
Abstract
Backround
The high morbidity and mortality in congenital diaphragmatic hernia (CDH) are attributed to severe pulmonary hypoplasia and persistent pulmonary hypertension (PH). PH is characterized by structural changes in pulmonary arteries, resulting in adventitial and medial thickness. These effects are triggered by abnormal apoptosis and proliferation of pulmonary vascular endothelial and smooth muscle cells (SMCs). Apelin (APLN), a target gene of bone morphogenic protein receptor 2 (BMPR2), is known to play an important and manifold role in regulating pulmonary homeostasis promoting endothelial cell (EC) survival, proliferation and migration. In addition to these autocrine effects of apelin, it displays a paracrine function attenuating the response of pulmonary SMCs to growth factors and promoting apoptosis. Apelin exerts its effect via its G-protein-coupled receptor (APLNR) and is solely expressed by pulmonary vascular EC, whereas APLNR is co-localized in pulmonary ECs and SMCs. Dysfunction of BMPR2 and downstream signalling have been shown to disturb the crucial balance of proliferation of SMCs contributing to the pathogenesis of human and experimentally induced PH. We designed this study to investigate the hypothesis that apelin and APLNR signalling are disrupted in the pulmonary vasculature of rats in nitrofen-induced CDH.
Methods
Pregnant rats were exposed to nitrofen or vehicle on D9 of gestation. Foetuses were sacrificed on D21 and divided into nitrofen and control group (n = 32). Pulmonary RNA was extracted and mRNA levels of APLN and APLNR were determined by quantitative real-time PCR. Protein expression of apelin and APLNR was investigated by western blotting. Confocal immunofluorescence double staining for apelin, APLNR and SMCs were performed.
Results
Relative mRNA level of APLN and APLNR were significantly decreased in the CDH group compared to control lungs. Western blotting and confocal microscopy confirmed the qRT-PCR results showing decreased pulmonary protein expression of apelin and APLNR in lungs of nitrofen-exposed foetuses compared to controls.
Conclusion
This study provides striking evidence of markedly decreased gene and protein expression of apelin and its receptor APLNR in the pulmonary vasculature of nitrofen-induced CDH. The disruption of the apelin–APLNR signalling axis in the pulmonary vasculature may lead to extensive vascular remodelling and contribute to PPH in the nitrofen-induced CDH model.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Approximately 1 in 2,000–3,000 neonates experiences a congenital diaphragmatic hernia (CDH). Despite recent advances in neonatal intensive care, morbidity and mortality rates for CDH still remain high. These are attributed to severe pulmonary hypoplasia and persistent pulmonary hypertension (PH). PH is histologically characterized by a loss of the microcirculation and structural changes in pulmonary arteries, resulting in adventitial and medial thickness [1]. This effect leading to elevated pulmonary artery resistance and muscularization of distal pulmonary arteries is caused by dysfunctional endothelial cells (ECs) as well as disrupted apoptosis and proliferation of pulmonary vascular smooth muscle cells (SMCs) and arterial fibroblasts [2–4]. Although multiple genetic, cellular and molecular functions are known to be involved in adult idiopathic PH, the exact molecular mechanisms of neonates with CDH-associated pulmonary hypertension remain concealed [5, 6].
Loss of bone morphogenic protein receptor 2 (BMPR2) signalling promotes EC apoptosis and results in reduced EC survival. Furthermore, disrupted BMPR2 expression has been shown to lead to attenuation of angiogenic capacity of pulmonary ECs [7]. Apelin (APLN), a transcriptional target gene of BMPR2 in ECs is known to play an important and manifold role responsible for EC homeostasis, regulating survival, proliferation and migration [8]. In addition to these autocrine effects of apelin, it displays a paracrine function attenuating the response of SMCs to growth factors and promoting apoptosis [8–10].
While apelin exerts its effects via its G-protein-coupled receptor (APLNR) and is expressed by pulmonary vascular ECs, the APLNR is co-localized in pulmonary ECs and SMCs. Both apelin and APLNR expression are known to be regulated by BMPR-2 via its peroxisome proliferator-activated receptor γ (PPARγ) signalling pathway [11–13]. Dysfunction of BMPR2 and downstream signalling have been shown to disturb the crucial balance of proliferation of SMCs contributing to the pathogenesis of human and experimentally induced PH [6, 8]. Serum and pulmonary endothelium apelin levels of subjects with PH have been reported to be significantly reduced [8, 14, 15]. Furthermore, apelin knockout mice developed exacerbated PH in response to hypoxia [15].
At present, the nitrofen-induced CDH is the most widely accepted animal model to study the pathogenesis and clinical consequences of CDH in accordance of the striking similarities to the human condition [16–18]. We hypothesized that the apelin–APLNR signalling axis is disrupted in the pulmonary vasculature of CDH-associated PH and therefore designed this study to investigate the gene and protein expression of apelin and its receptor APLNR.
Materials and methods
Animals and drugs
Timed-pregnant, adult Sprague–Dawley rats (Harlan Laboratories, Shardlow, UK) were randomly divided into two experimental groups (“CDH” and “control”). The presence of spermatozoids in the vaginal smear was considered as proof of pregnancy; the day of observation was determined as day 0 (D0). On D9, animals in the CDH group received 100 mg of nitrofen (2,4-dichloro-p-nitrophenyl ether, WAKO Chemicals, Osaka, Japan) intragastrically dissolved in 1 ml of olive oil. Animals in the control group received only vehicle. On D21, dams were anesthetized with 2 % volatile isofluran (Piramal Healthcare UK, Morpeth, UK), followed by delivery of the foetuses via caesarean section. The foetuses were killed by decapitation and divided according to the two experimental groups. In the CDH group laparotomy was performed for inspection of CDH. Left lungs with a diaphragmatic defect and controls were dissected via thoracotomy and stored native either at −80 °C, in formalin or in TRIzol reagent (Invitrogen, Carlsbad, CA, USA) at −20 °C. All animal experiments were carried out according to the current guidelines for management and welfare of laboratory animals. The experimental protocol was approved by the Department of Health and Children (Ref. B100/4378) under the Cruelty to Animals act 1876 (as amended by European Communities Regulations 2002 and 2005).
RNA isolation from left lungs (D21)
TRIzol reagent (invitrogen) was used for the acid guanidinium thiocyanate–phenol–chloroform extraction method to isolate total RNA from D21 lungs (n = 8) according to the manufacturer’s protocol. Spectrophotometrical quantification of total RNA was performed using a NanoDrop ND-1000 UV–Vis spectrophotometer (Thermo Scientific Fisher, Wilmington, USA). The RNA solution was stored at −20 °C until further use.
cDNA synthesis and quantitative polymerase chain reaction
Reverse transcription of total RNA was carried out at 85 °C for 3 min (denaturation), at 44 °C for 60 min (annealing) and at 92 °C for 10 min (reverse transcriptase inactivation) using a Transcriptor High Fidelity cDNA Synthesis Kit (Roche Diagnostics, West Sussex, UK) according to the manufacturer’s instruction. The resulting cDNA was used for quantitative real-time polymerase chain reaction (qRT-PCR) using a LightCycler 480 SYBR Green I Master (Roche Diagnostics, Mannheim, Germany) in a total reaction mix of 20 μl per well. Gene-specific primer pairs are listed in Table 1. After 5 min of initial denaturation at 95 °C, 55 cycles of amplification for each primer were carried out. Each cycle included denaturation at 95 °C for 10 s, annealing at 60 °C for 15 s, and elongation at 72 °C for 10 s. Relative mRNA levels of gene expression were determined using a LightCycler 480 System (Roche Diagnostics) and the relative changes in gene expression levels of APLN and APLNR were normalized against the level of GAPDH gene expression in each sample (∆∆C T method). Experiments were carried out in duplicate for each sample and primer.
Western blot
Fresh frozen left lungs (n = 4 for each group) were thawed, sonicated and proteins were isolated in lysis buffer containing 25 mM Tris–HCl, 50 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1 % NP-40, 10 % glycerol and 1 % protease inhibitor cocktail (Sigma-Aldrich Ireland, Wicklow, Ireland). Protein concentrations were determined by Bradford assay (Sigma-Aldrich Ireland). Protein concentrations were equalized by dilution with distilled water. For gel electrophoresis, a concentration of 20 μg of total protein was used for every specimen and denatured in Laemmli sample buffer (Sigma-Aldrich Ireland). Gel electrophoresis for protein separation was performed using precast 10 % SDS polyacrylamide gels (NuPAGE Novex Bis–Tris gels, Invitrogen) in NuPAGE MES SDS running buffer (invitrogen). Proteins were then transferred to 0.45-μm nitrocellulose membranes (Millipore Corporation, Billerica, USA) by Western blotting. Following Western blotting, the membranes were blocked in 3 % BSA + 0.05 % Tween for 30 min or overnight before antibody detection. Primary antibodies against apelin (rabbit polyclonal, sc-33804, dilution 1:200, Santa Cruz Biotechnology, Santa Cruz, USA) and APLNR (rabbit polyclonal, sc-33823 dilution 1:200, Santa Cruz Biotechnology) were incubated overnight at 4 °C. On the next day, followed by extensive washing (4 h), the membranes were incubated with the secondary antibodies in a dilution of 1:10,000 (anti-rabbit: #7074 S, Cell Signaling Technology, USA) followed again by extensive washing. Detection was performed with a PIERCE chemiluminescence kit (Thermo, Fisher Scientific, Dublin, Ireland).
Immunofluorescence staining and confocal microscopy
Foetal left lungs (n = 4 for each group) were fixed with 10 % buffered formalin (Santa Cruz Biotechnology) overnight. Whole organs were washed overnight in PBS, embedded in O.C.T. Mounting Compound (VWR International, Leuven, Belgium) and frozen at −80 °C. Frozen blocks were sectioned transversely at a thickness of 15 μm and mounted on SuperFrost Plus slides (VWR International). After washing with PBS, only the sections for apelin underwent cell membrane permeabilization with 1 % Triton X-100 for 20 min at room temperature. All sections were then washed and subsequently blocked with 3 % BSA for 30 min to avoid nonspecific absorption of immunoglobulin. Blocking solution was rinsed off and sections were incubated with primary antibodies against apelin (rabbit polyclonal, sc-33804, 1:200 dilution in PBST, Santa Cruz Biotechnology), APLNR (rabbit polyclonal, sc-33823, 1:200 dilution in PBST, Santa Cruz Biotechnology) and alpha smooth muscle actin (α-SMA, mouse monoclonal, M0851, 1:400 dilution in PBST, DAKO Diagnostics Ireland, Dublin, Ireland) overnight at 4 °C. After washing, apelin as well as APLNR sections were incubated with corresponding secondary antibodies (donkey anti-rabbit Alexa-A150067 and donkey anti-mouse Alexa 488-A150109, Abcam, UK) for 1 h at room temperature. After washing, sections were counterstained with DAPI antibody (1:1,000 in PBST, Roche), washed again and mounted using Sigma Mounting Medium (Sigma-Aldrich, St. Louis, MO, USA). Sections were scanned with a ZEISS LSM 700 confocal microscope (Carl Zeiss MicroImaging GmbH, Jena, Germany) and evaluated independently by two investigators.
Statistical analysis
All numerical data are presented as mean ± standard error of the mean. Student’s t test was used for evaluation of differences between two normal distributed groups on D21. The confidence interval was set at 95 %.
Results
Relative mRNA expression levels of APELIN and APLNR in lungs
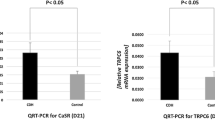
The relative mRNA expression levels of APELIN and APLNR were significantly decreased (p < 0.05) in lungs (D21) of the CDH group, compared to lungs of control animals (Fig. 1).

qRT-PCR revealed significantly decreased relative mRNA expression levels of a APELIN (p < 0.05) and b APLNR in nitrofen-exposed lungs (D21) compared to normal lung tissue. Western blot results show that the decreased APELIN (a) and APLNR (b) transcripts in nitrofen-exposed lungs compared to controls resulted in decreased amounts of apelin (a) and APLNR (b) protein expression
Western blot for apelin and APLNR
To confirm the qRT-PCR result we analyzed the protein expression of APELIN and additionally APLNR. Our Western blot results showed decreased pulmonary protein expression for both apelin and APLNR (Fig. 1) in the nitrofen-treated group when compared with controls. Equal loading of electrophoresis gels was confirmed by beta-actin and GAPDH staining of the stripped membranes (Fig. 1).
Immunofluorescence evaluation of apelin, APLNR and α-SMA
Confirmation by western blotting of the decreased apelin and APLNR protein expression was followed by immunofluorescence evaluation of pulmonary tissue. First noticeable difference was the increased medial and adventitial thickness of pulmonary arteries in the CDH group. Confocal microscopy validated the qRT-PCR and the Western blot findings showing decreased vascular apelin and APLNR expression in ECs and SMCs of nitrofen-exposed lungs compared to normal lung tissue (Fig. 2).

Immunofluorescence evaluation of pulmonary tissue for a apelin and b APLNR (1:200) as well as (a, b) alpha smooth muscle actin (α-SMA) (1:400). α-SMA was used to identify pulmonary arteries. Confocal microscopy further revealed decreased vascular apelin and APLNR expression in the pulmonary vasculature of nitrofen-exposed lungs compared to normal lung tissue (a, b)
Discussion
The development of adult idiopathic as well as CDH-associated pulmonary hypertension involves multiple and complex pathomechanisms [5]. Recently, several authors have outlined the role of endothelial dysfunction, impaired vasculogenesis and angiogenesis on the one hand and the imbalance of proliferation and apoptosis of pulmonary vascular SMCs on the other [6, 19, 20]. Endothelial dysfunction leads to an imbalance of vasodilators and vasoconstrictors resulting in pathological changes of the pulmonary microcirculation. In addition, the homeostasis of SMC proliferation and apoptosis is impaired, which concurrently mediates thickening of the pulmonary vasculature, leading to vasoconstriction, vascular remodelling, increased pulmonary vascular resistance and a rise in pulmonary arterial pressure [6, 21]. These changes seen in adult idiopathic PH are strikingly similar to those seen in infants with CDH, where pulmonary hypertension also manifests as abnormal proliferation of pulmonary SMCs and EC dysfunction [19]. Common existing models of experimental PH, such as the monocrotaline model or hypoxia-induced PH, mainly focus on adult or postnatal PH [22, 23]. However, the underlying molecular mechanisms involved in the pathogenesis of PH in CDH resulting in pulmonary vascular remodelling in utero remain unclear.
To further investigate these pathomechanisms, we used the well-established nitrofen-induced CDH model, which has been shown to reliably reproduce intrauterine abnormalities of the pulmonary vasculature [19].
Apelin, a downstream target gene of BMPR2 secreted by ECs, has been identified to regulate pulmonary vascular homeostasis. It exerts its function via the APLNR, which is co-localized on ECs and pulmonary vascular SMCs. Apelin contributes to EC homeostasis by promoting pulmonary EC survival, proliferation and migration. These are functionally significant features in the preservation and regeneration of the pulmonary vasculature. In addition to these autocrine effects, apelin also displays a paracrine function attenuating the response of pulmonary SMCs to growth factors and promoting apoptosis [8].
Different experimental models of PH have demonstrated the significance of the apelin–APLNR signalling axis for pulmonary vascular homeostasis. Piairo et al. [24] recently provided strong evidence for the presence of an apelinergic system operating in the developing lung. Mice lacking apelin are more sensitive to hypoxia-induced pulmonary hypertension thus exhibiting more distinct pulmonary vascular remodelling than wild-type mice [15]. Moreover, administration of exogenous apelin leads to a reversal of disease in the monocrotaline model of pulmonary hypertension [8, 25]. With apelin located in the pulmonary endothelium and its receptor situated on both SMCs and ECs, the apelin–APLNR signalling axis presents a novel target in the pulmonary vasculature of diseased lungs [5, 26–28].
In the present study, we investigated the pulmonary APLN and APLNR gene expression levels as well as protein expression and distribution in lungs of nitrofen-induced CDH. We demonstrated that APLN and APLNR gene expression was significantly downregulated in nitrofen-treated hypoplastic lungs with CDH when compared with controls. These findings are consistent with human data, where isolated pulmonary microvascular ECs (PMVECs) from patients with idiopathic PH showed reduced APLN mRNA and apelin protein expression when compared with PMVECs of healthy patients [8]. Furthermore, our western blotting and confocal microscopy findings showed decreased Apelin and APLNR protein expression in pulmonary artery ECs and SMCs of CDH lungs when compared with controls. These results therefore confirm that the quantitative decrease of APLN and APLNR mRNA transcripts in CDH lungs was also translated to the protein level, indicating that disturbance of the apelin–APLNR signalling axis contributes to the development of vascular remodelling in the nitrofen-induced CDH model.
Previous studies have shown BMPR2 signalling to regulate apelin and APLNR expression. Furthermore, they linked disrupted BMPR2 signalling to the pathogenesis of human and experimental PH [29–31]. Alastalo et al. [8] just recently identified apelin as a transcriptional target of the PPARγ/β-catenin complex that displays reduced expression in ECs of patients with deficient BMPR2 expression and resulting idiopathic PH. In accordance to these results, recent work from our laboratory has shown decreased pulmonary expression of PPARγ and BMPR2 signalling in the vasculature of nitrofen-induced CDH lungs on day 21 [6, 31]. Taking into account that loss of apelin function leads to reduction of EC and SMC apoptosis and to increased SMC proliferation [8, 15], these results corroborate our findings providing additional evidence that the apelin–APLNR signalling axis is downregulated in the pulmonary vasculature of nitrofen-treated rats.
Taken together, these observations strongly suggest the apelin–APLNR signalling axis to be involved in EC dysfunction, pathological SMC proliferation, migration and vasoconstriction, altogether playing an important role in the pathophysiology of vascular remodelling and contributing to PH in the nitrofen-induced CDH model.
References
Keijzer R, Puri P (2010) Congenital diaphragmatic hernia. Semin Pediatr Surg 19:180–185. doi:10.1053/j.sempedsurg.2010.03.001
Heath D, Smith P, Gosney J et al (1987) The pathology of the early and late stages of primary pulmonary hypertension. Br Heart J 58:204–213
Belik J, Davidge ST, Zhang W et al (2003) Airway smooth muscle changes in the nitrofen-induced congenital diaphragmatic hernia rat model. Pediatr Res 53:737–743. doi:10.1203/01.PDR.0000057986.74037.7B
Yamataka T, Puri P (1997) Active collagen synthesis by pulmonary arteries in pulmonary hypertension complicated by congenital diaphragmatic hernia. J Pediatr Surg 32:682–687
Andersen CU, Hilberg O, Mellemkjær S et al (2011) Apelin and pulmonary hypertension. Pulm Circ 1:334–346. doi:10.4103/2045-8932.87299
Gosemann J-H, Friedmacher F, Fujiwara N, et al (2013) Disruption of the Bone morphogenetic protein receptor 2 pathway in nitrofen-induced congenital diaphragmatic hernia. Birth Defects Res B. doi:10.1002/bdrb.21065 (Epub ahead of print)
de Jesus Perez VA, Alastalo T-P, Wu JC et al (2009) Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-beta-catenin and Wnt-RhoA-Rac1 pathways. J Cell Biol 184:83–99. doi:10.1083/jcb.200806049
Alastalo T-P, Li M, de Perez VJ et al (2011) Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest 121:3735–3746. doi:10.1172/JCI43382
Morrell NW, Yang X, Upton PD et al (2001) Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation 104:790–795
Zhang S, Fantozzi I, Tigno DD et al (2003) Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 285:L740–L754. doi:10.1152/ajplung.00284.2002
Bonnet S, Michelakis ED, Porter CJ et al (2006) An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113:2630–2641. doi:10.1161/CIRCULATIONAHA.105.609008
Glassford AJ, Yue P, Sheikh AY et al (2007) HIF-1 regulates hypoxia- and insulin-induced expression of apelin in adipocytes. Am J Physiol Endocrinol Metab 293:E1590–E1596. doi:10.1152/ajpendo.00490.2007
Morrell NW, Adnot S, Archer SL et al (2009) Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 54:S20–S31. doi:10.1016/j.jacc.2009.04.018
Goetze JP, Rehfeld JF, Carlsen J et al (2006) Apelin: a new plasma marker of cardiopulmonary disease. Regul Pept 133:134–138. doi:10.1016/j.regpep.2005.09.032
Chandra SM, Razavi H, Kim J et al (2011) Disruption of the apelin–APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol 31:814–820. doi:10.1161/ATVBAHA.110.219980
Greer JJ (2013) Current concepts on the pathogenesis and etiology of congenital diaphragmatic hernia. Respir Physiol Neurobiol. doi:10.1016/j.resp.2013.04.015
Beurskens N, Klaassens M, Rottier R et al (2007) Linking animal models to human congenital diaphragmatic hernia. Birth Defects Res Part A Clin Mol Teratol 79:565–572. doi:10.1002/bdra.20370
Mayer S, Metzger R, Kluth D (2011) The embryology of the diaphragm. Semin Pediatr Surg 20:161–169. doi:10.1053/j.sempedsurg.2011.03.006
Luong C, Rey-Perra J, Vadivel A et al (2011) Antenatal sildenafil treatment attenuates pulmonary hypertension in experimental congenital diaphragmatic hernia. Circulation 123:2120–2131. doi:10.1161/CIRCULATIONAHA.108.845909
Runo JR, Loyd JE (2003) Primary pulmonary hypertension. Lancet 361:1533–1544. doi:10.1016/S0140-6736(03)13167-4
Gurbanov E, Shiliang X (2006) The key role of apoptosis in the pathogenesis and treatment of pulmonary hypertension. Eur J Cardiothorac Surg 30:499–507. doi:10.1016/j.ejcts.2006.05.026
Keegan A, Morecroft I, Smillie D et al (2001) Contribution of the 5-HT(1B) receptor to hypoxia-induced pulmonary hypertension: converging evidence using 5-HT(1B)-receptor knockout mice and the 5-HT(1B/1D)-receptor antagonist GR127935. Circ Res 89:1231–1239
Guignabert C, Raffestin B, Benferhat R et al (2005) Serotonin transporter inhibition prevents and reverses monocrotaline-induced pulmonary hypertension in rats. Circulation 111:2812–2819. doi:10.1161/CIRCULATIONAHA.104.524926
Piairo P, Moura RS, Nogueira-Silva C, Correia-Pinto J (2011) The apelinergic system in the developing lung: expression and signaling. Peptides 32:2474–2483. doi:10.1016/j.peptides.2011.10.010
Falcão-Pires I, Gonçalves N, Henriques-Coelho T et al (2009) Apelin decreases myocardial injury and improves right ventricular function in monocrotaline-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 296:H2007–H2014. doi:10.1152/ajpheart.00089.2009
Habata Y, Fujii R, Hosoya M et al (1999) Apelin, the natural ligand of the orphan receptor APJ, is abundantly secreted in the colostrum. Biochim Biophys Acta 1452:25–35
Katugampola SD, Maguire JJ, Matthewson SR, Davenport AP (2001) [125I]-(Pyr 1)Apelin-13 is a novel radioligand for localizing the APJ orphan receptor in human and rat tissues with evidence for a vasoconstrictor role in man. Br J Pharmacol 132:1255–1260. doi:10.1038/sj.bjp.0703939
Ishida J (2004) Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J Biol Chem 279:26274–26279. doi:10.1074/jbc.M404149200
Du L, Sullivan CC, Chu D et al (2003) Signaling molecules in nonfamilial pulmonary hypertension. N Engl J Med 348:500–509. doi:10.1056/NEJMoa021650
Atkinson C, Stewart S, Upton PD et al (2002) Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 105:1672–1678
Gosemann J-H, Doi T, Kutasy B et al (2012) Alterations of peroxisome proliferator-activated receptor γ and monocyte chemoattractant protein 1 gene expression in the nitrofen-induced hypoplastic lung. J Pediatr Surg 47:847–851. doi:10.1016/j.jpedsurg.2012.01.038
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hofmann, A.D., Friedmacher, F., Takahashi, H. et al. Decreased apelin and apelin-receptor expression in the pulmonary vasculature of nitrofen-induced congenital diaphragmatic hernia. Pediatr Surg Int 30, 197–203 (2014). https://doi.org/10.1007/s00383-013-3450-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-013-3450-1