
Abstract
Congenital tracheal stenosis (CTS) is an uncommon condition that has challenged pediatric surgeons for decades. Patients with CTS can present with a wide spectrum of symptoms and varying degrees of severity. In addition, a variety of techniques have been devised to repair this malformation. A review of these procedures and our suggestions for clinical standards and practice guidelines will be presented in this paper. A retrospective review of the literature on CTS from 1964 to 31 March, 2006. There is not one standard technique for the repair of CTS, as individualized approach to each patient and airway lesion is necessary to optimize patient management; nevertheless there is a consensus about segmental resection and anastomosis being best for short segment stenosis while slide tracheoplasty is most effective for the long-segment ones. Conservative management is also an option for select group of patients with careful and close follow up. Survival following surgery over the years has improved, but mortality remained high, particularly in a specific subset of patients presenting at the age less than 1 month with associated cardiac malformations. In conclusion, CTS remains a significant challenge for pediatric surgeons. Additional research is required to improve our understanding of the pathogenesis of CTS, and to develop evidence-based treatment protocols for the entire spectrum of presentation including conservative management.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Tracheal morphogenesis is a complex developmental process that has evolved from the anatomic and descriptive domain of clinicians to the molecular and cellular understanding of organogenesis of the developmental biologists today. The complexity of tracheal morphogenesis mirrors the complexity and spectrum of presentations and characteristics associated with congenital tracheal stenosis (CTS). CTS represents a spectrum of lesions causing airway narrowing with diverse management options and patient outcomes. The diversity of clinical presentation and the rarity of these lesions present challenges to their timely and effective management. The objective of this report is to give a timely comprehensive review of the current knowledge of CTS, to present the different therapeutic options and their outcomes, as well as to delineate areas in which future efforts should be directed to improve patient treatment and outcomes.
Materials and methods
We reviewed all articles published between 1 January, 1964 and 31 March, 2006 that were reviewed from Medline database using the terms: “congenital tracheal stenosis”, “complete tracheal rings”, “tracheal malformation”, and “tracheoplasty”. The year of 1964 was chosen because of the first successful treatment for CTS was reported that year by Cantrell and Guild [1]. Secondary searches were conducted with “tracheal embryogenesis”, “trachea and embryo”, “trachea and imaging”, “trachea and diagnosis” and “endotracheal stent”. In total, 59 articles were included. In addition to the published data, we reviewed our institutional series of CTS patients over the past 5 years and report our updated outcomes since our last publication [2].
Definition and epidemiology
CTS is defined as a narrowing of the tracheal lumen, most commonly due to complete tracheal cartilage rings and absence of the membranous trachea, resulting in luminal constriction. A further obstruction of the lumen is caused by thickening of the tracheal mucosa from submucosal glands and connective tissue hyperplasia [1]. Airway obstruction due to other lesions such as subglottic stenosis, vascular tracheobronchial compression syndrome and tracheomalacia, will not be addressed in this review.
CTS is frequently associated with other anomalies. These associated conditions can interfere with diagnosis and treatment of CTS in two ways, either masking CTS-related respiratory symptoms, or increasing the severity of symptoms while compounding the operative risk at terms of eventual repair. Isolated CTS, without any associated malformation, is uncommon, it accounts for only 10–25% of patients [3–5]. Cardio-vascular anomalies are the most frequently associated and are reported to occur in up to 50% of patients [6]. The list of associated anomalies includes: cardiovascular anomalies (left pulmonary artery sling, patent ductus arteriosus, ventricular septal defects, double aortic arch, aberrant subclavian artery); respiratory anomalies (pulmonary agenesis or hypoplasia, tracheal bronchus); and others (gastrointestinal, renal, skeletal).
Given its acuity, rarity and the variability in its presentation there is no epidemiological data or population-based studies determining the incidence of CTS. However, we estimated this incidence based on our experience and the following assumptions:
-
(a)
Our institution is the only tertiary pediatric center to treating CTS patients in Canada;
-
(b)
The number of patients treated for this diagnosis over the last 5 years is known (patients with birth dates between January 2000 and December 2004);
-
(c)
An asymptomatic and undiagnosed population of CTS patients in a general presentation has been estimated to be 20% of the total of patients in several published series [5, 7, 8];
-
(d)
The birth rate and general population growth rate of Canada for the mentioned years.
Based on the above assumptions, using the following source: official data from HSC, and government statistical data, available through Statistics Canada: http://www.statcan.ca , we conclude that with 31.2 patients per 2,012,042 children under 5 years in the population of Canada, the population-based incidence of CTS is approximately 1 for every 64,500 live births. A five-year period was used to minimize the year-to-year variation due to the rarity of this condition.
Classification
CTS was classically described by Cantrell and Guild [1] and classified into three morphologically distinct groups according to anatomic criteria (Fig. 1). An additional classification system for CTS was introduced by Anton-Pacheco [5] in 2003, based on functional symptoms at the time of clinical presentation. In this classification of CTS, an additional sub-classification (A or B) has been added to indicate the presence or absence of associated malformations, respectively. (See details in Table 1). There is a trend toward clustering of anatomical types correlating with functional categories, so that generalized hypoplasia tends to be severely symptomatic, whereas segmental stenosis was categorized into milder categories. There is some overlap among different categories often complicated by infection involving airways that can easily precipitate crises and change mild and moderate categories into more severe ones. To date, these two classification systems have worked in a complementary manner in managing CTS patients. The utility of the functional classification has been demonstrated in the selection of operative candidates, whereas the anatomic classification largely influences the operative approach to the repair of CTS (See Therapeutic evolution and current options).

Anatomic classification from Cantrell and Gould; type I is generalized hypoplasia, type II is funnel type stenosis with one normal end and the other one stenotic, and type III is segmental stenosis with two or three cartilage rings involved
Embryology
Morphological/descriptive models
The developmental defects that result in CTS are not known. Clues to the pathogenesis of CTS rest in the organogenesis of the esophagus and trachea from their common origin, the primitive foregut. Our current understanding of tracheal development stems from the studies of animal models such as mice [9–13], rats [14] and chicks [15]. According to the classical model [16], based on the observation of human and other mammalian embryos, the respiratory bud emerges from the anterior wall of the foregut as the laryngo-tracheal groove (4th week, day 22–23, Carnegie stage 13–14). This groove elongates caudally and constricts laterally, forming two lateral ridges of mesenchyme that grow medially and fuse in the midline to partition the foregut into trachea and esophagus along the dorso-ventral axis. The trachea further divides at its caudal end into left and right main bronchi [16, 17] that further subsequently form the lungs. The chick embryo model [15], however, describes the existence of two pulmonary buds appearing as early as day 3–4 of chick embryo life, (Hamburger and Hamilton stage 16), one on each side of midline, originating from the lower part of the larynx. Trachea will appear later only by stage 19.
These two anatomic and descriptive models of foregut development can be merged into one if we view the foregut development in the developmental context of reciprocal inductive interaction between budding endoderm and surrounding mesoderm. This endodermal–mesenchymal inductive interaction occurs simultaneous to the branching of the airway into the mainstem bronchi such that airway and parenchymal tissues are closely integrated in development.
Pharmacologic model
The pathological basis for foregut malformations has also been extensively studied using teratogens that significantly affect the developing foregut. The Adriamycin-induced rat model for esophageal atresia results in the development of foregut anomalies in 20 to 100% of the exposed animals in a dose-dependant fashion [18–20]. Another model based on Vitamin A deficiency during rat embryogenesis, produces a predictable constellation of malformations in rats involving the eyes, respiratory tract, cardiovascular system, urogenital system and the diaphragm [21]. Although some genetic markers have been measured in this model, more detailed genetic analysis are needed to elucidate the underlying mechanisms including explanation for variability in penetrance of the phenotype.
Genetic models
Recent studies on the molecular basis of cellular differentiation and organ development have unveiled a number of molecular pathways and transcription factors that play a role in trachea and lung morphogenesis. Tracheal morphogenesis begins with the generation of an anterior foregut pouch and ends with the midline fusion of the upper portion of the lateral esophago-tracheal ridges from the carina to the larynx. Genetic experiments using targeted mutations of signaling molecules have helped to further elucidate the molecular and genetic mechanisms involved in the pathogenesis of these malformations. Molecular approaches to resolve the signaling pathways regulating foregut differentiation have shown Sonic Hedgehog (Shh) and Sox9 to be the prominent genes in controlling tracheal ring cartilage patterning and formation. Table 2 summarizes the main transcription factors identified to be related to the tracheo-bronchial tree formation, the phenotype when altered, and the chromosomal location of them.
Shh is a gene encoding a 49.6 kDa secreted glycoprotein widely expressed during different stages of embryogenesis. Its main role appears to be the coordination of axial patterning in different organs, right–left morphology, cell fate differentiation and apoptosis [12]. Shh knockout (Shh−/−) embryos are embryonic lethal and display a phenotype characterized by multiple pulmonary anomalies including hypoplastic bilobed lung and fused foregut anatomy (tracheo-esophageal tube with no cartilage rings, lined ventrally with respiratory columnar cells and dorsally with stratified squamous epithelium). In humans, Shh has been linked to a remarkable amount of different tumors, acting both as a proliferation inducer and as a secreted maintaining paracrine influence. It seems though that the carcinogenic potential is derived from mutations on the transcription factors [22]. In the developing airway of the mouse embryo, Shh gene expression on day E9.5 coincides with tracheo-esophageal septum (TES) formation. On day E10.5, there is a high level of expression of Shh protein on the ventral part of the foregut (floor of pharynx and laryngo-tracheal groove) with a sharp demarcation between expressing and non-expressing tissues. This Shh pattern is expressed throughout the foregut except the lung buds at the most dorsal endoderm. By day E11.5, the TES and posterior wall of the trachea form and a change in Shh expression pattern becomes evident, as the esophagus shows clear expression of the Shh protein, whereas the developing trachea shows down-regulation in Shh expression. This switch in the expression pattern occurs in a caudal to cranial “wave” analogous to the closing of a jacket zipper, and extends beyond or ahead of the septum formation, as preceding it. On day E12.5, the ventral formation of the TES is complete, but the dorsal Shh expression pattern remains unchanged [11].
The Shh expression pattern is also distorted in Adriamycin-treated mouse embryos. On day E10.5, some of the treated embryos fail to demonstrate the sharp demarcation of Shh expression along the ventral foregut, exhibiting a diffuse expression of Shh in the caudal part of the unpartitioned foregut [11]. The same alteration is observed in E11.5 embryos, in every mouse having Adriamycin-related tracheo-esophageal fistula, but this change in Shh expression is transient as the normal pattern is restored spontaneously on day E12.5, with a strong Shh for dorsal and negative ventral wall even in the mice that show the atresia and tracheo-esophageal fistula phenotype. However, with the dosage of Adriamycin used in this model only a minority of the treated mice developed foregut malformations as well as the Shh abnormal expression of Shh [11].
Sox9, on the other hand, a member of the Sox family of transcription factors is expressed during a specific stage of differentiation within the population of foregut cells destined to give rise to chondrocytes on the dorsal and lateral parts of the forming trachea. These cells eventually migrate to the anterior part of the airway in spaced intervals to form the final cartilage rings [23].
Although vitamin A is essential for TES formation, the loss of function of the vitamin A receptor knockout mice display “no TES” Phenotype [9]. Lung hypoplasia and agenesis has also been observed [9].
The FGF 7 seems to be related to peripheral lung maturation, since the overexpression model show a disordered multiplication of peripheral lung structures, lined with respiratory type epithelium similar to congenital cystic adenomatoid malformation [17, 24]. In contrast, FGF 10 seems to be related to lung differentiation, as knockout mice were stillborns, showing only trachea and no lungs, suggesting that the transcription factor is not involved in tracheal development.
The transgenic model utilizes the overexpression of FGF-18 in epithelial cells to investigate the pathogenesis of tracheobronchial anomalies [13]. The investigators found that FGF-18 overexpression altered the length, caliber and epithelial lining of the trachea and bronchi, increased the size of peripheral pulmonary vessels and induced the ectopic formation of cartilage in peripheral lung. It blocked the “peripheral maturation” of lung tissue.
Clinical presentation and outcome
The anatomic spectrum of CTS, ranging from short-segment stenosis to complete tracheobronchial hypoplasia, results in a wide range of clinical presentations and patient outcomes. Although there is no absolute correlation between luminal diameter and prognosis, the severity of symptoms in general correlates with degree of airway obstruction, with the diameter in the more symptomatic and operative groups being significantly smaller than the group successfully treated by observation alone [25].
Based on the pattern of symptoms, three cohorts of CTS patients can be categorized based on their clinical presentations:
-
(1) Asymptomatic or minimally symptomatic children The diagnosis of CTS is made incidentally during another event, such as elective surgery, in which any signs of airway stenosis are first recognized. Another common presentation for mildly symptomatic patients is the presence of inspiratory and expiratory wheeze secondary to biphasic airflow resistance, with very mild symptoms after treatment with bronchodilators. In such cases, CTS diagnosis is established while being investigated for other respiratory concerns like asthma [26–28]. Some patients can go undiagnosed until late childhood or early adolescence, if the stenosis was mild enough to let them go unnoticed through their earlier years of life. Later on, at their teenage, some patients may develop respiratory difficulties with exercise. Careful and thorough workup is warranted and treatment options of these patients have to be individualized [7].
-
(2) Early symptomatic An infant who develops respiratory difficulty, stridor and wheezing in the first hours of life. Symptoms may include frequent respiratory infections, recurrent croup, cyanotic spells, or a coarse, barking cough as a result of congenital narrowing within the small caliber airway of the neonate. Treatment options for these patients depend on the severity of respiratory compromise, co-morbid factors such as pre-existent infections and associated malformations, especially cardiac anomalies for which surgical treatment is required (see Table 3). This group of patients presents the greatest challenge for surgical management due to their small size and acute clinical status. Although other institutions have reported better outcomes [29], our institutional experience points towards the highest mortality rate, close to 100%, and outcomes would indicate a cautionary tale when counseling parents.
In the absence of cardiac malformations, an overall mortality of 73% was observed in this category of patient cohort [30].
-
(3) Late symptomatic These patients present with symptoms of CTS at the end of the first year of life, as their physical activity increases, causing airway flow restriction to manifest by wheezing, exertional shortness of breath or intercostal-supraclavicular retractions. There are data to suggest that nonsurgical management of this patient population is an option, as these children may outgrow their CTS by “catch-up” tracheal growth that increases the diameter of the entire main airway, including the stenotic segment. The children who survive beyond infancy have been shown to have a tracheal growth pattern that is faster than the normal population until the age of 7 or 8 years [25]. However, even a minor respiratory viral infection may “tip over” this group of patients, causing acute respiratory distress from the mucosal edema superimposed on to an already narrowed airway lumen [2]. Their operative mortality still ranges from 16 to 20% [30].
According to the morphologic classification of CTS, patients with Cantrell type I display the most severe symptoms and typically present early in infancy, with a high rate of associated intracardiac anomalies [31]; this correlation was observed in our data as well [25]. Cantrell Types II and III tend to present later at an older age with mild respiratory symptoms and are less likely to have associated intracardiac anomalies. The Cantrell anatomic categorization of CTS has significant prognostic value, as it has been shown to correlate with prognosis independently of the intervention for the treatment of CTS [25].
Diagnosis and investigation
Various approaches have been proposed for the definitive diagnosis of CTS, guided by specific practice patterns and the characteristics of local institutions [32].
Rigid bronchoscopy (RB) under general anesthesia remains the gold standard for definitive diagnosis of CTS, with complete cartilage rings being the diagnostic milestone (Fig. 2). RB provides accurate information about the length and minimum diameter of the involved segment by directly observing the compromised area from within. The drawbacks of this diagnostic tool are the need for general anesthesia and the possibility of minor mucosal damage resulting in mucosal edema and further narrowing of the airway. RB should be judiciously used with utmost care, since it may precipitate the obstructive event. This may further compromise the airway and result in the need for emergency thoracotomy or sternotomy, so that the surgical team should be always available during these procedures should emergency airway access become necessary. There is however a size limitation on the use of RB in the evaluation of CTS in lesions too narrow to allow passage of the smallest rigid Hopkins telescope available (2.5 mm in diameter).

Bronchoscopic view of complete cartilage rings (left). As opposed to normal C shaped tracheal cartilages (right)
One alternative is the insertion of an ultra-thin flexible bronchoscope through a rigid bronchoscope sheath, to allow ventilation and anesthesia, or bronchography. Although bronchography is advocated by some, many clinicians are cautious of employing bronchography, because of the potential for respiratory mucosal edema and its worsening. This extremely narrow airway group might also benefit from Virtual Endoscopy imaging to determine the size of post-stenosis bronchi and distal airway. Virtual Endoscopy is a modality of upper, segmental and subsegmental airway reconstruction; where using the high-resolution images obtained by multirow detectors on CT scan, and reformatting the data into three-dimensional images it resembles a dynamic video image obtained by flexible bronchoscopy [33].
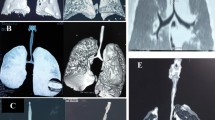
Recent technological advances in the field of computerized axial tomography (CAT) scanning have resulted in the widespread acceptance and use of “spiral” or “helical” scanning using multiple detectors (MDCT). This technology permits more anatomically accurate CT reconstructions in almost any plane desired to portray the child’s anatomy, including representations in “three dimensions” (3D MDCT) (Fig. 3) and “virtual endoscopy” (Fig. 4) which have proven to be more sensitive and specific (90.0 and 96.6%, respectively), superseding even flexible endoscopy in the diagnosis of tracheal narrowing secondary to neoplastic diseases in adults [33]. However, there are no similar studies validating the superiority of CT imaging to rigid bronchoscopy for the diagnosis of CTS in children. These techniques at reproducing the originally acquired axial images in different planes and projections are extremely useful to image the airway from both within (virtual endoscopy) and without (3D rendering techniques), but prove to be more challenging in the very tight congenital stenoses of the infants, as the computerized algorithms were designed for the adult patient. In these children, reliance on the original axial images is important to verify the accuracy of computerized reconstructions. With improving resolution, shorter acquisition times, improving tree construction pediatric algorithms and non invasiveness benefits, these studies have an excellent profile to become new gold standards in defining the extent and severity of airway narrowing in the years to come; but for the time being, there are still considerable limitations for its clinical use. At present, helical MDCT is the radiographic examination of choice for displaying complex combined anatomy of the major thoracic vessels and airways. However CT often underestimates the degree and the extent of narrowing in our experience [30, 34].

Posterior view of a computed tomography reconstruction of the airway (blue) and the heart and great vessels (red), subtracting all the surrounding tissues, including bone from spine, ribs and sternum. These images can be freely rotated on the computer in 360 grades for better visualization of anatomic details

Picture taken from a virtual bronchoscopy; tomographic reconstruction of airway lumen that comes in the form of a video, similar to the image you would get from flexible bronchoscopy
MRI has recently proven to be an elegant and non-invasive imaging modality to evaluate the associated cardiac and, in some cases, vascular malformations that may accompany a congenital tracheal stenosis. It can, as well, be useful for imaging the trachea but proves to be more challenging when imaging bronchial anatomy and secondary parenchymal changes. As well, a general anesthetic is often required for MRI of the infant or child. Patients with severe CTS are often already intubated and in critical condition prior to invasive investigation, making the imaging of the native airway even more difficult. However, rapid advances in MRI technology and scan sequences may soon prove to be an easier and safer modality to accurately and non-invasively image the cardiac anatomy as well as the airway of these complex patients.
Echocardiography still plays an important role in ruling out associated malformations in those patients with mild or no symptoms, in whom an MRI would seem too difficult (general anesthesia) or too expensive as a workup completion test.
Temptation to use every investigative modality available for imaging the airway and cardiovascular anatomy of a critically sick patient to gain as much information prior to intervention is strong. But time and effort required, particularly in acute clinically labile patients with unstable airway, may not be feasible or effective from a cost–benefit point of view.
A protocol for the investigation of the whole spectrum of CTS patients has not been established but would be welcome in order to best utilize resources and minimize invasive procedures or radiation exposure in this delicate patient group. We present a reasonable algorithm in Fig. 5, utilized by the ART at the Hospital for Sick Children in Toronto as a potential guideline.

This is a schematic representation for a proposed algorithm for the investigation of a neonate or infant presenting with respiratory distress, and inspiratory wheeze. The investigation of any child with symptoms suggesting CTS should have airway imaging performed by flexible or rigid bronchoscopy. The role of adjunctive investigation for assessment of associated anomalies should be performed in the context of any surgical repair of CTS needing careful cardio-vascular anatomy definition prior to clinical categorization
Therapeutic evolution and current options
Over the past decades different medical groups have attempted to treat this complex and very diverse disease through medical and surgical approaches.
Conservative treatment
Prior to 1980 the treatment for CTS was largely conservative with only a few case reports of successful surgical repairs [1, 26, 35]. There was a pervasive notion that CTS was not a surgically-treatable condition initially as evidenced by the list of publications showing poor operative outcomes and recommendations to avoid operative repairs in favor of palliative treatment [3]: Tracheostomy was a viable alternative, with the hope that the patient would eventually outgrow the stenosis. Now, it has become evident that there is a subset of CTS patients who can be managed nonoperatively and can be safely followed clinically, as they will outgrow their restrictive tracheal caliber by the age of 7–9 years [25]. There is however another subset of CTS patient who will clearly require surgery, often urgently, to manage both tracheal and cardiac malformations within the first month of life. This group is associated with the highest risk of mortality [34]. There is an intermediate group of CTS patients who are initially managed conservatively, but eventually require surgical intervention because of increasing symptoms [5, 7, 25].
Surgical management
Following reluctance for surgical repair and variable outcomes in the 1960s and 1970s, there is now an increasing optimism towards operative treatment of CTS as increasing surgical experience resulted in a significant improvement in patient survival since 1982. In general, the surgical approach to CTS can be divided into segmental resection, grafting (patch tracheoplasty) and slide tracheoplasty as dictated by the location of the stenosis, extent of disease and the surgeon’s experience. It is the actual tracheal pathology (anatomical classification) that dictates the choice of surgical decision.
Up to 50% of the length of trachea could be resected with acceptable complication rates in animals. In humans it is generally recognized that up to one-third of the trachea can be safely resected, before tension in the suture line becomes critical producing low perfusion, compromising the vitality of suture edges resulting in dehiscence and mediastinal air leak [36]. Moreover, granulation or re-stenosis of the anastomosis site develops delayed complications particularly if there is a tension in the suture line. Adjuvant techniques to reduce tension such as chin suturing and lung hilar mobilization are recommended when extensive resection bording up to one-third of the length is required.
Short segment stenosis (Cantrell type III) is amenable for resection and anastomosis. First done by Cantrell in 1964, then by Carcassone in 1973 and later reported by Harrison [26]. Additional use of cardio-pulmonary bypass (CPB) support brought additional stability to the patient, particularly small neonates, during anesthesia and surgery. The basic techniques however have changed little over the years.
Longer segment stenosis, including patients that have one-third of the length of the trachea compromised up to complete hypoplasia, cannot be resected because extensive mobilization of the trachea carry a significant amount of complications [29]. In these longer segment patients surgical repair has been approached with increasing enthusiasm since 1982, as reports showed increasing success in patient outcomes [37].
The first reported approach to the surgical management of long-segment CTS was the use of tracheal grafts to widen the airway diameter. A number of tissues have been used to graft the trachea after longitudinal incision of the stenotic segment, such as cartilage [30, 37], pericardium [38], periostium [39] in the anterior or lateral wall and esophagus as a long patch for the whole posterior wall [40]. Although the short-term results initially appeared very promising with rapid stabilization and re-epithelization of the graft surface, most of these procedures were eventually complicated by re-stenosis resulting from scarring and shrinkage of the grafted tissues or from granulation tissue [39, 41]. Typically, these patients would be symptom-free for a few weeks only to develop recurrent respiratory compromise necessitating repeated endoscopic investigation [8, 42]. In particular, the use of periostium for tracheal graft proved to be very disappointing because of bone ingrowths into the lumen of the trachea. This complication was very difficult to manage as no bronchoscopic instrumentation could successfully remove the bony spur from the tracheal lumen [6]. Another way of approaching the congenitally stenotic trachea was initially described in 1989 by Tsang and Goldstraw [43] and later augmented by Grillo [28] in 1994. As an alternative to tracheal grafting, slide tracheoplasty increased airway caliber without the need for extensive mobilization or the risk for tension at the suture line, thereby minimizing the risks of stricture and granulation tissue formation (Fig. 6). As it doesn’t need additional structural support as opposed to the grafting technique, it has the advantage of post-repair early extubation. CPB on the other hand has been considered unnecessary by some authors [28], but currently has become the standard of care in many centers for tracheal reconstruction for long segment patients.

Diagram of slide plasty for generalized hypoplasia: a shows a native stenotic trachea involving the whole length of the trachea, b the stenotic segment is divided in two parts, the upper half is divided on the posterior wall and the lower half in the anterior wall, according to the modification by Grillo et al. [28]. c, d the edges of both superior and inferior parts are trimmed away, so to leave round ends that can be sutured together evenly. e Final aspect of a shorter and wider appearance of trachea with the slide-plasty in place
From an historical perspective, the initial reluctance towards surgical treatment of CTS in the 1970s and 1980s turned into a burst of surgical activity in the 1990s; however the remaining challenge for treatment of this particular disease is to clarify the roles of various conservative and surgical treatment for each patient and each approach has been successful for the management of CTS.
For grouping complex patients with wide spectrum of pathology, presentation and treatment options, multidisciplinary team approach with understanding of the pathology has been one of the most important options in the quest for a favorable outcome since various combinations and alternatives of treatment bear a significant risk of mortality and morbidity along with a potentially low quality of life despite the best treatments.
Cardio-pulmonary bypass
Some authors promote the idea of selective use [28] of cardiopulmonary bypass in patients with a concomitant vascular or intracardiac malformation that needs to be repaired [36]. We think of the use of cardio-pulmonary bypass to be a standard part of surgical repair. Particularly in neonates and infants, the use of cardio-pulmonary bypass provides great stability and safety during the repair, regardless of the repair technique being chosen, although some morbidity associated to CPB are recognized. Although CPB has been examined as a potential risk factor by comparing patients that had CTS repair with and without CPB, there is no definitive answer albeit there is a trend in favor of using CPB [30] (Table 4) (Modified from Chiu et al. [30]).
Post-operative care
All patients with tracheal reconstruction regardless of the anatomy or the technique chosen need stenting for stability purposes. They require a suture stabilization period and are managed with deep sedation, intra tracheal intubation and mechanical ventilation for 7–days after surgery in our institution. Clinical expectation for an air leak should be maximal at the end of the first week as edema of the suture edges resorts. Intravenous Dexamethasone 1–2 mg/kg per day is divided and administered every 4 h for the first 24 h and then for the 24 h prior to extubation. For the next 6 weeks inhalable steroids are used to lower the risk of granulation in the suture line.
Prognosis
In this report, our recent experience with the management of these complex patients prompted a recent review of the prognostic factors for CTS patients [34]. Based on this review the clinical outcomes of CTS patients can be categorized into four distinct groups classified by their initial presentation, presence of associated co-morbidities and patient age [5, 25, 30] (Table 3). According to our data, CTS that has associated cardiac anomaly requiring surgical repair is associated with a higher risk factor for mortality, as is the patient aged less than one month at surgery. In combination, these prognostic features place this category of patients at the highest risk reaching almost 100%. This risk decreases to about 73% if the patient is older than 1 month (beyond the neonatal period). On the other hand if the patient is less than 2 years old, with isolated CTS without any cardiac malformation, the mortality in this group with surgical repair is about 16%, and rises to about 20% if respiratory failure has ever occurred at the time prior to surgery.
The usual clinical course after slide tracheoplasty repair is that patients improve with surgery, even though the doubled airway caliber is still smaller than the rest of the trachea and the norm for the age-matched tracheal diameter. The repaired trachea continues to grow postoperatively over the subsequent years as from the number of hospital admissions and respiratory infections per year or per winter season, the most critical period. They tend to recover in their weight gain curve and increase dramatically their spontaneous activity. The noisy breathing stays with them for a long time, though, because of the lumen irregularities after the repair. Some of the patients will need further airway interventions according to their post repair respiratory status sometimes due to the occurrence of granulation tissue. With our recent experience over 20 cases, we have not experienced any granulation tissue build up requiring a further intervention. Possible long-term complications such as re-stenosis at the suture line should be evaluated if symptoms recur and be treated accordingly.
Conclusions and future direction
Congenital tracheal stenosis remains an exciting open chapter in the textbooks of pediatric surgery. Our limited understanding of the pathogenesis of CTS highlights the need to develop embryologic models, which will shed light on tracheal cartilage development and pattern of segmentation. Although multiple centers have reported surgical success in the management of CTS, the best outcomes result from the management of CTS patients in specialized centers with focused interest and multi disciplinary approach.
The surgical treatment of CTS includes segmental resection and anastomosis, which is the best choice for short-segment stenosis, while slide tracheoplasty seems to be the best treatment for funnel-shaped and long-segment CTS. Stents may be used as an adjuvant therapy but they carry additional morbidity on their own and are used as salvage options [44, 45]. Last, there are increasing data to suggest that children can outgrow their CTS if they manage to survive beyond the first year of life [5, 7, 25]. This is more likely to occur if there are no associated cardiac malformations and with Cantrell types II and III of CTS.
References
Cantrell JR, Guild H (1964) Congenital stenosis of the trachea. Am J Surg 108:297–305
Chiu P, Airway Reconstruction Team (2005) Recent challenges in the management of congenital tracheal stenosis: an individualized approach. J Pediatr Surg 40(5):774–780
Benjamin B, Pitkin J, Cohen D (1981) Congenital tracheal stenosis. Ann Otolaryngol 90:364–371
Heimansohn D (1991) Anterior pericardial tracheoplasty for congenital tracheal stenosis. J Thoracic Cardiovasc Surg 102:710–716
Anton-Pacheco J, Cano I, Garcia A, Martinez A, Cuadros J, Berchi F (2003) Patterns of management of congenital tracheal stenosis. J Pediatr Surg 38(10):1452–1458
Loeff D, Filler R, Vinograd I, Ein SH, Williams WG, Smith CR, Bahoric A (1988) Congenital tracheal stenosis: a review of 22 patients from 1965 to 1987. J Pediatr Surg 23(8):744–748
Rutter M, Willging P, Cotton R (2004) Non-operative management of complete tracheal rings. Arch Otolaryngol Head Neck Surg 130:450–452
Kim HK, Kim YT, Sung SW, Park JD, Kang CH, Kim JH, Kim YJ (2004) Management of congenital tracheal stenosis. Eur J Cardio-Thoracic Surg 25:1065–1071
Felix JF, Keijzer R, Van Dooren MF, Rottier RJ, Tibboel D (2004) Genetics and developmental biology of oesophageal atresia and tracheo-esophageal fistula: lessons from mice relevant for paediatric surgeons. Pediatr Surg Int 20:731–736
Perl AK, Wert S, Nagy A, Lobe C, Whitsett JA (2002) Early restriction of peripheral and proximal cell lineages during formation of the lung. Dev Biol 99(16):10482–10487
Ioannides A, Henderson D, Spitz L, Copp AJ (2003) Role of sonic hedgehog in the development of the trachea and oesophagus. JPS 38(1):29–33
Miller L, Wert S, Clark J, Xu Yan, Perl AK, Whitsett J (2004) Role of sonic hedgehog in patterning of tracheal–bronchial cartilage and the peripheral lung. Dev Dyn 231(1):51–71
Whitsett J, Clark JC, Picard L, Tichelaar J, Wert S, Itoh N, Perl AK, Stahlman M (2002) Fibroblast growth factor 18 influences proximal programming during lung morphogenesis. J Biol Chem 277(25):22743–22749
Crisera C, Marosky J, Longaker M, Gittes G (2003) Organogenesis particularly relevant to fetal surgery. World J Surg 27(1):38–44
Kluth D, Fliegel H (2003) The embryology of the foregut. Semin Pediatr Surg 12(1):3–9
Gray and Skandalakis (1972) Embryology for surgeons. Saunders, Philadelphia, pp 293–298
Groenman F, Unger S, Post M (2005) The molecular basis for abnormal lung development. Biol Neonate 87:164–177
Orford J, Cass DT (1999) Dose response relationship between adriamycin and birth defects in a rat model of VATER association. J Pediatr Surg 34(5):392–398
Franca WM, Goncalves A, Moraes SG, Pereira LA, Sbragia L (2004) Esophageal atresia and other visceral anomalies in a modified Adriamycin rat model and their correlations with amniotic fluid volume variations. Pediatr Surg Int 20(8):602–608. Epub 2004 Aug 26
Diez-Pardo JA, Baoquan Q, Navarro C, Tovar JA (1996) A new rodent experimental model of esophageal atresia and tracheoesophageal fistula: preliminary report. J Pediatr Surg 31(4):498–502
Warkany J, Roth TB, Wilson J (1948) Multiple congenital malformations: a consideration of etiologic factors. Pediatrics 1:462–471
Magnaldo T (2002) SHH (Sonic hedgehog). Atlas genet cytogenet oncol haematol. February2002. URL: http://www.infobiogen.fr/services/chromcancer/Genes/SHHID378.html
Elluru R, Whitsett J (2004) Potential role of Sox9 in patterning tracheal cartilage ring formation in an embryonic mouse model. Arch Otolaryngol Head Neck Surg 130:732–736
Schnitzer JJ (2004) Control and regulation of pulmonary hypoplasia associated with congenital diaphragmatic hernia. Semin Pediatr Surg 13(1):37–43
Cheng W, Manson D, Forte V, Ein S, McKlusky I, Papsin B, Hectcher S, Kim PCW (2006) The role of conservative management in congenital tracheal stenosis: an evidence-based long term follow-up study. The role of conservative management in congenital tracheal stenosis: an evidence-based long-term follow-up study. J Pediatr Surg 41(7):1203–1207
Harrison MR, Heldt GP, Brasch RC, deLorimier A, Gregory G (1980) Resection of distal tracheal stenosis in a baby with agenesis of the lung. J Pediatr Surg 15(6):938–943
Grillo HC, Zannini P (1984) Management of obstructive tracheal disease in children. J Pediatr Surg 19(4):414–416
Grillo H (1994) Slide tracheoplasty for long-segment congenital tracheal stenosis. Ann Thorac Surg 58:613–621
Anton-Pacheco JL, Cano I et al (2006) Management of congenital tracheal stenosis in infancy. Eur J Cardiothorac Surg 29(6):991–996. Epub 2006 May 3
Chiu P, Kim PCW (2006) Prognostic factors in the surgical treatment of congenital tracheal stenosis: a multicenter analysis of the literature. J Pediatr Surg 41(1):221–225
Backer C, Mavroudis C, Dunham M, Holinger L (1998) Repair of congenital tracheal stenosis with a free tracheal autograft. Thorac Cardiovasc Surg 869–874
Elliot M, Roebuck D, Noctor C, McLaren C, et al (2003) The management of congenital tracheal stenosis. Int J Otolaryngol 67S1:S183–S192
Hoppe H, Dinkel H-P, Walder B, von Allmen G, Gugger M, Volk P (2004) Grading airway stenosis down to the segmental level using virtual bronchoscopy. Chest 125(2):704–711
Chiu PP, Kim PCW (2006) Prognostic factors in the surgical treatment of congenital tracheal stenosis: a multicenter analysis of the literature. J Pediatr Surg 41(1):221–5; discussion 221–225
Kleinhaus S, Winslow PR, Sheran M, Boley SJ (1978) Evolution of individualized management of tracheal obstruction. J Pediatr Surg 13(6):669–676
Grillo Hermes C (2004) Surgery of the trachea and bronchi. Decker, Hamilton, pp 665–666
Kimura K, Mukohara N, Tsugawa C, Matsumoto Y, Sugimura C, Murata H, Itoh H (1982) Tracheoplasty for congenital stenosis of the entire trachea. J Pediatr Surg 17(6):869–871
Idriss F, De Leon S, Ilbawi M, Gerson C, Tucker G, Holinger L (1984) Tracheoplasty with pericardial patch for extensive tracheal stenosis in infants and children. J Thorac Cardiovasc Surg 88:527–536
Cohen R, Filler R, Konuma K, Bahoric A, Kent G, Smith C (1986) A new model of tracheal stenosis and its repair with free periosteal grafts. J Thorac Cardiovasc Surg 92(2):296–304
Ein S, Friedberg J, Williams W, Rearon B, Barker G, Mancer K (1982) Tracheoplasty—a new operation for complete tracheal stenosis. J Pediatr Surg 17(6):872–877
Cheng A, Backer C, Holinger L, Dunham M, Mavroudis C, Gonzalez-Crussi F (1997) Histopathologic changes after pericardial patch tracheoplasty. Arch Otolaryngol Head Neck Surg 123:1069–1072
Cunningham M, Eavey R, Vlahakes G, Grillo H (1998) Slide tracheoplasty for long segment tracheal stenosis. Arch Otolaryngol Head Neck Surg 124:98–103
Tsang V, Murday A, Gilbe C, Goldstraw P (1989) Slide tracheoplasty for congenital funnel-shapeed tracheal stenosis. Ann Thoracic Surg 48:632–635
Vinograd I, Keidar S, Weinberg M, Silbiger A (2004) Treatment of airway obstruction by metallic stents in infants and children. J Thoracic Cardiovasc Surg 130(1):146–150
Filler R, Forte V, Chait P (1998) Tracheobronchial stenting for the treatment of airway obstruction. J Pediatr Surg 33(2):304–311
Tsugawa C, Kimura K, Muraji T, Nishijima E, Matsumoto Y, Murata H (1998) Congenital stenosis involving a long segment of the trachea: further experience in reconstructive surgery. J Pediatr Surg 23(5):471–475
Loukanov T, Sebening C, Springer W, Ulmer H, Hagl S (2005) Simultaneous management of congenital tracheal stenosis and cardiac anomalies in infants. J Thorac Cardiovasc Surg 130(6):1537–1541
Acknowledgement
We would like to kindly thank Dr. Sigmund H. Ein, for his endless sense of humor and his valuable and insightful comments on this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Herrera, P., Caldarone, C., Forte, V. et al. The current state of congenital tracheal stenosis. Pediatr Surg Int 23, 1033–1044 (2007). https://doi.org/10.1007/s00383-007-1945-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-007-1945-3