osmolytes [47, 48, 63, 91].

Schematic arrangement of the chief constituents of the endothelial glycocalyx and the established endothelial surface layer. Glycoproteins include intercellular and vascular adhesion molecules, selectins and integrins. These all extend at most 10–20 nm from the lipid double layer and are, consequently, normally sequestered beneath a healthy surface layer [5]. Shed components of the glycocalyx are to be found in normal plasma [11, 88]. CD44 membrane receptor binding hyaluronan (adapted from Pries et al. [86] and Tarbell and Prahakis [103])
The situation is dramatically different in vivo when natural proteins are present at the endothelial surface. Albumin, for instance, has been shown to be tightly attached to the endothelial surface, even after a protein-free washout procedure lasting several minutes [47, 54]. Obviously, many proteins, peptides (including cytokines and chemokines) and even lipids are bound to and intercalated in the endothelial glycocalyx, together constituting the so-called “endothelial surface layer” (ESL) [114]. This seems to possess a functional thickness of at least 1 μm [85], but general consensus is that the width of the glycocalyx proper and that of the ESL varies in different regions of the vascular system. Large vessels seem to support a wider ESL than microvessels. A dynamic equilibrium in vivo between the proteins of the flowing blood and those bound to or within this structure has been described before (Fig. 6) [85, 86].
The question exactly how this layer might be able to generate an inwardly directed oncotic gradient even when interstitial and intravascular protein concentrations are comparable was answered in 2004 using isolated rat mesenteric microvessels as model [2]. Adamson et al. [2] suggested a small space of much less than 100 nm width situated directly beneath the endothelial surface layer to remain practically protein-free when plasma is forced outwards hydrostatically as proteins are excluded or retained within the structure. Because the fluid, nonetheless, passing through will be extremely low in protein, an inwardly directed oncotic gradient will be generated between this small space and the protein-loaded endothelial surface layer (Fig. 7) [47]. This force limits the net outflow of ultrafiltrate towards the interstitial space. A further limitation of hydraulic conductivity occurs on the basis of the endothelial anatomy in continuous arteriolar and capillary vessel segments. As described in detail by Adamson et al. [2] as well as by Curry and Adamson [26] in a recent review, fluid can only pass into the interstitial space in these vessel segments via small infrequent breaks within the junction strands of narrow clefts between the endothelial cells. This normally small flow of ultrafiltrate is important, because it prevents back-diffusion of colloid from the interstitial space (see below) into the sub-glycocalyx zone, a process which would obliterate the oncotic gradient developed across the luminal glycocalyx.

The modern low filtration–(low) no resorption concept of fluid homeostasis in the coronary system. In the capillary segments there is a relatively high hydrostatic pressure gradient, but this is opposed by a large oncotic pressure gradient established between the endothelial surface layer and the underside of the glycocalyx. In addition, there is a high resistance to flow through the narrow interendothelial clefts. In the venular sections the hydrostatic pressure gradient is small and owing to the easy egress of colloids, there is hardly any oncotic pressure gradient. For details, see [47]. The enlargement of the interendothelial cleft illustrates the ability of the endothelial glycocalyx to establish an oncotic gradient and the maintenance of the protein-poor zone under the glycocalyx by the low flow of ultrafiltrate through the narrow cleft. This convection prevents interstitial proteins from diffusing up to the luminal side of the endothelial cells. P hydrostatic pressure, Π oncotic pressure, c capillary lumen, e endothelial surface layer (ESL), g underside of the glycocalyx, t tissue, IS interstitial space, v venular lumen
As formulated by Levick and Michel [63], the revised Starling equation for continuous endothelia should read as
where F/A is filtration rate per area, C H hydraulic conductivity, P HV − P HI hydrostatic pressure (P H) gradient between the vascular lumen (V) and the interstitial space (I), σ reflection coefficient for the respective colloids at the vascular barrier and πOV − πOg is oncotic pressure (πO) gradient between the vascular (V) lumen and the underside of the glycocalyx (g).
The coronary venules, in contrast, appear to be more porous than arterioles and capillaries. Qualitative observations in isolated heart preparations have shown that highly charged positive ionic species such as lanthanum ions, which should be retained by the strongly negatively charged glycocalyx, and also plasma proteins such as albumin pass into the interstitial space within minutes after onset of intracoronary infusion, but only around venules [47]. This relatively facile passage of colloids explains why lymphatic fluid of the heart and the so-called “transudate” appearing on the epicardial surface of perfused heart preparations contain colloid in such high concentrations in the steady state (~90% of the arterial value) [91]. As a consequence, there is practically no oncotic gradient hindering outflow of plasma in the venular vessel segments, or favouring resorption of interstitial fluid. On the other hand, there is only a very small hydrostatic pressure gradient across the vascular wall in this coronary section, limiting fluid extravasation. Accordingly, the actual amount of colloid passing into the interstitial space of the myocardium via passive transport should be quite small in a healthy coronary bed. Colloid that does extravasate will do so largely via diffusion.
The present concept describing fluid homeostasis in the intact cardiac microvascular bed is schematically outlined in Fig. 7 [47]. In the arteriolar and capillary segments we have a low-flow situation of protein-poor plasma filtrate; in the venular section we have a low-flow exchange of protein-rich plasma with little or no direct resorption from the interstitium, but with ready exchange (via diffusion) of all dissolved constituents of the intra- and extravascular fluids. Allowing such exchange answers the age-old problem of how large molecules and essential nutrients such as low-density lipoprotein particles gain access to the parenchymal cells of the heart under physiological conditions. It, of course, also explains why cardiac lymphatic fluid is potentially able to coagulate.
Though neither σ, the colloid reflection coefficient, nor πOg, the oncotic pressure at the underside of the endothelial glycocalyx, of the revised Starling formula will generally be known, consequences of damaging or degrading the endothelial surface layer (see below) are readily apparent: Loss of the ability to form a transglyceal oncotic gradient will heighten filtration forces in arteriolar and capillary segments. Figure 7 also allows one to appreciate the “double barrier concept”, exemplified in experiments with histamine by Rehm et al. [91]. Histamine applied to isolated heart preparations perfused at constant flow with a purely crystalloid solution (Krebs-Henseleit buffer) did not elevate net filtration of coronary perfusate unless the glycocalyx had been enzymatically degraded beforehand. Conversely, using heparinase to cleave off the heparan sulphates causes collapse of the glycocalyx [22], but does not elevate net filtration under constant flow conditions unless histamine is applied to widen the endothelial clefts [91].
Vascular permeability of colloidal and electrically charged substances
With respect to short-term transport of substances across the vascular wall, one must distinguish between changes in vascular permeability and changes in hydraulic conductance. Alteration of the latter will influence solvent drag (provided permeability is not zero); alteration alone of the former will affect both diffusional transport and solvent drag. Transport via solvent drag is discussed in detail in recent reviews [46, 102].
Permeability of plasma proteins and of xenogenic colloids in the arterial and capillary sections of the coronary bed is governed by the glycocalyx according to selectivity based on size and charge. Especially, charge has been implicated to play a major role [64]. Because of the preponderance of heparan and chondroitin sulphate side chains, negatively charged molecules are excluded from entry, while positively charged species will be bound and retained. This has been shown for charged dextrans [7, 95, 110]. We have observed that infused antithrombin III, a molecule with negative charge, lines the luminal vessel wall, but does not enter the interstitial space of the heart unless the glycocalyx is removed [19]. Albumin is a special case; though the molecule carries a bulk negative charge at physiological pH, there are also positively charged groups along the protein chain. This amphoteric nature allows albumin to interact particularly well with the glycocalyx. Consequences are increased hydraulic resistance [43, 44, 68, 91, 112], stabilisation against degradation [54], and facilitated transmission of shear stress as evidenced by enhancement of flow-mediated dilatation of coronary vessels [55].
Even in the relatively permeable venular region (large-pore endothelial zone), the glycocalyx extends into the pore and exerts a limiting action on protein and colloid extravasation [112]. Equilibration of infused colloids (albumin and hydroxyethyl starches) with the interstitial space requires at least 20 min in isolated heart preparations [91] and is, thus, far from being instantaneous. Similarly, neither lanthanum ions nor antithrombin pass into the perivenular interstitial space readily as long as the glycocalyx is intact [19].
Testing the “low-filtration, venular egress” concept
An immediate consequence of plasma proteins being able to leave, albeit slowly, the coronary intravascular space via large venular pores and of bulk extravasation not being offset by high resorptive flow of interstitial fluid is that cardiac lymphatic fluid will also contain plasma proteins in appreciable concentrations. This is the case [4, 61]. While this also satisfies the observation concerning coagulability of lymphatic fluid, there is an evident problem: myocardial tissue is rich in tissue factor, the strongest initiator of the extrinsic pathway of coagulation [38, 101]. What then prevents coagulation from starting in the interstitial spaces of the heart under physiological conditions? Clearly, this would be a devastating scenario.
With this in mind, we have recently examined human heart tissue with respect to the distribution, in relation to the vasculature, of coagulation-initiating (tissue factor) and coagulation-inhibiting factors (thrombomodulin and activated protein C) [52]. Significantly, tissue factor was never found in the vicinity of venules or small veins of the heart. Instead, it was present subendothelially in arterioles and capillaries (Fig. 8). This distribution corresponds with that of pericytes in the vascular system, cells which have recently been shown to express tissue factor exclusively in the vessel wall under physiological conditions [56]. The considerable intra-organ heterogeneity of distribution observed supports the opinion that tissue factor forms a haemostatic envelope preferably around high-pressure segments of the vasculature to limit bleeding also in the face of rupture or trauma [34]. Thrombomodulin, in contrast, was present on all endothelial surfaces of the coronary system, indiscriminate of the vessel type or magnitude, and particularly high concentrations were localised in the interendothelial clefts [52]. Thus, any thrombin being carried towards the interstitial space would be expected to initiate the strongly anticoagulatory protein C system [36]. Interestingly, there was little activated protein C anywhere in the heart tissue. In those instances in which it was detected, there were discrete amounts in areas where tissue factor had typically been found, i.e., near capillaries and arterioles [52].

Immunohistology showing the distribution of tissue factor (left panel brown granular deposits) and thrombomodulin (right panel brown luminal lining) in explanted human heart tissue. Arrows in the left panel indicate some of the capillaries; asterisks show some veins and venules. For details, see [52]
Altogether, the inhomogeneous distribution of tissue factor, with its noteworthy absence around venules, and the strategic localisation of thrombomodulin at endothelial junctions are in full accordance with the proposed easy access of plasma constituents into the myocardial interstitial space because there is a minimised risk of generating fibrin. The result is also gratifying in the sense of Karl Popper (1902–1994). This great philosopher of science maintained that theories should be validated by asking questions and designing experiments potentially likely to disprove them. Fittingly, evidence for the important role of the endothelial glycocalyx in regulating coronary permeability comes from experiments designed to damage this sensitive structure.
Deterioration and shedding of the endothelial glycocalyx
Pathophysiological situations clearly leading to damage of the glycocalyx are ischaemia/reperfusion, hypoxia, volume loading and heart surgery and will be addressed in the following. Deterioration due to sepsis and chronic alteration as in atherosclerosis and diabetes mellitus are likely but have not been expressly studied in the heart. There are several recent publications and reviews dealing with these aspects [5, 16, 70, 74, 76, 78, 81, 93, 105, 108].
Ischaemia/reperfusion and hypoxia
The hypoxia-induced increase in coronary permeability was, surprisingly for early investigators, not visualised in ultrastructural examinations of the endothelium [113]. However, these exact and detailed electron microscopical studies did not conserve the endothelial glycocalyx during fixation, so that it escaped notice.
Degradation of the endothelial glycocalyx due to ischaemia/reperfusion and hypoxia has been demonstrated in various experimental models. Rat intravital microscopy revealed that intestinal ischaemia/reperfusion led to a significant reduction of glycocalyx thickness due to shedding of glycosaminoglycan chains [73]. Intravital microscopy of the mouse cremaster muscle showed a rapid deterioration of the endothelial glycocalyx after reperfusion [82, 95]. Postischemic destruction of the endothelial glycocalyx could also be demonstrated in isolated heart models [3, 6, 10, 14, 15, 17, 19, 28, 48, 54, 91, 106]. Indicators for evaluation of disruption of the endothelial glycocalyx included rate of formation of coronary transudate (a direct measure of net fluid filtration), colloid permeability, shedding and washout of major components of the glycocalyx (syndecan, heparan sulphate and hyaluronan) and direct visualisation of the glycocalyx by electron microscopy [3, 12, 14, 17, 21, 54]. Hypoxia also disrupted the endothelial glycocalyx of isolated rat hearts [112].
Recently, damage of the endothelial glycocalyx was indirectly demonstrated in patients undergoing global or regional ischaemia [88]. The level of two components of the endothelial glycocalyx, syndecan and heparan sulphate, increased multifold in arterial blood of patients undergoing major vascular and heart surgery. Surprisingly, quantitatively identical increases in shed constituents were detected in plasma of patients undergoing coronary artery bypass, irrespective of whether the operation was performed with or with cardiac arrest (on- vs. off-pump mode, see below) [11].
Much remains unclear about the mechanisms involved in ischaemia/reperfusion injury. Possible mediators of this type of injury include free radicals [13, 23, 32, 95], complement activation [23], TNF-α expression [59, 118] and mast-cell degranulation [39, 57, 92]. The latter may be induced by elevated levels of endogenous adenosine, via adenosine A3 receptors [81], as recently discussed in a review of van Teeffelen et al. [107]. Leucocyte activation and adhesion have also been reported to contribute to reperfusion injury [13, 23, 41, 60, 87]. Chappell et al. [14] have recently demonstrated that intracoronary adhesion of human polymorphonuclear leucocytes in the coronary system, as in postischemic reperfusion, is only possible if the glycocalyx has been largely shed (see also [5, 54]).
Postischemic or TNF-α-induced mast-cell degranulation liberates cytokines such as TNF-α [39, 92, 117], demonstrated to induce shedding of the glycocalyx [15], and proteases such as tryptase and cathepsin B [3], which could cleave syndecans and hyaluronan from the endothelial membrane. Of particular note is our hitherto unpublished finding that the resident mast cells are the sole store of the enzyme heparanase in the human myocardium. This is shown in Fig. 9. Liberation of heparanase would be required to explain postischemic and postinflammatory shedding and washout of heparan sulphates from the heart as observed ex vivo [3, 10, 12, 14, 15, 17, 19, 54]. Heparanase acts just like heparinase, employed by us and others to selectively degrade the glycocalyx [10, 17, 22, 31, 48, 91]. Interestingly, heparinase has been applied to human patients undergoing coronary artery bypass grafting as an alternative means of abrogating heparin anticoagulation [99]. This clinical study was terminated ahead of time because of an inferior safety profile to protamine, hardly surprising in view of the strong degradative potential of heparinase versus glycocalyx [22].

Mast cells and heparanase in human heart tissue. Left panel Alcian blue stain shows mast cells (arrows) in an explanted human heart with the typical perivascular localisation (for details, see Gilles et al. [39]). Right panel immunohistological detection shows heparanase (intense brown stain) exclusively in the granules of a cell in the same perivascular localisation as mast cells (rabbit anti-human heparanase antibody A00078, Genscript Corp., dilution 1:100 in formalin-fixed and paraffin embedded ventricular myocardium; for further details, see [52])
Hypervolemia
Atrial natriuretic peptide (ANP), a small peptide released by the heart upon hypervolemia due to atrial stretch, acts acutely to reduce plasma volume by renal excretion of salt and water, vasodilatation and increasing vascular permeability [25]. While several studies demonstrated that ANP increased vascular permeability [45, 96, 104], and Curry et al. [27] have since been able to show that the permeabilising effect is mediated via endothelial ANP guanylyl cyclase type-A receptors, the exact mechanism remained undefined. In an isolated perfused heart model, we have been able to identify a general feature, namely that an increase in endothelial cyclic GMP always leads to an increase in coronary leak, irrespective of whether the agent is bradykinin, acetylcholine, nitroprusside or ANP [20].
In this context, our group investigated a possible connection between the ability of ANP to acutely shift volume from the intra- to the extravascular space and the integrity of the endothelial glycocalyx [10]. Low-dose infusion of ANP (10−9 M) in isolated guinea pig hearts resulted in an increase in fluid leak and an accelerated extravasation of colloid. Moreover, ANP caused rapid shedding of syndecan core protein and histologically detectable degradation of the coronary glycocalyx [10]. Thus, the ANP-induced increase in vascular permeability described in vivo might be related to changes in the integrity of the endothelial glycocalyx.
The results of this investigation are in accordance with the findings of previous double-label blood volume measurements in humans [89, 90]. As a substitute during acute bleeding, iso-oncotic colloids had volume effects of more than 90%, provided normovolaemia was carefully maintained [90]. In contrast, volume loading of normovolaemic patients with the same iso-oncotic colloid led to volume effects of only approximately 40% [89]. Consequently, volume effects of colloids seem to depend on the “context”, i.e., the volume and hydration state of the patient [18, 50, 51]. The most likely explanation for this phenomenon is that volume loading resulted in an increased level of plasma ANP, thereby degrading the endothelial glycocalyx. Furthermore, a greater decrease in large vessel haematocrit (measured by centrifugation of arterial blood samples) in relation to whole body haematocrit (derived by double-label measurements of erythrocyte and plasma volume) was found during volume loading [89]. This observation leads one to suspect that a considerable decrease in the volume of the endothelial surface layer occurred during volume loading, leading to a larger distribution space for red blood cells. Again, ANP-induced alteration of the endothelial surface layer might be the underlying cause.
The double-tracer dilution technique based on indocyanin green (as label for the total plasma space) and fluorescein-labelled red blood cells (RBC) yielded a whole body volume of the endothelial surface layer in man amounting to about 730 ml, i.e., approximately 25% of the total plasma volume [89]. This volume was diminished to 250–350 ml upon volume loading [89]. Double-tracer dilution using fluorescent dextran and RBC has been applied by others to assess the glycocalyx volume in studies on goats and mice [7, 95, 110]. However, some of the results were physiologically rather unlikely and have been strongly criticised by Curry and Michel [71] for methodological reasons, but also on principle. The methodological shortcomings of the animal studies, for example redistribution of plasma space tracer into other compartments or inhomogeneous molecular size, were excluded in the measurements on humans [49, 53]. The principal criticism centred on whether a large molecular weight tracer is at all able to distribute fully into a space filled by the branched chains of glycocalyx molecules. In other words, the tracer dilution values must fall short of the mark. The validity of this argument is surely given, but the extensive calculations performed to establish the magnitude of the under-representation of endothelial surface layer volume far overrate the error. Curry and Michel assume the glycocalyx to exist as a rigid, highly ordered geometrical structure. However, this is not the case in vivo, where fluctuations in blood flow and pressure and passage of cells, especially that of leucocytes, constantly deform and rearrange the glycocalyx fringe lining the endothelial cells [70, 75, 109].
Cardiac bypass surgery and reperfusion after acute myocardial infarction
Mechanical manipulation of the heart is another adequate stimulus for the release of ANP, and robust handling inadvertently takes place both in pump-assisted and off-pump bypass surgeries. Indeed, systemic shedding of the glycocalyx of equal magnitude was noted by us in patients irrespective of whether they were subjected to the on- or off-pump procedure, i.e., irrespective of whether the heart had experienced ischaemia/reperfusion or not [11]. At present it is not known whether shedding of the coronary glycocalyx itself occurs in bypass patients, but there is no reason to believe that the coronary system is exempt from the systemic vascular phenomenon.
As a point of interest, ANP was applied pharmacologically to patients during reperfusion by percutaneous coronary intervention after acute myocardial infarction [72]. The so-called J-WIND study was unable to show any benefit 6–12 months after ANP treatment versus control although the treatment had seemingly produced a 15% decrease in infarct size. Also in the AMISTAD II trial, in which adenosine was applied during reperfusion, there was no benefit for the patients at 6-month follow-up despite early pronounced reduction in infarct size [94]. It might be a coincidence, but both ANP and adenosine have the potential to deteriorate the coronary glycocalyx, thereby enhancing oedema as well as adhesion of leucocytes and, thus, secondary myocardial damage [87, 116].
Pathophysiological implications of damage to the glycocalyx
As outlined above, volume overload can induce impressive fluid and protein shifting towards the interstitium by deteriorating the vascular barrier [18, 51]. Consequently, and in accordance with clinical studies, volume loading in normovolaemic patients should most likely be abolished in favour of demand-related fluid regimens [8, 65, 77]. Using fluid in adequate amounts seems to have the power to improve patient outcome by minimising perioperative fluid shifting.
Interstitial oedema is a common problem in the critically ill and in the surgical patients and appears to be related to mortality [66, 74]. Therefore, protecting vascular barrier competence is an important therapeutical issue. Until today, it remains unclear what the best strategy might be in practise to combine a sufficient cardiac preload with limiting oedema, especially in patients already suffering from capillary leakage. The introduced endothelial surface layer concept might provide some new aspects in this standoff. Therapeutic strategies targeting the endothelial glycocalyx are in view, but clinically unproven at this stage [5].
In the heart, oedema leads to a loss of myocardial compliance [30, 33]. As depicted in Fig. 10, this increase in wall stiffness is equivalent to a shift-to-the-left and steepening of the passive filling pressure curve of the ventricles. Accordingly, the ventricular pressure–volume loop for given pre- and afterload and inotropy will also be shifted to the left, amounting to a loss of stroke volume. To maintain cardiac output, preload enhancement, afterload reduction, inotropic stimulation and heart rate increase are required, either singly or in combination, to compensate. Thus, prevention of myocardial oedema must be awarded high priority in cardioprotection.

Pressure–volume loop of the left ventricle (schematic drawing), illustrating the consequence of tissue oedema. The passive filling pressure curve becomes steeper, shifting the loop to the left and, thus, reducing stroke volume at given filling and aortic pressures and unaltered inotropic state
Improved magnetic resonance imaging techniques are allowing the detection of myocardial oedema early after cardiac infarction and coronary microembolisation in vivo [9, 58]. It is tempting to relate this formation of oedema to damage of the glycocalyx by ischaemia and secondary inflammatory responses. Unfortunately, to date, it is (still) impossible to simultaneously and repetitively assess the state of the coronary endothelial glycocalyx by any means of non-invasive imaging. Concerning functional consequences, it is of interest that sites of myocardial oedema matched areas of contractile dysfunction [9].
Jacob et al. [54] have examined experimentally the ability of albumin, given as supplement to histidine–tryptophan–ketoglutarate (HTK) cardioplegic solution, to prevent postischemic damage in a glycocalyx-directed manner in a transplantation setting. Guinea pig hearts were arrested in situ by perfusion with either cold unaltered HTK solution or HTK solution containing 1% human albumin and stored for 4 h at 4°C. The hearts were reperfused with Krebs-Henseleit buffer containing 4% albumin at 37°C. Pertinently, albumin supplementation lessened shedding of syndecan and heparan sulphate, reduced myocardial oedema and postischemic adhesion of granulocytes in the coronary system, and improved pump function, especially that of the intrinsically weaker right ventricle.
Apart from albumin, other experimentally promising procedures aimed at preserving the coronary glycocalyx from damage under various conditions include cardiac (pre)treatment with hydrocortisone and antithrombin [5, 15, 17, 19]. Since full regeneration of the glycocalyx seems to require 5–7 days in vivo [84], prevention is certainly better than cure. However, partial recovery may occur more rapidly because the endothelial cavaeolae are filled with glycocalyx which could be externalised on short notice [21]. The restitution of fluid homeostasis of septic patients within just days in the advent of successful anti-infectious therapy, a familiar clinical observation, suggests pathways of quick repair of the glycocalyx. Indeed, nature shows that extremely rapid turnover is possible. Trypanosomes avoid immunological destruction by weaving a dense coat of surface glycoproteins of constantly changing composition to ward off attack by human antibodies [37].
Caveats and conclusions
-
1.
Per definition, one may distinguish between the endothelial glycocalyx proper, and the layer formed when plasma constituents bind to and intercalate in the structure. Although there are basic differences between the endothelial glycocalyx and the endothelial surface layer, it is the latter we are concerned with in vivo.
-
2.
Serious differences exist between endothelial cells in situ and cultured endothelial cells. This pertains, for one, to the dimensions of the glycocalyx (200–2,000 nm in situ, only 20–40 nm in culture [21, 55, 83]). For another, when studying permeability, responses of cultured endothelial layers and endothelial cells in situ differ so much that they cannot really be compared. Curry and Adamson [26] have listed a great number of discrepant results and are adamant in the claim that cultured cells are at best a model for endothelium in a chronically inflamed state.
-
3.
Gender may influence coronary permeability responses [46]. However, hard data for the human heart is still needed. Furthermore, the fundamental mechanisms regarding the role of the endothelial glycocalyx can hardly be different for males and females.
-
4.
The main contradictions of the historical principle of Ernest Starling with the modern Endothelial Surface Layer Concept for the coronary system are:
-
The endothelial glycocalyx is the basal structure which creates, together with plasma proteins, the physiological oncotic gradient at the vascular barrier.
-
The interstitial protein concentration is of little consequence with respect to arteriolar and capillary fluid filtration under physiological circumstances.
-
Bulk colloid exchange between the vascular and the interstitial spaces is substantial, but not indiscriminate, and occurs mainly in the venular segments. Tissue factor is sequestered away from extravasating coagulation factors in intact heart tissue.
-
The endothelial cell bodies act as a second barrier, the width of interendothelial clefts and pores determining hydraulic conductivity, especially if the endothelial glycocalyx is altered.
-
-
5.
The endothelial glycocalyx is important for regulating vascular permeability and fluid exchange in many organs besides the heart [1, 35, 97, 98, 115]. However, details are often still lacking, and the systemic importance is just beginning to emerge.
-
6.
These findings are interesting from a physiological standpoint as they challenge old knowledge. More importantly, however, they are crucial for the treating physician. It is time to forcefully sound possibilities to protect the endothelial surface layer in critically ill patients [5].
References
Adamson RH, Clough G (1992) Plasma proteins modify the endothelial cell glycocalyx of frog mesenteric microvessels. J Physiol 445:473–486
Adamson RH, Lenz JF, Zhang X, Adamson GN, Weinbaum S, Curry FE (2004) Oncotic pressures opposing filtration across non-fenestrated rat microvessels. J Physiol 557:889–907
Annecke T, Chappell D, Chen C, Jacob M, Welsch U, Sommerhoff CP, Rehm M, Conzen PF, Becker BF (2010) Sevoflurane preserves the endothelial glycocalyx against ischaemia-reperfusion injury. Br J Anaesth 104:414–421
Areskog NH, Arturson G, Grotte G, Wallenius G (1964) Studies on heart lymph. Arch Dis Child 39:182–186
Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M (2010) Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc Res 87:300–310
Beresewicz A, Czarnowska E, Maczewski M (1998) Ischemic preconditioning and superoxide dismutase protect against endothelial dysfunction and endothelium glycocalyx disruption in the postischemic guinea-pig hearts. Mol Cell Biochem 186:87–97
Brands J, Spaan JA, van den Berg BM, Vink H, VanTeeffelen JW (2010) Acute attenuation of glycocalyx barrier properties increases coronary blood volume independently of coronary flow reserve. Am J Physiol Heart Circ Physiol 298:H515–H523
Brandstrup B, Tonnesen H, Beier-Holgersen R, Hjortso E, Ording H, Lindorff-Larsen K, Rasmussen MS, Lanng C, Wallin L, Iversen LH, Gramkow CS, Okholm M, Blemmer T, Svendsen PE, Rottensten HH, Thage B, Riis J, Jeppesen IS, Teilum D, Christensen AM, Graungaard B, Pott F (2003) Effects of intravenous fluid restriction on postoperative complications: comparison of two perioperative fluid regimens: a randomized assessor-blinded multicenter trial. Ann Surg 238:641–648
Breuckmann F, Nassenstein K, Bucher C, Konietzka I, Kaiser G, Konorza T, Naber C, Skyschally A, Gres P, Heusch G, Erbel R, Barkhausen J (2009) Systematic analysis of functional and structural changes after coronary microembolization: a cardiac magnetic resonance imaging study. JACC Cardiovasc Imaging 2:121–130
Bruegger D, Jacob M, Rehm M, Loetsch M, Welsch U, Conzen P, Becker BF (2005) Atrial natriuretic peptide induces shedding of endothelial glycocalyx in coronary vascular bed of guinea pig hearts. Am J Physiol Heart Circ Physiol 289:H1993–H1999
Bruegger D, Rehm M, Abicht J, Paul JO, Stoeckelhuber M, Pfirrmann M, Reichart B, Becker BF, Christ F (2009) Shedding of the endothelial glycocalyx during cardiac surgery: on-pump versus off-pump coronary artery bypass graft surgery. J Thorac Cardiovasc Surg 138:1445–1447
Bruegger D, Rehm M, Jacob M, Chappell D, Stoeckelhuber M, Welsch U, Conzen P, Becker BF (2008) Exogenous nitric oxide requires an endothelial glycocalyx to prevent postischemic coronary vascular leak in guinea pig hearts. Crit Care 12:R73
Carden DL, Granger DN (2000) Pathophysiology of ischaemia-reperfusion injury. J Pathol 190:255–266
Chappell D, Doerfler N, Jacob M, Rehm M, Welsch U, Conzen P, Becker BF (2010) Glycocalyx protection reduces leukocyte adhesion following ischemia/reperfusion. Shock 34:133–139
Chappell D, Hofmann-Kiefer K, Jacob M, Rehm M, Briegel J, Welsch U, Conzen P, Becker BF (2009) TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res Cardiol 104:78–89
Chappell D, Jacob M, Becker BF, Hofmann-Kiefer K, Conzen P, Rehm M (2008) Expedition glycocalyx: a newly discovered “Great Barrier Reef”. Anaesthesist 57:959–969
Chappell D, Jacob M, Hofmann-Kiefer K, Bruegger D, Rehm M, Conzen P, Welsch U, Becker BF (2007) Hydrocortisone preserves the vascular barrier by protecting the endothelial glycocalyx. Anesthesiology 107:776–784
Chappell D, Jacob M, Hofmann-Kiefer K, Conzen P, Rehm M (2008) A rational approach to perioperative fluid management. Anesthesiology 109:723–740
Chappell D, Jacob M, Hofmann-Kiefer K, Rehm M, Welsch U, Conzen P, Becker BF (2009) Antithrombin reduces shedding of the endothelial glycocalyx following ischaemia/reperfusion. Cardiovasc Res 83:388–396
Chappell D, Jacob M, Paul O, Mehringer L, Newman W, Becker BF (2008) Impaired glycocalyx barrier properties and increased capillary tube haematocrit. J Physiol 586:4585–4586
Chappell D, Jacob M, Paul O, Rehm M, Welsch U, Stoeckelhuber M, Conzen P, Becker BF (2009) The glycocalyx of the human umbilical vein endothelial cell: an impressive structure ex vivo but not in culture. Circ Res 104:1313–1317
Chappell D, Jacob M, Rehm M, Stoeckelhuber M, Welsch U, Conzen P, Becker BF (2008) Heparinase selectively sheds heparan sulphate from the endothelial glycocalyx. Biol Chem 389:79–82
Collard CD, Gelman S (2001) Pathophysiology, clinical manifestations, and prevention of ischemia-reperfusion injury. Anesthesiology 94:1133–1138
Curry FE, Michel CC (1980) A fiber matrix model of capillary permeability. Microvasc Res 20:96–99
Curry FR (2005) Atrial natriuretic peptide: an essential physiological regulator of transvascular fluid, protein transport, and plasma volume. J Clin Invest 115:1458–1461
Curry FR, Adamson RH (2010) Vascular permeability modulation at the cell, microvessel, or whole organ level: towards closing gaps in our knowledge. Cardiovasc Res 87:218–229
Curry FR, Rygh CB, Karlsen T, Wiig H, Adamson RH, Clark JF, Lin YC, Gassner B, Thorsen F, Moen I, Tenstad O, Kuhn M, Reed RK (2010) Atrial natriuretic peptide modulation of albumin clearance and contrast agent permeability in mouse skeletal muscle and skin: role in regulation of plasma volume. J Physiol 588:325–339
Czarnowska E, Karwatowska-Prokopczuk E (1995) Ultrastructural demonstration of endothelial glycocalyx disruption in the reperfused rat heart. Involvement of oxygen free radicals. Basic Res Cardiol 90:357–364
Danielli JF (1940) Capillary permeability and oedema in the perfused frog. J Physiol 98:109–129
Desai KV, Laine GA, Stewart RH, Cox CS Jr, Quick CM, Allen SJ, Fischer UM (2008) Mechanics of the left ventricular myocardial interstitium: effects of acute and chronic myocardial edema. Am J Physiol Heart Circ Physiol 294:H2428–H2434
Desjardins C, Duling BR (1990) Heparinase treatment suggests a role for the endothelial cell glycocalyx in regulation of capillary hematocrit. Am J Physiol 258:H647–H654
Dhalla NS, Elmoselhi AB, Hata T, Makino N (2000) Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc Res 47:446–456
Dongaonkar RM, Stewart RH, Geissler HJ, Laine GA (2010) Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Cardiovasc Res 87:331–339
Drake TA, Morrissey JH, Edgington TS (1989) Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol 134:1087–1097
Dull RO, Mecham I, McJames S (2007) Heparan sulfates mediate pressure-induced increase in lung endothelial hydraulic conductivity via nitric oxide/reactive oxygen species. Am J Physiol Lung Cell Mol Physiol 292:L1452–L1458
Esmon CT (2002) Protein C pathway in sepsis. Ann Med 34:598–605
Field MC, Lumb JH, Adung’a VO, Jones NG, Engstler M (2009) Macromolecular trafficking and immune evasion in african trypanosomes. Int Rev Cell Mol Biol 278:1–67
Gebhard C, Akhmedov A, Mocharla P, Angstenberger J, Sahbai S, Camici GG, Luscher TF, Tanner FC (2010) PDGF-CC induces tissue factor expression: role of PDGF receptor alpha/beta. Basic Res Cardiol 105:349–356
Gilles S, Zahler S, Welsch U, Sommerhoff CP, Becker BF (2003) Release of TNF-alpha during myocardial reperfusion depends on oxidative stress and is prevented by mast cell stabilizers. Cardiovasc Res 60:608–616
Gotte M (2003) Syndecans in inflammation. FASEB J 17:575–591
Heindl B, Reichle FM, Zahler S, Conzen PF, Becker BF (1999) Sevoflurane and isoflurane protect the reperfused guinea pig heart by reducing postischemic adhesion of polymorphonuclear neutrophils. Anesthesiology 91:521–530
Huxley VH, Curry FE (1985) Albumin modulation of capillary permeability: test of an adsorption mechanism. Am J Physiol 248:H264–H273
Huxley VH, Curry FE (1987) Effect of superfusate albumin on single capillary hydraulic conductivity. Am J Physiol 252:H395–H401
Huxley VH, Curry FE (1991) Differential actions of albumin and plasma on capillary solute permeability. Am J Physiol 260:H1645–H1654
Huxley VH, Tucker VL, Verburg KM, Freeman RH (1987) Increased capillary hydraulic conductivity induced by atrial natriuretic peptide. Circ Res 60:304–307
Huxley VH, Wang J (2010) Cardiovascular sex differences influencing microvascular exchange. Cardiovasc Res 87:230–242
Jacob M, Bruegger D, Rehm M, Stoeckelhuber M, Welsch U, Conzen P, Becker BF (2007) The endothelial glycocalyx affords compatibility of Starling’s principle and high cardiac interstitial albumin levels. Cardiovasc Res 73:575–586
Jacob M, Bruegger D, Rehm M, Welsch U, Conzen P, Becker BF (2006) Contrasting effects of colloid and crystalloid resuscitation fluids on cardiac vascular permeability. Anesthesiology 104:1223–1231
Jacob M, Chappell D, Conzen P, Finsterer U, Krafft A, Becker BF, Rehm M (2008) Impact of the time window on plasma volume measurement with indocyanine green. Physiol Meas 29:761–770
Jacob M, Chappell D, Rehm M (2007) Clinical update: perioperative fluid management. Lancet 369:1984–1986
Jacob M, Chappell D, Rehm M (2009) Third space—fact or fiction? Best Pract Res Clin Anaesthesiol 23:145–157
Jacob M, Chappell D, Stoeckelhuber M, Welsch U, Rehm M, Bruegger D, Kaczmarek I, Conzen P, Becker BF (2010) Perspectives in microvascular fluid handling: does distribution of coagulation factors in human myocardium comply with plasma extravasation in venular coronary segments? J Vasc Res (in press)
Jacob M, Conzen P, Finsterer U, Krafft A, Becker BF, Rehm M (2007) Technical and physiological background of plasma volume measurement with indocyanine green: a clarification of misunderstandings. J Appl Physiol 102:1235–1242
Jacob M, Paul O, Mehringer L, Chappell D, Rehm M, Welsch U, Kaczmarek I, Conzen P, Becker BF (2009) Albumin augmentation improves condition of guinea pig hearts after 4 hours of cold ischemia. Transplantation 87:956–965
Jacob M, Rehm M, Loetsch M, Paul JO, Bruegger D, Welsch U, Conzen P, Becker BF (2007) The endothelial glycocalyx prefers albumin for evoking shear stress-induced, nitric oxide-mediated coronary dilatation. J Vasc Res 44:435–443
Juchem G, Weiss DR, Gansera B, Kemkes BM, Mueller-Hoecker J, Nees S (2010) Pericytes in the macrovascular intima: possible physiological and pathogenetic impact. Am J Physiol Heart Circ Physiol 298:H754–H770
Kanwar S, Hickey MJ, Kubes P (1998) Postischemic inflammation: a role for mast cells in intestine but not in skeletal muscle. Am J Physiol 275:G212–G218
Kellman P, Aletras AH, Mancini C, McVeigh ER, Arai AE (2007) T2-prepared SSFP improves diagnostic confidence in edema imaging in acute myocardial infarction compared to turbo spin echo. Magn Reson Med 57:891–897
Kleinbongard P, Heusch G, Schulz R (2010) TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol Ther 127:295–314
Kupatt C, Habazettl H, Zahler S, Weber C, Becker BF, Messmer K, Gerlach E (1997) ACE-inhibition prevents postischemic coronary leukocyte adhesion and leukocyte-dependent reperfusion injury. Cardiovasc Res 36:386–395
Langdell RD, Bowersox LW, Weaver RA, Gibson WS (1960) Coagulation properties of canine thoracic-duct lymph. Am J Physiol 199:626–628
Levick JR (1991) Capillary filtration-absorption balance reconsidered in light of dynamic extravascular factors. Exp Physiol 76:825–857
Levick JR, Michel CC (2010) Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res 87:198–210
Lieleg O, Baumgartel RM, Bausch AR (2009) Selective filtering of particles by the extracellular matrix: an electrostatic bandpass. Biophys J 97:1569–1577
Lobo DN, Bostock KA, Neal KR, Perkins AC, Rowlands BJ, Allison SP (2002) Effect of salt and water balance on recovery of gastrointestinal function after elective colonic resection: a randomised controlled trial. Lancet 359:1812–1818
Lowell JA, Schifferdecker C, Driscoll DF, Benotti PN, Bistrian BR (1990) Postoperative fluid overload: not a benign problem. Crit Care Med 18:728–733
Luft JH (1966) Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed Proc 25:1773–1783
McDonagh PF, Rauzzino MJ (1993) Stimulated leukocyte adhesion in coronary microcirculation is reduced by a calcium antagonist. Am J Physiol 265:H476–H483
Mehlhorn U, Geissler HJ, Laine GA, Allen SJ (2001) Myocardial fluid balance. Eur J Cardiothorac Surg 20:1220–1230
Meuwese MC, Mooij HL, Nieuwdorp M, van LB, Marck R, Vink H, Kastelein JJ, Stroes ES (2009) Partial recovery of the endothelial glycocalyx upon rosuvastatin therapy in patients with heterozygous familial hypercholesterolemia. J Lipid Res 50:148–153
Michel CC, Curry FR (2009) Glycocalyx volume: a critical review of tracer dilution methods for its measurement. Microcirculation 16:213–219
Miura T, Miki T (2008) Limitation of myocardial infarct size in the clinical setting: current status and challenges in translating animal experiments into clinical therapy. Basic Res Cardiol 103:501–513
Mulivor AW, Lipowsky HH (2004) Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol 286:H1672–H1680
Nelson A, Berkestedt I, Schmidtchen A, Ljunggren L, Bodelsson M (2008) Increased levels of glycosaminoglycans during septic shock: relation to mortality and the antibacterial actions of plasma. Shock 30:623–627
Nieuwdorp M, Meuwese MC, Mooij HL, Ince C, Broekhuizen LN, Kastelein JJ, Stroes ES, Vink H (2008) Measuring endothelial glycocalyx dimensions in humans: a potential novel tool to monitor vascular vulnerability. J Appl Physiol 104:845–852
Nieuwdorp M, van Haeften TW, Gouverneur MC, Mooij HL, van Lieshout MH, Levi M, Meijers JC, Holleman F, Hoekstra JB, Vink H, Kastelein JJ, Stroes ES (2006) Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes 55:480–486
Nisanevich V, Felsenstein I, Almogy G, Weissman C, Einav S, Matot I (2005) Effect of intraoperative fluid management on outcome after intraabdominal surgery. Anesthesiology 103:25–32
Noble MI, Drake-Holland AJ, Vink H (2008) Hypothesis: arterial glycocalyx dysfunction is the first step in the atherothrombotic process. QJM 101:513–518
Pahakis MY, Kosky JR, Dull RO, Tarbell JM (2007) The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem Biophys Res Commun 355:228–233
Parish CR (2006) The role of heparan sulphate in inflammation. Nat Rev Immunol 6:633–643
Platts SH, Duling BR (2004) Adenosine A3 receptor activation modulates the capillary endothelial glycocalyx. Circ Res 94:77–82
Platts SH, Linden J, Duling BR (2003) Rapid modification of the glycocalyx caused by ischemia-reperfusion is inhibited by adenosine A2A receptor activation. Am J Physiol Heart Circ Physiol 284:H2360–H2367
Potter DR, Damiano ER (2008) The hydrodynamically relevant endothelial cell glycocalyx observed in vivo is absent in vitro. Circ Res 102:770–776
Potter DR, Jiang J, Damiano ER (2009) The recovery time course of the endothelial cell glycocalyx in vivo and its implications in vitro. Circ Res 104:1318–1325
Pries AR, Kuebler WM (2006) Normal endothelium. Handb Exp Pharmacol 1:1–40
Pries AR, Secomb TW, Gaehtgens P (2000) The endothelial surface layer. Pflugers Arch 440:653–666
Raschke P, Becker BF, Leipert B, Schwartz LM, Zahler S, Gerlach E (1993) Postischemic dysfunction of the heart induced by small numbers of neutrophils via formation of hypochlorous acid. Basic Res Cardiol 88:321–339
Rehm M, Bruegger D, Christ F, Thiel M, Conzen P, Jacob M, Chappell D, Stoeckelhuber M, Welsch U, Reichart B, Peter K, Becker BF (2007) Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation 116:1896–1906
Rehm M, Haller M, Orth V, Kreimeier U, Jacob M, Dressel H, Mayer S, Brechtelsbauer H, Finsterer U (2001) Changes in blood volume and hematocrit during acute preoperative volume loading with 5% albumin or 6% hetastarch solutions in patients before radical hysterectomy. Anesthesiology 95:849–856
Rehm M, Orth V, Kreimeier U, Thiel M, Haller M, Brechtelsbauer H, Finsterer U (2000) Changes in intravascular volume during acute normovolemic hemodilution and intraoperative retransfusion in patients with radical hysterectomy. Anesthesiology 92:657–664
Rehm M, Zahler S, Lotsch M, Welsch U, Conzen P, Jacob M, Becker BF (2004) Endothelial glycocalyx as an additional barrier determining extravasation of 6% hydroxyethyl starch or 5% albumin solutions in the coronary vascular bed. Anesthesiology 100:1211–1223
Reil JC, Gilles S, Zahler S, Brandl A, Drexler H, Hultner L, Matrisian LM, Welsch U, Becker BF (2007) Insights from knock-out models concerning postischemic release of TNFalpha from isolated mouse hearts. J Mol Cell Cardiol 42:133–141
Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG (2007) The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch 454:345–359
Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW (2005) A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J Am Coll Cardiol 45:1775–1780
Rubio-Gayosso I, Platts SH, Duling BR (2006) Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 290:H2247–H2256
Sabrane K, Kruse MN, Fabritz L, Zetsche B, Mitko D, Skryabin BV, Zwiener M, Baba HA, Yanagisawa M, Kuhn M (2005) Vascular endothelium is critically involved in the hypotensive and hypovolemic actions of atrial natriuretic peptide. J Clin Invest 115:1666–1674
Salmon AH, Neal CR, Sage LM, Glass CA, Harper SJ, Bates DO (2009) Angiopoietin-1 alters microvascular permeability coefficients in vivo via modification of endothelial glycocalyx. Cardiovasc Res 83:24–33
Singh A, Satchell SC, Neal CR, McKenzie EA, Tooke JE, Mathieson PW (2007) Glomerular endothelial glycocalyx constitutes a barrier to protein permeability. J Am Soc Nephrol 18:2885–2893
Stafford-Smith M, Lefrak EA, Qazi AG, Welsby IJ, Barber L, Hoeft A, Dorenbaum A, Mathias J, Rochon JJ, Newman MF (2005) Efficacy and safety of heparinase I versus protamine in patients undergoing coronary artery bypass grafting with and without cardiopulmonary bypass. Anesthesiology 103:229–240
Starling E (1896) On the absorption of fluid from the connective tissue spaces. J Physiol 19:312–326
Szotowski B, Antoniak S, Rauch U (2006) Alternatively spliced tissue factor: a previously unknown piece in the puzzle of hemostasis. Trends Cardiovasc Med 16:177–182
Tarbell JM (2010) Shear stress and the endothelial transport barrier. Cardiovasc Res 87:320–330
Tarbell JM, Pahakis MY (2006) Mechanotransduction and the glycocalyx. J Intern Med 259:339–350
Tucker VL (1996) Plasma ANP levels and protein extravasation during graded expansion with equilibrated whole blood. Am J Physiol 271:R601–R609
van den Berg BM, Spaan JA, Vink H (2009) Impaired glycocalyx barrier properties contribute to enhanced intimal low-density lipoprotein accumulation at the carotid artery bifurcation in mice. Pflugers Arch 457:1199–1206
van den Berg BM, Vink H, Spaan JA (2003) The endothelial glycocalyx protects against myocardial edema. Circ Res 92:592–594
VanTeeffelen JW, Brands J, Vink H (2010) Agonist-induced impairment of glycocalyx exclusion properties: contribution to coronary effects of adenosine. Cardiovasc Res 87:311–319
Vink H, Constantinescu AA, Spaan JA (2000) Oxidized lipoproteins degrade the endothelial surface layer: implications for platelet-endothelial cell adhesion. Circulation 101:1500–1502
Vink H, Duling BR (1996) Identification of distinct luminal domains for macromolecules, erythrocytes, and leukocytes within mammalian capillaries. Circ Res 79:581–589
Vink H, Duling BR (2000) Capillary endothelial surface layer selectively reduces plasma solute distribution volume. Am J Physiol Heart Circ Physiol 278:H285–H289
Vogel J, Sperandio M, Pries AR, Linderkamp O, Gaehtgens P, Kuschinsky W (2000) Influence of the endothelial glycocalyx on cerebral blood flow in mice. J Cereb Blood Flow Metab 20:1571–1578
Ward BJ, Donnelly JL (1993) Hypoxia induced disruption of the cardiac endothelial glycocalyx: implications for capillary permeability. Cardiovasc Res 27:384–389
Ward BJ, Firth JA (1989) Effect of hypoxia on endothelial morphology and interendothelial junctions in the isolated perfused rat heart. J Mol Cell Cardiol 21:1337–1347
Weinbaum S, Tarbell JM, Damiano ER (2007) The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng 9:121–167
Yuan W, Li G, Zeng M, Fu BM (2010) Modulation of the blood-brain barrier permeability by plasma glycoprotein orosomucoid. Microvasc Res 80:148–157
Zahler S, Becker BF, Raschke P, Gerlach E (1994) Stimulation of endothelial adenosine A1 receptors enhances adhesion of neutrophils in the intact guinea pig coronary system. Cardiovasc Res 28:1366–1372
Zhang C (2008) The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol 103:398–406
Zhang C, Wu J, Xu X, Potter BJ, Gao X (2010) Direct relationship between levels of TNF-alpha expression and endothelial dysfunction in reperfusion injury. Basic Res Cardiol 105:453–464
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Becker, B.F., Chappell, D. & Jacob, M. Endothelial glycocalyx and coronary vascular permeability: the fringe benefit. Basic Res Cardiol 105, 687–701 (2010). https://doi.org/10.1007/s00395-010-0118-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-010-0118-z