
Abstract
Pediatric airway surgery is a challenging field in pediatric surgery. Laryngotracheal stenosis has a variety of congenital and acquired conditions that require precise assessment and tailored treatment for each individual patient. About 90% of acquired conditions are represented by subglottic stenosis (SGS) resulting as a complication of tracheal intubation. Congenital tracheal stenosis (CTS) is a rare and life-threatening malformation, usually associated with complete tracheal rings along a variable length of the trachea. Tracheomalacia (TM) is a process characterized by flaccidity of the supporting tracheal cartilage, widening of the posterior membranous wall, and reduced anterior-posterior airway caliber. The clinical presentation can vary from almost asymptomatic patients to near fatal airway obstruction. There is considerable variation in both the morphologic subtypes and the prognosis of pediatric airway. The patients are divided into three clinical groups (mild, moderate, and severe). A further division was proposed according to the presence or absence of associated anomalies. The definitive diagnosis of pediatric airway was made by means of rigid bronchoscope and computed tomography scan with three-dimensional reconstruction (3D-CT). Rigid bronchoscopy and 3D-CT confirmed the diagnosis in all the cases. Other associated anomalies include congenital heart disease, vascular anomalies, and BPFM (maldevelopment of aerodigestive tract). After definitive diagnosis of pediatric airway lesions, surgical intervention should be considered. Surgical strategy was presented on each lesion.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pediatric airway surgery is significant challenge for pediatric surgeon over many years due to the complexity of the disease, often requiring careful diagnostic investigation and urgent and demanding reconstructive treatments gained to improve the surgical outcome for children suffering from a variety of compromised airways (Table 1). This paper focuses on the most common pediatric airway disorders to present the technical aspects of diagnosis and treatment not only with well-established treatment modalities, but also with new concepts of pediatric airway management.
Embryological aspects
At the 3rd week, the laryngotracheal groove or sulcus appears in the proximal foregut and progresses caudally alongside the cephalad progression of the lateral ridges, there by forming the primordial trachea. Anomalies may occur at any point along the tract, the most common being failures in separation of the airway and the alimentary tracts resulting in tracheoesophageal fistulas distally and laryngotracheoesophageal clefts superiorly. Segmental failure leads to atresia or complete agenesis that is typically fatal. The separate development of the laryngeal, trachea/esophagus, bronchial, and pulmonary complexes allows a diverse configuration of anomalies that may include agenesis and/or stenosis of the airway and alimentary tract, either as isolated segments or as combinations of these four components [35].
Subglottic stenosis (SGS)
Laryngeal stenosis may most commonly occur in the subglottis. In infants and children, the subglottis is the narrowest section of the airway [1]. The subglottic region begins just below the vocal folds and extends to the lower border of the cricoid cartilage. It is composed of the cricothyroid membrane, the cricoid cartilage, and surrounding soft tissues (Fig. 1).

Subglottic stenosis (endoscopic findings)
Etiology and diagnosis
Congenital SGS
Congenital SGS results from an embryogenic abnormality. The cricoid cartilage is a derivative of the sixth branchial arch and its development occurs early in the process of gestation. By the 10th week of gestation, the laryngeal lumen is recanalized. The failure of this recanalization may result in the laryngeal anomalies [1].
Acquired SGS
Prior to the1960s, acquired SGS was predominantly associated with laryngeal infections, such as diphtheria, which showed SGS by the release of an exotoxin that causes necrosis of the mucosa of airway [2]. With widespread vaccination in the early twentieth century, this infection became rare. Following the introduction of prolonged intubation for respiratory support in premature infants, the rates of acquired SGS increased and became the most common cause of SGS [2, 4]. The incidence of combined congenital and acquired SGS remains unclear. The incidence of SGS reported from 0 to 2% in intubated neonates, [7].
Factors thought to play a main factor for development of SGS are the size of the endotracheal tube, duration of intubation, traumatic intubation, presence of an infection (MRSA?), and gastroesophageal reflux [5, 6]. The incidence of acquired SGS has decreased in the past few decades. This decrease may be associated with the advance in neonatal care.
Endoscopic evaluation
Endoscopic assessment is the gold standard for airway evaluation and a perquisite for the successful management of SGS. Laryngoscopy and rigid bronchoscopy may clear the levels of obstruction in pediatric airway. In subglottis, any scarring or stenosis should be documented as the length of the stenosis and its proximity to the vocal folds. The Myer–Cotton grading system for SGS is widely used in the pediatric community (Fig. 2) [3]. If a tracheotomy is in place, attention may be paid to the evaluation of the supra stomal area, such as suprastomal collapse or a suprastomal granuloma.

Grading system for SGS (Mayer & Cotton)
Treatment
Endoscopic management
Endoscopic approaches are used both of the primary treatment and an adjuvant treatment after open reconstruction. Children with grade I or II stenosis may be suitable for endoscopic management, whereas those with more severe SGS are more likely to require open surgery [8, 9]. Balloon dilatation of the airway has evolved in recent years and is now considered an invaluable addition to the tools used to manage SGS [10–14], reducing the need for open airway reconstruction [13].
Balloons show a number of advantages with the radial force over the circumference of the stenosis minimizing the risk of airway rupture or mucosal trauma. Selection of balloon size is based on the expected size of the normal airway. Repeated dilatation at 1–3-week intervals on up to four occasions may result in good outcomes. Adjunctive treatment, such as steroid injection and scar division, may further enhance this approach.
Open airway reconstruction
A wide variety of open airway reconstructive procedures are used to manage patients with laryngotracheal stenosis. Open airway reconstruction may be performed by a single- or double-stage procedure in the severity of the stenosis and the overall severity of the patient’s comorbidities. A single-stage procedure may not be advisable in children with a history of difficult intubation, poor pulmonary function, and a previous history of reconstruction failures [10]. In a child with multiple levels of obstruction and when there are concerns regarding coexisting morbidities, a double-stage procedure is recommended [10].
Anterior cricoid split
The anterior cricoid split was originally described in 1980 as an alternative to performing a tracheotomy in premature infants with prolonged intubation [16]. The advance in neonatal care over the past several decades (introduction of nasal high flow cannulae or nasopharyngeal CPAP), the need for this operation has been minimized.
Laryngotracheal reconstruction (LTR)
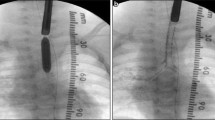
Costal cartilage is the most widely used graft material and has been shown to yield excellent long-term results (Fig. 3) [15]. The costal cartilage is easily accessible, easily carved, and easily amenable to flange formation, thus minimizing the risk of graft prolapse into the airway [15].

Laryngotracheal reconstruction (costal cartilage graft)
Although LTR with anterior costal cartilage grafting was the operation of choice for children with severe SGS, it is also used in patients who have failed endoscopic management or those with suprastomal collapse. The anterior graft LTR is now typically performed as a single-stage procedure using costal cartilage. Reported success rates exceed 90% [17].
Partial cricotracheal resection (PTCR)
Partial cricotracheal resection (PTCR) is an alternative to LTR. The goal of this operation is to remove the stenotic segment of the airway and reconnect the healthy superior and inferior segments [18]. This procedure is indicated in patients with severe SGS or a structurally inadequate subglottis and in patients who have undergone the previous airway reconstruction. Relative contraindication to PTCR include low-grade SGS, stenosis within 3 mm of the vocal folds, or conditions that impair mobilization of the trachea [8].
Disadvantages of PTCR include the risk of potentially catastrophic dehiscence of the repair and risk to the recurrent laryngeal nerves.
Congenital tracheal stenosis (CTS)
Etiology and presentation
Congenital tracheal stenosis (CTS) is a rare disease estimated to affect 1 in 64,500 live births. It was characterized by complete tracheal rings (Fig. 4), localized to any part of the large airway. It is classified according to the length of the affected area as either short segment or long segment [19]. The percentage of trachea affected >50% is considered long-segment disease [20, 21].

Complete tracheal rings
A total of 60% of children born with CTS often have other associated malformations, particularly cardiovascular anomalies. It is the combination of both airway and cardiovascular disease that often leads to life-threatening compromise [20]. Other concomitant congenital malformations are seen in approximately 40% [20]. The most common cardiovascular anomaly is the presence of an aberrant left pulmonary artery or “pulmonary artery sling,” which is noted in approximately 50% of patients [38]. Other congenital malformations include gastrointestinal malformations and anorectal anomalies.
Classification
A variety of classification systems have been reported based on the extent and severity. However, no consensus on classification has been achieved due to the heterogeneous nature of the condition. Speggiorin and Elliott showed that the patterns have been classified as normal bifurcation, anomalous right upper lobe or porcine bronchus (tracheal bronchus), bronchial trifurcation, or as single lung [36, 37]. This is further classified into congenital tracheal rings limited to the trachea only or involving the bronchi.
Clinical presentation
Patients present according to age and stenosis. Tracheal diameter is a particularly significant factor as symptoms will only be apparent when there is >50% stenosis, and dyspnea at rest is only evident when there is >75% tracheal occlusion [39]. If the stenosis is mild and there is no other pulmonary or cardiac anomalies, children may have minimal or mild respiratory symptoms, and so diagnosis may be delayed or made incidentally [20]. In some cases, the tracheal stenosis may be so severe, the child will require extracorporeal membrane oxygenation (ECMO) until a definitive repair can be performed [41].
Diagnosis
Endoscopic assessment is the gold standard for airway evaluation (Fig. 5) and a perquisite for the successful management of CTS. CT gives 3-D imaging with a strong understanding of cardiovascular–airway relationships (especially pulmonary artery slings) (Fig. 6). Bronchography is controversial even among radiologists.

Bronchoscopic finding (complete tracheal rings and absent of pars membranacea)

3D-CT segmental stenosis with bronchial anomaly (tracheal bronchus)
Treatment strategy
Treatment strategies for children with CTS have evolved over the past two decades, such that life expectancy has significantly improved [22]. Original strategies are focused upon end-to-end resection and primary anastomosis. However, due to tensions placed on the anastomosis, such an approach was only possible on short-segment disease [22, 41]. Alternative procedures were required for long-segment congenital tracheal stenosis (LCTS), and many surgeons attempted an augmentation with an anterior patch (pericardial, cartilage, etc.) combined with suspension sutures or stents [23–25]. This surgical technique would overcome the problems of the immediate stenosis, and long-term results were often complicated by persistent granulation and restenosis [26, 27]. Other strategies included tracheal replacement with aortic and tracheal homograft, but these were associated with significant morbidity and variable outcomes [28–30]. Slide tracheoplasty was first used for CTS in 1989 [31] (Fig. 7) and has subsequently proved to be extremely versatile. This procedure has been adopted by many surgeons as the surgical treatment for LCTS. The large series confirming it to be a safe and reliable technique with survival rates of over 88% and low associated morbidity and mortality [32–34, 41]. Ono et al. introduced Balloon tracheoplasty as the initial treatment for neonates with symptomatic CTS [40].

Slide tracheoplasty (Tsuang 1989)
Clinical outcomes
Hewitt et al. [41] showed that the outcomes are based on 127 patients who present in the neonatal and early infant period with a median age of 4 months and a median weight of 4 kg. A total of 12% of patients either arrived on ECMO support or were rapidly started on ECMO following arrival. Survival rates have risen with increasing institutional experience—survival at 110 months was 89.9%. The presence of preoperative distal bronchomalacia significantly increases mortality risk (71.5 vs. 94.5% survival at 200 months), as does bronchial stenosis (74.8 vs. 94.1% survival at 140 months). Preoperative ECMO and ventilation, the presence of syndromes or a single lung, and prematurity and low birth weight are also likely to emerge as important risk factors [41].
Tracheomalacia (TM)
Tracheomalacia (TM) is a process characterized by flaccidity of the supporting tracheal cartilage, widening of the posterior membranous wall, and reduced anterior–posterior airway caliber (Fig. 8). These factors cause tracheal collapse, especially during times of increased airflow, such as coughing, crying, or feeding.

Tracheomalacia
During normal inspiration, the intrathoracic tracheal lumen dilates, and during expiration, it narrows, because the airway diameter is in part determined by the difference between intrathoracic and intraluminal airway pressures [45]. Even when very high intrathoracic pressures are generated, the rigidity of the cartilage in the tracheal wall prevents complete airway collapse [46].
In patients with TM, the abnormal collapsibility of the trachea accentuates the physiological airway narrowing that occurs during expiration, and in severe cases, clinically obvious obstruction of the airway may result when greater intrathoracic pressure increases occur, as during forced expiration or coughing [47, 48]. In addition, the lack of normal tracheal stiffness in TM allows tracheal collapse from compression by adjacent thoracic structures, mainly the aortic arch and innominate artery anteriorly and the esophagus posteriorly [49]. The size of the esophagus increases with swallowing, gastroesophageal reflux, and in the presence of stenotic or obstructive lesions of the lower esophagus, [50] which may compress the trachea posteriorly. Therefore, patients with TM may have their most severe symptoms during or shortly after eating [51].
Clinical presentation
Many infants with TM do not show symptoms until 2 to 3 months of age [50]. In some infants, the presence of serious TM is evident soon after repair of EA, and they cannot be extubated. In a small number of cases with long-segment TM, symptoms may begin at birth [54].
A barking cough and expiratory stridor are present in most patients with TM [55]. Recurrent respiratory distress, wheezing, cyanosis, and spontaneous hyperextension of the neck are also seen. “Dying spells”, are the most common life-threatening events (ALTE) in severe group, and responsible for cases of sudden infant death [57]. They usually occur during feeding or within 5–10 min of a meal [56].
Diagnosis
The diagnosis of TM should be suspected by a clinical history of noisy respiration, tracheal rhonchi, harsh barking cough, apneic spells, recurrent pneumonia, or inability to extubate the airway [47, 48].
Because most symptoms are not specific for TM, radiographic and endoscopic evaluations must be performed [52, 59]. Many methods have been used through the years to diagnose TM [42]; there is no standardized diagnostic criteria [52, 59]. A narrowing of the air-filled trachea on exhalation can be noted on the lateral view of a plain chest roentgenogram in almost all children with severe TM. Although the fluoroscopy is highly specific for TM (97–100%), it is poorly sensitive for this diagnosis (23–62%) [51, 52].
In recent years, contrast-enhanced multidetector computed tomography (MDCT) has become an alternative modality to aid in the diagnosis of TM in children [57]. Using the MDCT, the surgeon can better understand not only the region of TM but also the relevant surrounding structures, such as location of the aorta and the innominate artery relative to the trachea, and the presence of an aberrant right subclavian artery [53].
Direct visualization of the airway with either flexible or rigid bronchoscopy remains the “gold standard” for the diagnosis of TM [43, 50, 52, 53, 59]. Flexible bronchoscopy may obviate the need for general anesthesia and is the modality for direct visualization and dynamic assessment of the airway [55].
Advantages of rigid bronchoscopy include improved clarity of visualization, particularly in infants. In addition, the ability to ventilate and place probes and contrast in the airway markedly improves the identification of tracheoesophageal fistulas and other airway anomalies.
More than 50% narrowing of the lumen with forced exhalation is diagnosis of TM. The majority of infants with TM have more than 75% lumen collapse, with complete tracheal collapse in up to 33% of infants [45].
Treatment
Many children with mild airway collapse may be treated non-surgically. The abnormally soft trachea tends to become more rigid with continued growth, and minor symptoms can be expected to improve by age 1 to 2 years [47, 48, 64–66]. Continuous positive airway pressure (CPAP) is an effective treatment for moderate-to-severe TM [68, 69].
The primary indications for surgery are dying spells, recurrent pneumonia, intermittent respiratory obstruction, and inability to extubate the malacic airway [50, 57]. According to the 2012 Cochrane review [67], there is no evidence supporting one therapy over another for the treatment of severe TM.
Surgical treatment
Surgical treatment may be necessary to improve symptoms and sequelae for severe TM. Options for the treatment include open aortopexy, thoracoscopic aortopexy, tracheal resection, or external stabilization [70].
Aortopexy
Aortopexy has been the main stay of surgical therapy for pediatric intrathoracic TM [44, 47, 49, 71]. The technique of aortopexy was first used by Filler et al. [48] and it was derived from the operation described by Gross in 1948 to treat the “innominate artery compression syndrome”.
With the airway loosely attached anteriorly to the major vessels and posteriorly to mediastinal structures by connective tissue, suture elevation of the aorta and the innominate artery and possibly pericardium to the back of the sternum may support maintain an adequate airway [51]. By this indirect mechanism, aortopexy has been found to be relieving the consequences of severe TM [59].
Sometimes, these maneuvers have proven insufficient, and direct suture placement into the tracheal and/or bronchial cartilages has been required to provide adequate airway support [63]. The ability to assess the effects of the elevation of the vessels on the airway by bronchoscopy, while the procedure is being carried out is extremely valuable to ensure the desire results [40].
The use of pledgets on the sutures passed through the adventitia of the aorta was proposed by Kimura et al. [58] to allow the aortic wall to tolerate major tension without the risk of tears. Applebaum and Wooley [79] have suggested the use of a pericardial flap to suspend the aorta (Fig. 9).

Modification of aortopexy
Several surgical approaches have been used to access the major arteries. Right or left anterior thoracotomies as well as more lateral thoracotomy incisions have been used, and also cervical, mediastinal, and thoracoscopic approaches have been reported [47, 59, 60, 72, 73].
Partial sternotomy
A 3-cm transverse incision is made at the manubrial sternal junction and a partial upper sternotomy along with partial thymectomy allowed the upper portion of the pericardium to be opened to reveal the innominate artery, ascending aorta, and main pulmonary artery [59, 60, 74].
Thoracoscopic aortopexy
Three ports are typically used, and the non-absorbable sutures are placed along the most anterior portion of the upper ascending aorta and into the sternum. After the sutures have been placed and pulled against the posterior surface of the sternum, bronchoscopy is done to determine the efficacy of the aortopexy [75–77].
Thoracotomy aortopexy
A left or right anterior thoracotomy can be used to gain access to the anterior mediastinum for aortopexy. The pectoralis major muscle is elevated and the chest entered through the third inter costal space [50, 51, 59].
Whatever approach or technique used, the efficacy of the aortopexy in treating clinical symptoms of TM was reported in more than 80% of patients [87], but there is a significant mortality (6%) and complications rate (16.6%). The most common related complications are pericardial effusion, phrenic nerve palsy, chylothorax, and vocal cord palsy [62].
Splinting
In some patients, TM is so diffuse that aortopexy may not sufficiently alleviate the symptoms, and other methods have to be used [61]. External splinting with autologous materials and prosthetic materials have been used to support the flaccid trachea [80, 81]. Morrison et al. recently described the successful implantation of external airway splints for children with severe TM [82].
Internal tracheal stent placement by endoscopy has been attempted to treat pediatric TM. The advantages of internal stents include their less invasive nature and shorter recovery times. Airway stenting with silicone and metallic expandable stents has been reported, but remains problematic due to the small size of the pediatric airway and the need for growth [83–85]. The stents for TM are used in very limited situations in which non-surgical or surgical treatments have failed [43, 62]. However, absorbable biopolymer stents currently in development may become a more attractive treatment option in the future [86].
Conclusion
Surgery remains a very important component in the seamless collaboration of a multiple disciplinary team to provide the best care of children with airway disorders.
References
Jefferson ND, Cohen AP, Rutter MJ (2016) Subglottic stenosis. Semin Pediatric Surg 25:138–143
Santos D, Mitchell R (2010) The history of pediatric airway reconstruction. Laryngoscope 120:815–820
Myer CM, O’Connor DM, Cotton RT (1994) Proposed grading system for subglottic stenosis based on endotracheal tube sizes. Ann Otol Rhinol Laryngol 103:319–323
Fearon B, Ellis D (1971) The management of long term airway problems in infants and children. Ann Otol 80:669–677
Walner DL, Stern Y, Gerber ME et al (1998) Gastroesophageal reflux in patients with subglottic stenosis. Arch Otolaryngol Head Neck Surg 124:551–555
Halstead LA (1999) Gastroesophageal reflux: a critical factor in pediatric subglottic stenosis. Otolaryngol Head Neck Surg 120:683–688
Walner DL, Loewen MS, Kimura RE (2001) Neonatal subglottic stenosis—incidence and trends. Laryngoscope 111:48–51
Hart CK, Yang CJ, Rutter MJ (2015) Reconstruction of the airway. In: Thompson JW, Vieira FO, Rutter MJ (eds) Managing the difficult airway: a handbook for surgeons. J P Medical Publishers, London, pp 115–124
Simpson GT, Strong MS, Healy GB et al (1982) Predictive factors of success or failure in the endoscopic management of laryngeal and tracheal stenosis. Ann Otol Rhinol Laryngol 91:384–388
Quesnel AM, Lee GS, Nuss RC et al (2011) Minimally invasive endoscopic management of subglottic stenosis in children: success and failure. Int J Pediatr Otorhinolaryngol 75:652–656
Maresh A, Preciado DA, O’Connell AP et al (2014) A comparative analysis of open surgery vs endoscopic balloon dilation for pediatric subglottic stenosis. J Am Med Assoc. Otolaryngol Head Neck Surg 140:901–905
Lang M, Brietzke SE (2014) A systematic review and meta-analysis of endoscopic balloon dilation of pediatric subglottic stenosis. Otolaryngol Head Neck Surg 150:174–179
Hautefort C, Teissier N, Viala P, et al. (2012) Balloon dilatation laryngoplasty for subglottic stenosis in children. Arch Otolaryngol Head and Neck Surg 138:235–240
Balakrishnan K, Rutter MJ (2015) Balloon dilation of the airway. In: Thompson JW, Vieira FO, Rutter MJ (eds) Managing the difficult airway: a handbook for surgeons. JP Medical Publishers, London, pp 125–136
Zalzal GH, Cotton RT, McAdams AJ (1986) The survival of costal cartilage grafts in laryngotracheal reconstruction. Otolaryngol Head Neck Surg 94:204–211
Cotton RT, Seid AB (1980) Management of the extubation problem in the premature child: anterior cricoid split as an alternative to tracheotomy. Ann Otol 89:508–511
Gustafson LM, Hartley BE, Liu JH et al (2000) Single-stage laryngotracheal reconstruction in children: a review of 200 cases. Otolaryngol Head Neck Surg 123:430–434
Jacquet Y, Lang F, Pilloud R et al (2005) Partial cricotracheal resection for pediatric subglottic stenosis: long-term outcome in 57patients. J Thorac Cardiovasc Surg 130:726–732
Herrera P, Caldarone C, Forte V, Campisi P, Holtby H, Chait P et al (2007) The current state of congenital tracheal stenosis. Pediatr Surg Int 23(11):1033–1044
Elliott M, Roebuck D, Noctor C, McLaren C, Hartley B, Mok Q et al (2003) The management of congenital tracheal stenosis. Int J Pediatr Otorhinolaryngol 67(Suppl1):S183–S192
Grillo HC (1994) Slide tracheoplasty for long-segment congenital tracheal stenosis. Ann Thorac Surg 58(3):613–619
Anton-Pacheco JL, CanoI, Comas J, Galletti L, Polo L, Garcia A et al (2006) Management of congenital tracheal stenosis in infancy. Eur J Cardiothorac Surg 29(6):991–996
Elliott MJ, Haw MP, Jacobs JP, Bailey CM, Evans JN, Herberhold C (1996) Tracheal reconstruction in children using cadaveric homograft trachea. Eur J Cardiothorac Surg 10(9):707–712
Dayan SH, Dunham ME, Backer CL, Mavroudis C, Holinger LD (1997) Slide tracheoplasty in the management of congenital tracheal stenosis. Ann Otol Rhinol Laryngol 106(11):914–919
Idriss FS, DeLeon SY, Ilbawi MN, Gerson CR, Tucker GF, Holinger L (1984) Tracheoplasty with pericardial patch for extensive tracheal stenosis in infants and children. J Thorac Cardiovasc Surg 88(4):527–536
Cosentino CM, Backer CL, Idriss FS, Holinger LD, Gerson CR, Mavroudis C (1991) Pericardial patch tracheoplasty for severe tracheal stenosis in children: intermediate results. J Pediatr Surg 26(8):879–884
Backer CL, Mavroudis C, Dunham ME, Holinger LD (1997) Reoperation after pericardial patch tracheoplasty. J Pediatr Surg 32(7):1108–1111
Grillo HC (1990) Tracheal replacement. Ann Thorac Surg 49(6):864–865
Jacobs JP, Quintessenza JA, Andrews T, Burke RP, Spektor Z, DeliusRE et al (1999) Tracheal allograft reconstruction: the total North American and world wide pediatric experiences. Ann Thorac Surg 68(3):1043–1051
Jacobs JP, Elliott MJ, Haw MP, Bailey CM, Herberhold C (1996) Pediatric tracheal homograft reconstruction: a novel approach to complex tracheal stenosis in children. J Thorac Cardiovasc Surg 112(6):1549–1558
Tsang V, Murday A, Gillbe C, Goldstraw P (1989) Slide tracheoplasty for congenital funnel-shaped tracheal stenosis. Ann Thorac Surg 48(5):632–635
Manning PB, Rutter MJ, Border WL (2008) Slide tracheoplasty in infants and children: risk factors for prolonged postoperative ventilator support. Ann Thorac Surg 85(4):1187–1191
Manning PB, Rutter MJ, Lisec A, Gupta R, Marino BS (2011) One slide fits all: the versatility of slide tracheoplasty with cardiopulmonary bypass support for airway reconstruction in children. J Thorac Cardiovasc Surg 141(1):155–161
Butler CR, Speggiorin S, Rijnberg FM, Roebuck DJ, Muthialu N, Hewitt RJ et al (2014) Outcomes of slide tracheoplasty in 101 children: a 17-year single-center experience. J Thorac Cardiovasc Surg 147(6):1783–1789
Adamiec E, Dzieciolowska-Baran E, Czerwinski F, Miklaszewska D, Teul I (2002) Prenatal development of the human trachea. Folia Morphol (Warsz) 61(2):123–125
Speggiorin S, Torre M, Roebuck DJ, McLaren CA, Elliott MJ (2011) Surgical outcome of slide tracheoplasty in patients with long congenital segment tracheal stenosis and single lung. Eur J Cardiothorac Surg 39(6):170–174
Speggiorin S, Torre M, Roebuck DJ, McLaren CA, Elliott MJ (2012) A new morphologic classification of congenital tracheobronchial stenosis. Ann Thorac Surg 93(3):958–961
vanSon JA, Hambsch J, Haas GS, Schneider P, Mohr FW (1999) Pulmonary artery sling: reimplantation versus antetracheal translocation. Ann Thorac Surg 68(3):989–994
Koletsis EN, Kalogeropoulou C, Prodromaki E, Kagadis GC, Katsanos K, Spiropoulos K et al (2007) Tumoral and non-tumoral trachea stenoses: evaluation with three-dimensional CT and virtual bronchoscopy. J Cardiothorac Surg 2:18
Ono S, Maeda K Baba K, Usui Y Tsuji Y et al (2014) Balloon tracheoplasty as initial treatment for neonateswith symptomatic congenital tracheal stenosis. Pediatr Surg Int 30:957–960
Hewitt RJ, Butler CR, Maughan EF, Elliott MJ (2016) Congenital tracheobronchial stenosis. Semin Pediatr Surg 25:144–149
Fraga JC, Jennings RW, Kim PCW (2016) Pediatric tracheomalacia. Semin Pediatr Surg 25:156–164
Carden KA, Boiselle PM, Waltz DA et al (2006) Tracheomalacia and tracheobroncho malacia in children and adults. Chest 127:964–1005
Benjamin B, Cohen D, GlassonM (1976) Tracheomalacia in association with congenital tracheoesophageal fistula. Surgery 79:504–508
Wittenborg MH, Gyepes MT, Crocker D (1967) Tracheal dynamic in infants with respiratory distress, stridor and collapsing trachea. Radiology 88:653–662
Vinograd I, Filler RM, Bahoric A (1987) Long-term functional results of prosthetic splinting intracheomalacia and bronchomalacia. J Pediatr Surg 22:38–41
Kiely EM, Spitz L, Brereton R (1987) Management of tracheomalacia by aortopexia. Pediatr Surg Int 2:13–15
Filler RM, Rosselo P, Lebowitz R (1976) Life-threating anoxic spells caused by tracheal compression after repair of esophageal atresia: correction by surgery. J Pediatr Surg 41:739–748
Rode H, Millar AJW, Vega M et al (1985) Oesophageal atresia—severe tracheomalacia and its correction by aortopexia. Z Kinderchir 40:282–286
Filler RM, Fraga JC (1994) Tracheomalacia. Semin Thorac Cardiovasc Surg 6:211–215
Filler RM (1991) Tracheomalacia. In: Fallis JC, Filler RM, Lemoine G, eds. Pediatric Thoracic Surgery. ElsevierScience, New York, pp 163–171
Hysinger EB, Panitch HB (2016) Paediatric tracheomalacia. Paediatr Respir Rev 17:9–15
Ngerncham M, Lee EY, Zurakowski D et al (2015) Tracheobronchomalacia in pediatric patients with esophageal atresia: comparison of diagnosis laryngoscope/bronchoscopy and dynamic airway multi detector computed tomography. J Pediatr Surg 50:402–407
Malone PS, Kiely EM (1990) Role of aortopexy in the management of primary tracheomalacia and bronchomalacia. Arch Dis Child 65:438–440
Pan W, Peng D, Luo J et al (2014) Clinical features of airway malacia in children: a retrospective analysis of 459patients. Int J Clin Exp Med 7:3005–3012
Schwartz MZ, Filler RM (1980) Tracheal compression as a cause of apnea following repair of tracheoesophageal fistula: treatment by aortopexia. J Pediatr Surg 15:842–848
Weber TR, Keller MS, Fiore A (2002) Aortic suspension(aortopexy) for severe tracheomalacia in infants and children. Am J Surg 184:573–577
Kimura K, Soper RT, Kao SCS et al (1990) Aortosternopexy for tracheomalacia following repair of esophageal atresia: evaluation by cine-CT and technical refinement. J Pediatr Surg 25:769–772
Jennings RW, Hamilton TE, Smithers CJ et al (2014) Surgical approaches to aortopexy for severe tracheomalacia. J Pediatr Surg 49:66–71
Briganti V, Oriolo L, Mangia G et al (2006) Tracheomalacia in esophageal atresia. Usefulness of preoperative imaging evaluation for tailored surgical correction. J Pediatr Surg 41:1624–1628
Filler RM (1995) Tracheal congenital anomalies. In: Pearson EG, Deslauriers J, Hiebert CA, Ginsberg RJ, McKneally MF, Urschel HC (eds) Thoracic Surgery. Churchill Livingstone Inc., New York, pp 235–250
Calkoen EF, Gabra HO, Roebuck DJ et al (2011) Aortopexy as treatment for tracheobronchomalacia in children: an 18-years ingle experience. Pediatr Crit Care Med 12:545–551
Bairdain S, Smithers CJ, Hamilton TE et al (2015) Direct tracheobronchopexy to correct airway collapse due to severe tracheobronchomalacia: short-term outcomes in a series of 20 patients. J Pediatr Surg 50:972–977
Vasco JS, Ahn C (1968) Surgical management of secondary tracheomalacia. Ann Thorac Surg 6:269–272
Picot C, Monnet P, Bethenod M et al (1969) Tracheomalacia in infants. Arch Pediatr 26:493–506
Cox WL, Shaw RR (1965) Congenital chondromalacia of the trachea. J Thorac Cardiovasc Surg 49:1033–1039
Goyal V, Master IB, Chang AB (2012) Interventions for primary (intrinsic) tracheomalacia in children. Cochrane Database Syst Rev 10:CD005304
Panitch HB, Allen JL, Alpert BE et al (1994) Effects of CPAP on lung mechanics in infants with acquired tracheobronchomalacia. Am J Resp Crit Care Med 150:1341–1346
Davis S, Jones M, Kisling J et al (1998) Effect in infants with tracheobronchomalacia. Am J Resp Crit Care Med 158:148–152
Kugler C, Stanzel F (2014) Tracheomalacia. Thorac Surg Clin 24:51–58
Corbally MT, Spitz L, Kiely E et al (1993) Aortopexy for tracheomalacia in oesophageal anomalies. Eur J Pediatr Surg 3:264–266
Abdal-Rahman U, Ahrens P, Fiegut H (2002) Surgical treatment of tracheomalacia by bronchoscopic monitoring, aortopexy in infants and children. Ann Thorac Surg 74:315–319
Brawn W, Huddart S (1991) Tracheoaortopexy via midline sternotomy in trachea malacia. J Pediatr Surg 26:660–662
Vaishnav A, Mackinnon AE (1986) New cervical approach for tracheopexy. Br J Surg 73:441–442
Van der Zee DC, Straver M (2015) Thoracoscopic aortopexy for tracheomalacia. World J Surg 39:158–164
Arnaud AP, Rex D, Elliot MJ et al (2014) Early experience of thoracoscopic aortopexy for severe tracheomalacia in infants after esophageal atresia and tracheoesophageal fistula repair. J Laparoendosc Adv Surg Tech A 7:508–512
Perger L, Kim HB, Jaksic T et al (2009) Thoracoscopic aortopexy of tracheomalacia in infants and children. J Laparoendosc Adv Surg Tech A 19:5249–5254
Spitz L (1986) Dacron-patch aortopexy. Prog Pediatr Surg 19:117–119
Applebaum H, Wooley MM (1990) Pericardial flap aortopexy for tracheomalacia. J Pediatr Surg 25:30–32
Johnston MR, Loeber N, Hillyer P et al (1980) External stent for repair of secondary tracheomalacia. Ann Thorac Surg 30:291–298
Bianchi A, Greenhough SG (1992) Repair of long-segment tracheomalacia with free autologous cartilage ring grafts. Pediatr Surg Int. 7:236–239
Morrison RJ, Hollister SJ, Niedner MF et al (2015) Mitigation of tracheobronchomalacia with 3D-printed personalized medical devices in pediatric patients. Sci Transl Med 29:285–295
Furman RH, Bacher CL, Dunham ME et al (1999) The use of balloon-expandable metallic stent in the treatment of pediatric tracheomalacia and bronchomalacia. Arch Otolaryngol Head Neck Surg 125:203–207
Fraga JC, Filler RM, Forte V et al (1997) Experimental trial of balloon-expandable metallic Palmaz stent in the trachea. Arch Otolaryngol Head Neck Surg 123:522–528
Filler RM, Forte V, Fraga JC et al (1995) The use of expandable metallic airway stents for tracheobronchial obstruction in children. J Pediatr Surg 30:1050–1055
Sommer D, Forte V (2000) Advances in the management of major airway collapse: the use of airway stents. Otolaryngol Clin North Am 33:163–167
Torre M, Carlucci M, Speggiorin S et al (2012) Aortopexy for the treatment of tracheomalacia in children: review of the literature. Ital J Pediatr 38:62–68
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Maeda, K. Pediatric airway surgery. Pediatr Surg Int 33, 435–443 (2017). https://doi.org/10.1007/s00383-016-4050-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-016-4050-7