
Abstract
Purpose
The roles of commensal bacteria after intestinal ischemia and reperfusion (IIR) are unclear. In current study, we aim to investigate the effects and underlying mechanisms of commensal bacteria in injury and epithelial restitution after IIR.
Methods
Commensal gut bacteria were deleted by broad-spectrum antibiotics in mice. IIR was induced by clamping superior mesenteric artery. Intestinal injury, permeability, epithelial proliferation, and proinflammatory activity of mesenteric lymph were investigated.
Results
Commensals deletion improved mice survival in the early phase, but failed to improve the overall survival at 96 h after IIR. Commensals deletion reduced proliferation of intestinal epithelial cells (IEC) and augmented proinflammatory activity of mesenteric lymph after IIR. Lipopolysaccharides (LPS) supplement promoted IEC proliferation and improved survival in mice with commensals deletion after IIR. LPS induced production of prostaglandin E2 (PGE2) in mucosa via toll-like receptor 4-NFκB-cyclooxygenase 2 pathway. PGE2 enhanced IEC proliferation in vivo, which was preceded by activation of Akt and extracellular signal-regulated kinase (ERK) 1/2. Blocking of EGFR, PI3K/Akt activity abolished LPS-induced IEC proliferation.
Conclusions
Commensal bacteria are essential for epithelial restitution after IIR, which enhance IEC proliferation via induction of PGE2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intestinal ischemia and reperfusion (IIR) occurs in a wide range of diseases [1]. If severe enough, it may result in systemic inflammatory response syndrome (SIRS) or multiple organ dysfunction syndrome (MODS) [1, 2]. Reported IIR-associated mortality was up to 30–40% [1].
The orthodox theory regarding the role of intestine is that IIR induces disruption of mucosal barrier, allowing translocation of bacteria and endotoxins to distal organs, which further triggers SIRS [3]. Recent evidence supports that post-shock mesenteric lymph is responsible for the development of SIRS and MODS [3, 4]. The intestinal epithelium separates the lumen from the underlying intestinal mucosa [3]. Compromised mucosal barrier after IIR results in direct contact between commensal bacteria and immune system to produce proinflammatory products carried out by mesenteric lymph [5].
Yoshiya found that deletion of commensal bacteria reduced intestinal expression of Toll-like receptor 2 (TLR2) and TLR4, production of inflammatory mediators, and attenuated intestinal damage [6]. Germ-free mice were reported to have less local and remote injury following IIR [7]. Intestine decontamination with antibiotics reduced IIR-induced lung injury [8]. Recent studies have shown that the interaction between intestinal epithelium and bacteria mediates mucosal defense and homeostasis. These knowledge have led to new hypotheses to explain the SIRS and MODS development after IIR. Chen found that lipopolysaccharides (LPS), a ligand for TLR4, decreased IIR injury-induced intestine damage through tumor necrosis factor a (TNF-a) signaling [9]. Similarly, luminal LPS was found to ameliorate IIR injury of the intestinal wall in germ-free pigs [10]. Tatum found neonatal mice deficient in TLR4, either alone or also deficient in TLR2 as well as those lacking normal commensals, were more susceptible to IIR [11]. Importantly, selective intestinal decontamination has not been consistently shown to increase the survival of patients with IIR [12]. These findings indicate the possibilities that the interactions between intestinal mucosa and commensal bacteria play multiple roles during IIR.
We hypothesized that commensal bacteria were essential in maintenance of intestinal homeostasis and epithelial restitution after IIR. In this study, we investigated the effects of commensals deletion with or without LPS supplement in intestinal injury and restitution, using a murine model of IIR. We also explored the mechanisms of LPS in promoting IEC proliferation. Our study provides a new perspective on intestinal epithelium–commensal bacteria symbiosis in the pathophysiology of IIR, which may be helpful to develop new therapeutic strategy for the treatment of IIR and related SIRS and MODS.
Methods
Mice
Specific pathogen-free male TLR4−/− knockout (KO) mice on a C57BL/10 background and TLR4+/+ wild type (WT) C57BL/10 mice were maintained in the specific pathogen-free animal facility. All studies were approved by the Institutional Animal Care and Use of the Committee at Tongji Medical College (Wuhan, China) (Permit Number 2012-AR0122).
Commensal bacteria deletion and LPS supplement
Commensal bacteria were deleted using an established protocol [13]. Briefly, C57BL/10 mice were provided ampicillin (1 g/L), vancomycin (500 mg/L), neomycin sulfate (1 g/L), and metronidazole (1 g/L) in drinking water from 4 to 8 weeks of age. Fecal sample was collected from colon using a sterile technique. Both aerobic and anaerobic cultures were performed to confirm the commensals deletion.
To study the effects of bacteria on injury and restitution after intestinal ischemia and reperfusion (IIR), LPS was given to the mice with commensals deletion. At the 4th week of antibiotics treatment, LPS (1, 5 or 25 µg/mL) was added to drinking water for 1 week. After 4 weeks’ antibiotics and 1 week LPS treatment, the mice were subjected to IIR.
Mouse model of intestinal ischemia and reperfusion (IIR)
Eight-weeks-old male mice were anesthetized and performed a midline laparotomy. The superior mesenteric artery (SMA) and the collateral branch from the celiac axis were identified and occluded with non-traumatic vascular clamps. The clamps were removed after 45 min of ischemia to induce reperfusion. In the sham-operated group, the SMA and collateral branches were identified, but were not occluded. Mice were provided drinking water containing antibiotics until the end point was reached. At endpoints, mice were killed and their intestine, lung, and blood samples were harvested.
Histologic injury of ileum was graded using a histologic scoring system. Intestinal permeability of the distal jejunum was assessed using the everted sac method [14]. Apoptotic epithelial cells were detected by TUNEL staining. Proliferating enterocytes were detected by immunoperoxidase staining for the thymidine analog bromodeoxyuridine (BrdU).
Western blot and qRT-PCR analysis was carried out to study the cell signaling proteins. PGE2 levels were determined by enzyme-linked immunosorbent assay.
Survival study
Mice were randomized into the following groups (n = 40 for each group): (1) control group: the mice with normal commensals were subjected to IIR. (2) AT group: after 4 weeks’ antibiotics treatment (AT), the mice with commensals deletion were subjected to IIR. (3) AT + LPS1μg/mL: after commensals deletion, mice were fed with LPS (1 μg/mL) for 1 week and subjected to IIR. (4) AT + LPS5μg/mL: after commensals deletion, mice were fed with LPS (5 μg/mL) for 1 week and subjected to IIR. (5) AT + LPS25μg/mL: after commensals deletion, mice were fed with LPS (25 μg/mL) for 1 week and subjected to IIR. Death of mice was recorded every 4 h until the endpoint.
Pulmonary injury secondary to IIR
Tissue sections were stained with H and E. Injury was assessed using an established scoring system. Myeloperoxidase (MPO) activity in homogenates of the lung and Evans blue dye assay for pulmonary vascular permeability was determined as previously described [15].
Determination of neutrophil respiratory burst induced by mesenteric lymph
Whole blood was obtained via cardiac puncture from naïve male mice. PMNs were isolated. Medium or the collected mesenteric lymph was then added and PMN respiratory burst was measured by flow cytometry [14].
Cell proliferation assay
Caco-2 cells (1 × 106 cells) were plated on 96-well plate. After LPS or specific blockers treatment, proliferation was assessed after 36 h incubation using Quick Cell Proliferation Assay kit [16].
Statistical analysis
The Student t test or Wilcoxon two-sample test was used to compare the differences between two groups and one-way analysis of variance or Kruskal–Wallis test was used to compare differences, among multiple groups. For survival determination, Kaplan–Meier analysis was used. Statistical analyses were performed using SAS software (SAS 9.2, SAS Institute, NC, USA). p values less than 0.05 were considered statistically significant in all the studies.
Results
Intestinal ischemia and reperfusion injury
Prior to IIR, bacterial cultures were performed to confirm the complete deletion of commensals. After IIR, histologic injuries were manifested as: vacuolization of IEC, presence of a sub-epithelial space, separation of epithelium and basal membrane, mucosal ulceration, and necrosis (Fig. 1a). Injuries to jejunum, ileum, and colon peaked at 4 h after IIR. Most severe IIR was found at ileum. However, at 72 h, integrity of epithelium was almost restored, indicating accomplished epithelial restitution.

Intestinal ischemia and reperfusion injury (IIR). a Injury of jejunum, ileum, and colon at 4 h, 24 h, 48 h, 72 h after IIR. b Histologic injury scoring system. Grade 0: no injury; Grade 1: presence of a sub-epithelial space; Grade 2: loss of mucosal lining of the villous tip; Grade 3: loss of less than half of the villous structure; Grade 4: loss of more than half of the villous structure; Grade 5: transmural necrosis; c intestinal epithelial cells (IEC) apoptosis at 4 h, 24 h, 48 h, 72 h after IIR (TUNEL staining); d intestinal permeability; e injury score of terminal ileum; f apoptotic index of terminal ileum; n = 6 per group. Antibiotics treatment: the mice were treated with antibiotics and subjected to IIR. Controls: the mice with normal commensals were subjected to IIR. *vs Antibiotics treatment, **vs Controls, p < 0.05
Injury scores of ileum (Fig. 1b, e), intestinal permeability (FD4 clearance) (Fig. 1d), and apoptosis of IEC (Fig. 1c, f) significantly increased after IIR. Importantly, intestinal permeability and apoptotic indices peaked at 4 h, and decreased thereafter until 72 h.
Commensals deletion failed to improve the overall survival after IIR
After IIR, 22 (55%) of the 40 mice died during the 96 h observational period. None of the 40 sham-operated mice died. Among the 40 mice with commensals deletion, 20 (50%) died during 96 h, which was not significantly different from the mice with normal commensals (Fig. 2a). However, in the first 36 h, mortality of the mice with normal commensals was 47.5% (19/40), which was significantly higher than that of the mice with commensals deletion (27.5%, 11/40) (Fig. 2b). Because intestinal injury peaked in the first 36 h, during which most mortality occurred, we defined the first 36 h as the early phase after IIR and the subsequent period as the restitution phase. After IIR, commensals deletion reduced mortality in the early phase, but did not improve the overall survival.

Commensals deletion does not improve the overall survival after IIR. Mice were subjected to IIR. Death of mice was recorded every 4 h until 96 h after IIR. n = 40 per group. a Commensals deletion failed to improve the survival at 96 h; b commensals deletion improved the survival at the early phase after IIR. c Each mouse was injected intraperitoneally with 5-BrdU at 2 h (120 mg/kg) before being killed. BrdU immunohistochemistry demonstrates labelled crypt cells at 96 h after IIR in mice with normal commensals; d proliferative IEC in mice with commensals deletion; e quantitation of BrdU labelling was by determining per cent positive cells in 20–35 well-oriented villi and crypts measured per sample, n = 3 per group. *vs Controls, p < 0.05. Controls: the mice with normal commensals were subjected to IIR. Antibiotics treatment: the mice with commensals deletion were subjected to IIR. f Neutrophils purification was determined by flow cytometry; g neutrophils were treated with mesenteric lymph from the mice at 12 h after IIR. Proinflammatory activity of mesenteric lymph was measured by PMN respiratory burst; h neutrophils were treated with mesenteric lymph from the mice at 48 h after IIR. Controls the lymph from the mice with normal commensals, AT the lymph from the mice with commensals deletion, SO the lymph from the sham-operated control mice, IIR + LPS1, 5, 25 LPS (1, 5 or 25 µg/mL) was fed to the mice for 1 week before IIR. *vs Controls, p < 0.05; **vs AT, p < 0.05
Commensals deletion impeded intestinal epithelial restitution
At 4 h after IIR, intestinal injury scores of the mice with commensals deletion were lower than controls with normal commensals (4.25 ± 0.67 vs 3.5 ± 0.52, p < 0.05) (Fig. 1e).
At 4 h and 12 h, intestinal permeability and IEC apoptotic indices of the mice with commensals deletion were lower than controls with normal commensals (all p < 0.05) (Fig. 1f). However, at 48 h and 60 h, intestinal permeability of the mice with commensals deletion were higher than controls with normal commensals (all p < 0.05) (Fig. 1d).
Commensals deletion impaired proliferation of IEC, augmented pro-inflammatory activity of mesenteric lymph in restitution phase
In mice with commensals deletion, calculated proliferation (13.20 ± 1.41 vs 8.67 ± 2.16, p < 0.05) was lower than controls with normal commensals (Fig. 2c–e).
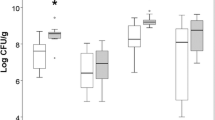
Naïve neutrophils were isolated from blood by density gradient centrifugation. The purity of PMNs was > 95%, as confirmed by flow cytometry (Fig. 2f). The mice with or without commensals deletion were subjected to IIR. At 12 h after IIR, mesenteric lymph was collected. In vitro respiratory burst of neutrophils was measured after treatment by mesenteric lymph (Fig. 2g). Neutrophil response induced by the lymph from the mice with commensals deletion was weaker than the lymph from controls.
Lymph collected at 48 h induced neutrophils response was weaker than those collected at 12 h after IIR (Fig. 2h). Neutrophil response induced by lymph from the mice with commensals deletion was stronger than the lymph from controls (Fig. 2h).
LPS supplement improved survival of mice with commensals deletion after IIR
After antibiotics treatment (AT), the relative mRNA expressions of TLR4 reduced from 1 to 0.20 ± 0.04 (Fig. 3a). Luminal LPS reduced from 4.68 ± 0.53 to 0.16 ± 0.08 µg/mL (Fig. 3b). Before LPS1µg/mL feeding, mRNA expression of TLR4 decreased to 0.20 ± 0.04. After LPS feeding, it increased to 0.89 ± 0.11 prior to IIR, and 1.13 ± 0.16 at 3 days after IIR (Fig. 3c). However, higher dose (5 or 25 µg/mL) of LPS did not further increase TLR4 expression (Fig. 3c).

LPS supplement alleviates intestinal injury and improves the survival of the mice with commensals deletion after IIR. a mRNA expression of TLR4 at terminal ileum of mice before antibiotics treatment (0 w), after treatment for 1 week (1 w), 2 weeks (2 w), and 3 weeks (3 w), *vs 0 w, p < 0.05. b LPS concentration at terminal ileum before antibiotics treatment (0 w), after treatment for 1 w, 2 w, and 3 w, *vs 0 w, p < 0.05. c The mice with commensals deletion were fed with LPS (1, 5 or 25 µg/mL) in drinking water. After LPS feeding for 1 w, the mice were subjected to IIR. mRNA expression of TLR4 at terminal ileum at 1 day, 2 days, and 3 days after IIR. n = 3 per group. *vs LPS (1, 5 or 25 µg/mL), p < 0.05. d LPS (1 μg/mL) supplement improved the survival of the mice with commensals deletion at 96 h. AT vs LPS1μg/mL, p < 0.05. e IEC apoptotic index at 0–96 h after IIR; f intestinal permeability. n = 6 per group. Controls: the mice with normal commensals; AT: the mice with commensals deletion; AT + LPS1, 5, 25 μg/mL: LPS1, 5, 25 μg/mL was fed to the mice with commensals deletion for 1 week prior to IIR. AT + LPS: LPS (1 μg/mL) was fed to the mice with commensals deletion for 1 week prior to IIR; * AT vs AT + LPS, p < 0.05
At 96 h, of the mice treated with antibiotics (AT mice) and fed with 1 µg/mL LPS, mortality was 7.5%. Mortality was 32.5% and 40% of in the AT mice fed with 5 µg/mL or 25 µg/mL LPS. Compared with AT mice, LPS1µg/mL feeding improved survival after IIR. However, higher dose LPS (5 µg/mL or 25 µg/mL) did not improve survival (Fig. 3d).
Compared with AT mice, IEC apoptotic indices of AT + LPS1µg/mL mice were not different after IIR (all p > 0.05) (Fig. 3e). At 24 h, 36 h, 48 h, and 60 h intestinal permeability of AT + LPS1µg/mL mice was significantly lower than AT mice (Fig. 3f). The findings suggested that LPS did not increased injury to intestine, but promoted intestinal barrier recovery.
LPS supplement elevated expression of TLR4 and production of PGE2 in mucosa
After antibiotics treatment, mucosal PGE2 (prostaglandin E2) reduced to 0.38 ± 0.10 pg/mg protein. After LPS (1, 5 or 25 μg/mL) feeding for 1 week, mucosal PGE2 increased (Fig. 4a). At 24 h after IIR, LPS (1, 5 or 25 μg/mL) feeding significantly increased mucosal PGE2 in mice with commensals deletion. No difference was found between the mice fed with LPS of different dose. Same results were found at 48 h and 72 h, LPS increased mucosal PGE2 in mice with commensals deletion, regardless of LPS dosage (all p > 0.05) (Fig. 4a). At 24 h, PGE2 production abolished TLR4 gene knockout or a COX2 inhibitor, Celecoxib treatment (Fig. 4b).

LPS supplement increases epithelial proliferation via inducing PGE2 in mucosa of the mice with commensals deletion. a Mucosal PGE2 examined by ELISA at 0 day, 1 day, 2 days, and 3 days after IIR. *vs AT, p < 0.05; **vs Controls, p < 0.05. b Mucosal PGE2 examined by ELISA at 0 day or 1 day after IIR. *vs AT, p < 0.05; **vs AT + LPS, p < 0.05. c, d Mucosal COX2 and PCNA expression examined by western blotting. *vs AT, p < 0.05; **vs AT + LPS, p < 0.05. e IEC proliferation in C57BL/10 mice at 48 h. n = 3 per group. *vs Control, p < 0.05; **vs AT, p < 0.05; #vs AT + LPS, p < 0.05. f IEC proliferation of TLR4−/− mice at 48 h. Controls: the mice with normal commensals; AT: the mice with commensals deletion; AT + LPS: LPS1, 5, 25 μg/mL was fed to the mice with commensals deletion for 1 week prior to IIR. AT + LPS: LPS1μg/mL was fed to the C57BL/10 mice with commensals deletion for 1 week prior to IIR. TLR4 KO + AT + LPS: LPS1μg/mL was fed to the TLR4−/− mice with commensals deletion for 1 week prior to IIR; AT + LPS + Celecoxib: LPS1µg/mL and Celecoxib100mg/kg was fed to the TLR4−/− mice with commensals deletion for 1 week prior to IIR. *vs Control, p < 0.05
The results suggested LPS feeding in AT mice increased mucosal PEG2 at 24 h, 48 h, and 72 h. However, increasing dose of LPS did not further increase PEG2.
LPS supplement promoted IEC proliferation via TLR4-COX2-PGE2 signaling pathway
Mucosal PGE2 was produced by COX2 (cyclooxygenase). Of the mice with commensal deletion, at 24 h and 72 h, mucosal COX2 increased. LPS supplement (1 µg/mL) further increased mucosal COX2 at 24 h and 48 h (Fig. 4c, d). Expression of PCNA did not change at 48 h and 72 h in the mice with commensals deletion. However, LPS treatment increased the expression of PCNA at 48 h and 72 h (Fig. 4c, d).
At 48 h after IIR, LPS1µg/mL increased proliferation of IEC in mice with commensals deletion, which was abolished by Celecoxib (Fig. 4e). Furthermore, LPS1µg/mL or LPS1µg/mL + Celecoxib treatment did not change IEC proliferation of the TLR4 knockout mice with commensals deletion (all p > 0.05) (Fig. 4f). The findings suggested LPS-induced IEC proliferation was TLR4- and COX2-dependent.
LPS supplement activated mucosal NFκB, EGFR, and PI3K/AKT signaling pathway
At 24 h after IIR, phosphorylation of IκB and expression of nuclear p65 in the mucosa of the AT mice increased, but they were lower than those of AT + LPS1µg/mL mice. Celecoxib did not alter phosphorylation of IκB and nuclear p65 expression in mice fed with LPS (Fig. 5a, b).

Western blotting of signaling pathway molecules. a Western blotting; LPS activates NFκB signaling pathway in mucosa, which could be abolished by knockout of TLR4. b Mucosal IκB phosphorylation and nuclear NFκB p65 level examined by western blotting at 24 h after IIR. Each protein was normalized to β-actin. Each lane is loaded with equivalent amounts of tissue extract pooled from n = 3 mice per point. *vs AT + IIR1d, p < 0.05. **vs AT + LPS + IIR1d, p < 0.05. c LPS activates EGFR, PI3K/AKT, and MAPK/ERK1/2 signaling pathway. Phosphorylation of EGFR, AKT, ERK1/2, P38, and JNK in mucosa examined by western blotting. *vs AT + IIR1d, p < 0.05. **vs AT + LPS + IIR1d, p < 0.05. AT: the mice with commensals deletion; AT + LPS: LPS1μg/mL was fed to the C57BL/10 mice with commensals deletion for 1 week prior to IIR. AT + IIR0d: the C57BL/10 mice with commensals deletion prior to IIR. AT + IIR1d: the C57BL/10 mice with commensals deletion at 1 day after IIR. AT + LPS + IIR1d: the C57BL/10 mice with commensals deletion and LPS supplement at 1 day after IIR. AT + LPS + Celecoxib + IIR1d: the C57BL/10 mice with commensals deletion and LPS1µg/mL and Celecoxib100mg/kg treatment at 1 day after IIR. TLR4 KO + AT + LPS + IIR1d: the TLR4−/− mice with commensals deletion and LPS supplement at 1 day after IIR
Phosphorylation of EGFR, AKT, and P38 in mucosa increased. Celecoxib abolished LPS-induced phosphorylation of EGFR, AKT, and P38 in AT mice. In AT + LPS mice, phosphorylation of EGFR, AKT, and P38 of TLR4 KO mice was lower than WT mice (Fig. 5a, c).
Phosphorylation of ERK1/2 in AT + LPS mice was higher than AT mice. Celecoxib abolished LPS-induced phosphorylation of ERK1/2 in AT mice. In AT + LPS mice, phosphorylation of ERK1/2 in TLR4 KO mice was lower than WT mice (Fig. 5a, c).
LPS promoted PGE2 expression via TLR4-NFκB signaling pathway
Caco-2 cells were treated with LPS (1 µg/mL). Expression of PGE2 increased at 1 h after treatment and peaked at 4 h (Fig. 6a). LPS-induced expression of PGE2 was abolished by MYD88 inhibitor Pepinh-MYD, NFκB inhibitor BAY117085 and COX2 inhibitor (Fig. 6b).

LPS promotes epithelial cells proliferation via PI3K/Akt and MAPK/ERK1/2 signaling pathway. a LPS promotes production of PGE2 in Caco-2 cells. Cells were treated with LPS1µg/mL for 4 h. *vs NC, p < 0.05. b Caco-2 cells were treated with 1 µg/mL LPS and signaling pathway blockers, including BAY 11–7085 (10 μM), AG1478 (500 nM), Celecoxib(100 µg/mL), LY294002 (20 mM), UO126 (0.07 µM), or SB203580 (10 Μm) for 4 h. PGE2 was examined by ELISA. **vs Controls, p < 0.05. *vs LPS, p < 0.05. c Proliferative assay of Caco-2 cells. **vs Controls, p < 0.05. *vs LPS, p < 0.05
EGFR inhibitor AG1478, PI3K inhibitor LY294002, p38 MAPK inhibitor SB203580 and ERK inhibitor UO126 did not alter PGE2 production induced by LPS (Fig. 6b).
LPS promoted IEC proliferation via PI3K/AKT signaling pathway
After incubation with LPS (1 µg/mL) for 36 h, Caco-2 cells’ proliferation was promoted, which could be reversed by NFκB inhibitor, BAY117085; EGFR inhibitor, AG1478; PI3K inhibitor, LY294002; and COX2 inhibitor, Celecoxib. P38 MAPK inhibitor, SB203580; and ERK inhibitor, UO126 did not reverse Caco-2 cells’ proliferation promoted by LPS (Fig. 6c).
Discussion
Commensal bacteria are recognized as important regulators of intestinal homeostasis and innate immunity [13]. Our study firstly confirmed that commensals were essential for intestinal epithelial restitution after IIR.
To study the role of the commensals in facilitating inflammatory responses after IIR, previous study employed germ-free mice [7]. Intestinal commensals are crucial for development of the intestine-associated lymphoid tissue (GALT) [17]. The inflammatory hyporesponsiveness in germ-free mice after IIR might be due to insufficient production of post-shock mesenteric lymph by defected GALT. Our model is more clinical-relevant because of normally developed GALT. Previous study deleted commensals with antibiotics and found attenuated intestinal inflammation and injury following IIR [9]. However, the mice were killed shortly after IIR. The results reflected the role of commensal bacteria at the early phase after IIR. Similarly, after commensals deletion, we found survival was better in the early phase. However, the overall survival was not improved, suggesting no reduced mortality in the restitution phase.
Human intestine serves as a reservoir of commensal bacteria and its products [17]. Translocation of bacteria and endotoxins after disruption of the gut barrier were thought to be responsible for SIRS and MODS [3]. However, it failed to find either bacteria or endotoxin in the portal blood of severe traumatic hypovolemic shock patients or experimental animals [8, 18-20]. Clinical trials in which anti-endotoxin antibodies were injected into patients have failed to show consistent benefit [21]. Recent studies suggested that the factors carried in the mesenteric lymph contributed to the development of SIRS after IIR [22]. The post-shock mesenteric lymph contains cells and a collection of unknown proinflammatory mediators [3, 22]. Loss of gut barrier is a major contributing factor leading to the production of proinflammatory mesenteric lymph [14]. At 48 h and 60 h, intestinal permeability was still higher in the mice with commensals deletion, indicating commensals deletion postponed recovery of barrier function. Crypt cell proliferation is the main mechanism involved in intestinal restitution after IIR [23]. At 48 h, commensals deletion compromised IEC proliferation. Moreover, the proinflammatory activity of mesenteric lymph was augmented at the restitution phase.
TLRs comprise a family of pattern-recognition receptors expressed on IEC and are critical for the initiation of inflammatory response [24]. Commensals deletion reduced intraluminal LPS and TLR4 expressions. Interestingly, LPS supplement restored TLR4 expression in a dose-independent manner. In mice with commensals deletion, 1 μg/mL LPS improved survival after IIR. Under normal conditions, LPS present in the intestine is harmless. However, elevated LPS in colon was able to initiate inflammatory responses in intestine, reflecting concentration-dependent effects of LPS between inflammation and intestinal homeostasis[9]. In this study, higher LPS did not confer better survival.
Prostaglandins (PG) are generated throughout the gastrointestinal tract and play critical roles in a series of physiologic and pathophysiologic conditions [25]. Administration of PGE2 stimulated IEC proliferation and the weight of the intestinal mucosa [26, 27]. Mucosal PGE2 reduced after commensals deletion, but could be recovered by LPS supplement. Importantly, at 48 h, LPS improved migration and proliferation of IEC, which was abolished by Celecoxib or TLR4 gene knockout. At 24 h and 48 h, LPS did not alter the level of activated Caspase-3, but increased the expression of PCNA. The results suggested that LPS-induced IEC proliferation was TLR4-, COX2- and PEG2-dependent. LPS activated mucosal NFκB, EGFR, PI3K/AKT, MAPK/ERK1/2, and P38MAPK signaling pathway. Celecoxib abolished LPS-induced phosphorylation of EGFR, AKT, P38, and ERK1/2. These findings revealed the signaling pathway involved in LPS-induced IEC proliferation.
To examine the biologic effects of these signaling pathways, in Caco-2 cells, expression of PGE2 increased at 1 h after LPS treatment and peaked at 4 h. LPS-induced production of PGE2 was abolished by Pepinh-MYD, BAY117085, and Celecoxib, suggesting LPS promoted PGE2 expression via TLR4-NFκB signaling pathway.
In conclusion, commensals deletion abolished inflammatory response in the early phase, but failed to improve the overall survival after IIR. Moreover, it impeded intestinal epithelial restitution by inhibiting IEC proliferation. LPS increased production of PGE2 in mucosa via TLR4-NFκB-COX2 pathway, by which it enhanced IEC proliferation and migration. A new TLR4 ligand that is originated from LPS structures, which can promote intestinal epithelial restitution, but without its proinflammatory ability could be developed in the future to improve clinical outcome of the patients with IIR.
References
Granger DNHL, Kvietys P (2015) The gastrointestinal circulation: physiology and pathophysiology. Compr Physiol 5:1541–1583. https://doi.org/10.1002/cphy.c150007
Deitch E, Xu D, Kaise V (2006) Role of the gut in the development of injury- and shock induced SIRS and MODS: the gut-lymph hypothesis, a review. Front Biosci 1:520–528. https://doi.org/10.2741/1816
Deitch E (2012) Gut-origin sepsis: evolution of a concept. Surgeon 10:350–356. https://doi.org/10.1016/j.surge.2012.03.003
Yi J, Slaughter A, Kotter C, Moore E, Hauser C, Itagaki K, Wohlauer M, Frank D, Silliman C, Banerjee A, Peltz E (2015) A "clean case" of systemic injury: mesenteric lymph after hemorrhagic shock elicites a sterile inflammatory response. Shock 44:336–340. https://doi.org/10.1097/SHK.0000000000000431
Kagnoff M (2014) The intestinal epithelium is an integral component of a communications network. J Clin Invest 124:2841–2843. https://doi.org/10.1172/JCI75225
Yoshiya K, Lapchak P, Thai T, Kannan L, Rani P, Dalle Lucca J, Tsokos G (2011) Depletion of gut commensal bacteria attenuates intestinal ischemia/reperfusion injury. Am J Physiol Gastrointest Liver Physiol 301:G1020–1030. https://doi.org/10.1152/ajpgi.00239.2011
Souza D, Vieira A, Soares A, Pinho V, Nicoli J, Vieira L, Teixeira M (2004) The essential role of the intestinal microbiota in facilitating acute inflammatory responses. J Immunol 173:4137–4146. https://doi.org/10.4049/jimmunol.173.6.4137
Sorkine P, Szold O, Halpern P, Gutman M, Greemland M, Rudick V, Goldman G (1997) Gut decontamination reduces bowel ischemia-induced lung injury in rats. Chest 112:491–495. https://doi.org/10.1378/chest.112.2.491
Chen L, Chang W, Chen P, Liu W, Hsu C (2008) TLR ligand decreases mesenteric ischemia and reperfusion injury-induced gut damage through TNF-alpha signaling. Shock 30:563–570. https://doi.org/10.1097/SHK.0b013e31816a3458
van der Hoven B, Nabuurs M, van Leengoed L, Groeneveld A, Thijs L (2001) Gut luminal endotoxin reduces ischemia-reperfusion injury of the small gut in germ-free pigs. Shock 16:28–32
Tatum PM Jr, Lorenz RG, Dimmitt RA (2010) Toll-like receptor 4 is protective against neonatal murine ischemia–reperfusion intestinal injury. J Pediatr Surg 45:1246–1255. https://doi.org/10.1016/j.jpedsurg.2010.02.093
Gatt M, Reddy B, MacFie J (2007) Review article: bacterial translocation in the critically ill-evidence and methods of prevention. Aliment Pharmacol Ther 25:741–757. https://doi.org/10.1111/j.1365-2036.2006.03174.x
Rakoff-Nahoum SPJ, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118:229–241. https://doi.org/10.1016/j.cell.2004.07.002
Zhang H, Besner G, Feng J (2016) Antibody blockade of mucosal addressing cell adhesion molecule-1 attenuates proinflammatory activity of mesenteric lymph after hemorrhagic shock and resuscitation. Surgery 159:1449–1460. https://doi.org/10.1016/j.surg.2015.12.013
James I, Chen C, Huang G, Zhang H, Velten M, Besner G (2010) HB-EGF protects the lungs after intestinal ischemia/reperfusion injury. J Surg Res 163:86–95. https://doi.org/10.1016/j.jss.2010.03.062
El-Assal O, Besner G (2005) HB-EGF enhances restitution after intestinal ischemia/reperfusion via PI3K/Akt and MEK/ERK1/2 activation. Gastroenterology 129:609–625. https://doi.org/10.1016/j.gastro.2005.05.054
Round J, Mazmanian S (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9:313–323. https://doi.org/10.1038/nri2515
Nezu Y, Tagawa M, Sakaue Y, Hara Y, Tsuchida S, Ogawa R (2002) Kinetics of endotoxin concentration and tumor necrosis factor-alpha, interleukin-1beta, and interleukin-6 activities in the systemic and portal circulation during small intestinal ischemia and reperfusion in dogs. Am J Vet Res 63:1682–1686
Cicalese L, Billiar T, Rao A, Bauer A (1997) Interaction between ischemia/reperfusion-induced leukocyte emigration and translocating bacterial enterotoxins on enteric muscle function. Transplant Proc 29:1815. https://doi.org/10.1016/s0041-1345(97)00080-8
Bauer P, Russell J, Granger D (1999) Role of endotoxin in intestinal reperfusion-induced expression of E-selectin. Am J Physiol 276:G479–484. https://doi.org/10.1152/ajpgi.1999.276.2.G479
Hardaway R (1998) Traumatic and septic shock alias post-trauma critical illness. Br J Surg 88:1473–1479. https://doi.org/10.1046/j.1365-2168.1998.00911.x
Deitch E (2010) Gut lymph and lymphatics: a source of factors leading to organ injury and dysfunction. Ann N Y Acad Sci 1207:E103–111. https://doi.org/10.1111/j.1749-6632.2010.05713.x
Tang Y, Swartz-Basile D, Swietlicki E, Yi L, Rubin D, Levin M (2004) Bax is required for resection-induced changes in apoptosis, proliferation, and members of the extrinsic cell death pathways. Gastroenterology 126:220–230. https://doi.org/10.1053/j.gastro.2003.10.077
Kubinak J, Round J (2012) Toll-like receptors promote mutually beneficial commensal-host interactions. PLoS Pathog 8:e1002785. https://doi.org/10.1371/journal.ppat.1002785
Im E, Riegler F, Pothoulakis C, Rhee S (2012) Elevated lipopolysaccharide in the colon evokes intestinal inflammation, aggravated in immune modulator-impaired mice. Am J Physiol Gastrointest Liver Physiol 303:G490–497. https://doi.org/10.1152/ajpgi.00120.2012
Shao J, Sheng G, Mifflin R, Powell D, Sheng H (2006) Roles of myofibroblasts in prostaglandin E2-stimulated intestinal epithelial proliferation and angiogenesis. Cancer Res 66:846–855. https://doi.org/10.1158/0008-5472.CAN-05-2606
Iwanaga K, Okada M, Murata T, Hori M, Ozaki H (2012) Prostaglandin E2 promotes wound-induced migration of intestinal subepithelial myofibroblasts via EP2, EP3, and EP4 prostanoid receptor activation. J Pharmacol Exp Ther 340:604–611. https://doi.org/10.1124/jpet.111.189845
Funding
This study was supported by grants from the National Natural Science Foundation of China (nos. 81200266 and 81270441).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, Hy., Wang, F., Chen, X. et al. Dual roles of commensal bacteria after intestinal ischemia and reperfusion. Pediatr Surg Int 36, 81–91 (2020). https://doi.org/10.1007/s00383-019-04555-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-019-04555-5