
Abstract
Introduction
Cavernous angiomas of the brain (CCM) are being increasingly diagnosed, especially in the paediatric age group. Though classic presentations with haemorrhage or seizures are well recognised, presentation as a large lesion with mass effect is rare and creates difficulty in diagnosis as well as management.
Methods
Our cases of paediatric giant CCMs that presented as a ‘mass lesion’ are reported here, and the PubMed database for giant CCMs in the paediatric population is reviewed. All articles where the size of the lesion was reported to be > 4 cm were selected for analysis to study the varying modes of presentation, treatment, and outcome; to gain a proper perspective on this distinct entity of ‘giant CCMs’.
Results
Analysis of a total of 53 cases (inclusive of our 3 cases) reported so far showed slight male preponderance (58.49%). The largest reported lesion was 14 cm in largest diameter. Most of the lesions (83.02%) occurred in the supratentorial region. In the infratentorial region, paediatric giant CCMs were more commonly seen in the cerebellum than in the brainstem. Seizures were observed in 47.17% at presentation. Features of mass effect were the mode of presentation in all our cases, and literature analysis has shown raised intracranial pressure in 37.74% (20 patients) and focal neurological deficit in 33.96% (18 patients) at presentation. Macrocephaly was seen in younger children up to the age of 7 years (16.98% or 9 patients). Gross total resection was carried out (with a good outcome) in all our cases and in 36 of the other 49 analysed patients who were operated on.
Discussion
About one-fourth of CCMs occur in paediatric patients. Giant CCMs are rare but can present in children even in the immediate post-natal period. Features of a mass lesion such as raised intracranial pressure, macrocephaly, and focal neurological deficit are much more common than their smaller counterparts. Their appearance on imaging also often causes diagnostic dilemmas with other intracranial mass lesions. Timely surgery with standard microsurgical principles leads to a favourable outcome in the majority.
Conclusion
Giant CCMs, though rare, often present as a diagnostic challenge. Presentation with mass effect is common, and complete microsurgical excision remains the mainstay of treatment. Though transient neurological deficits may be encountered with this strategy, the long-term outcome remains favourable.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Developmental malformations of the blood vessels supplying the brain are traditionally classified (McCormick et al.) based on histopathological features, into four categories namely, venous malformations, arteriovenous malformations, cavernous malformations (CCMs), and telangiectases [1]. CCMs account for about 5–13% of intracranial vascular malformations [2,3,4]. Their peak incidence varies between 20 and 40 years of age [2, 3, 5]. Their size usually ranges from 9 to 20 mm [6], and the reported mean size is about 1.4 cm in diameter [7,8,9,10]. Giant CCMs, measuring more than 4 cm in diameter, are rare [3, 5, 11, 12] and more so in the paediatric age group. They can cause diagnostic and therapeutic dilemmas. We describe three cases of giant paediatric CCMs and review the relevant literature.
Methods
Literature review
A search of the PubMed database was done for giant cavernous angiomas in the paediatric population. Keywords such as ‘giant’ or ‘huge’ and ‘cavernoma’ or ‘cavernous angioma’ or ‘cavernous malformation’ were used for the search. All the articles in English literature and one in Portuguese (which has been frequently referred to) where the size of the lesion was reported to be > 4 cm were selected for analysis.
Lawton et al. [13] defined a giant cavernous malformation as one that has a diameter of more than 6 cm [13]. However, Kan et al. in 2007, in their study on the radiographic features of tumefactive giant cavernomas, defined giant ones as those having a diameter greater than 4 cm [14]. For this study of giant CCMs, all lesions with a documented size of more than 4 cm in paediatric patients (up to 18 years of age) were included for the literature review. We think that this threshold of 4 cm [14] may be more apt for a series of paediatric patients and it is a commonly used threshold for ‘giant’ size in most brain tumours. Only cranial intraparenchymal lesions were included in the review; extraparenchymal lesions such as those in the cavernous sinus, orbit, and dural-based lesions were excluded. Spinal CCMs were also excluded.
After a review of the literature, a total of 31 publications reporting 53 cases (including our own three cases) were identified and analysed (Table 1) [2,3,4,5,6, 9, 10, 12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34].
Case studies
Case 1: A 2-year-old boy with a diagnosis of brain tumour was referred to us. He presented with headache and vomiting, with progressive right-sided weakness (grade 4/5 power) for 1 month, for which he was investigated, and an MRI of the brain was done, which showed a 4 cm × 3.8 cm T2-mixed intensity mass lesion in the left basal ganglia and thalamic regions, with a surrounding hypointense rim (Fig. 1A).

A Coronal T2W MRI (left) and axial FLAIR sequence view (right) of a 2-year-old boy with a large 4 cm × 3.8 cm mixed intensity mass lesion in the left basal ganglia and thalamic regions, with a surrounding hypointense rim. B Postoperative T2W MRI images showing complete excision
The indications for surgical treatment were the symptoms caused by mass effect due to the lesion, and the need for a histopathological diagnosis. A left temporal craniotomy and corticectomy through the middle temporal gyrus showed a solid-cystic lesion with few areas of calcification. Further dissection showed the honeycomb pattern of the lesion with surrounding hemosiderin capsule-like gliosis. Complete excision of the lesion was done, while leaving behind the peripheral hemosiderin gliotic capsule in the deeper plane adjacent to the basal ganglia. Postoperatively, the patient had a slight worsening of the weakness of the right upper and lower limbs (grade 3/5) which gradually improved over the next 6 months to normal power. A postoperative MRI was done which confirmed complete excision (Fig. 1B). At follow-up after 3 years, the patient is free of all preoperative symptoms and is doing well.
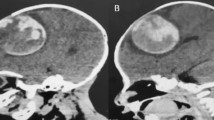
Case 2: A 16-year-old boy who had been operated on elsewhere twice earlier for a large brainstem cavernoma in the last 2 years came to us with progressive left-sided weakness and left facial palsy. Examination showed a left lower motor neuron facial paresis and a left lateral rectus paresis along with decreased sensations on the left side of his face. MRI showed a giant cavernoma measuring 3.5 cm × 4 cm in the left middle cerebellar peduncle becoming exophytic in the CPA (Fig. 2A).

A T2, FLAIR, and contrast (left to right) MRI images of a 16-year-old boy with a giant recurrent cavernoma in the left middle cerebellar peduncle. B Postoperative contrast MRI showing complete excision
The indication for surgical treatment was the mass effect as well as repeated re-bleeds due to incomplete excision inspite of two previous surgeries. He was taken up for a redo suboccipital exposure. The lesion was adherent to the lower cranial nerves on the left side, which were carefully separated. The lesion was excised completely, leaving behind the peripheral hemosiderin-ring at the brainstem. Post-operatively, the patient had transient swallowing dysfunction and hoarseness of voice. Postoperative MRI done at 3 months confirmed complete excision (Fig. 2B). At a follow-up of 8 years, the patient is free of his preoperative symptoms and is an active contributor to his society.
Case 3: A 14-year-old girl presented with a headache for a week, blurring of vision (both eyes) for 5–7 days, and history of having increased appetite and weight gain for the last 4–6 weeks. Her examination was unremarkable except for bilateral severe optic disc oedema. MRI showed a complex lesion, with mildly enhancing solid and non-enhancing cystic components, in the frontal horn and anterior body of the right lateral ventricle, measuring 6.3 cm × 4.3 cm × 5.2 cm, with dilatation of the right lateral ventricle and trans-ependymal seepage of cerebrospinal fluid due to obstruction at the level of the foramen of Monro, with a midline shift of 1.3 cm to the left. Inferomedially, the solid component was abutting the right optic nerve and chiasm (Fig. 3A). The lesion was reported by a radiologist to be an ependymoma or a craniopharyngioma, for which we evaluated her hormonal status, which was normal.

A T1W (top row), T2W (middle row), and post-contrast (bottom row) coronal MRI images showing a large complex lesion in the right frontal horn and body of the right lateral ventricle, with the solid component extending inferomedially up to the right optic nerve. The cystic component is hypointense on T1W and hyperintense on T2W. The solid component is hyperintense on T1W and shows mild post-contrast enhancement. There is dilatation of the lateral ventricles with periventricular seepage of cerebrospinal fluid, with shift of the midline to the left by 1.3 cm. B Intraoperative photograph of the mulberry-coloured solid component of the lesion. C Pre (left) and post-operative (right) CT images showing total excision of the solid portion. D Histopathology section showing large vascular sinusoidal spaces (VS) with newly formed thrombi (T), lacking a smooth muscle layer, and lacking intervening brain parenchyma. E Post-operative MRI-T2W (top row) and post-contrast (bottom-row) images showing complete excision of the lesion with complete resolution of the hydrocephalus and midline shift
Given her clinical and imaging features suggestive of raised intracranial pressure due to the presumed diagnosis of a craniopharyngioma with a large cyst and resultant hydrocephalus because of obstruction at the level of the right Foramen of Monro, she was taken up early for surgery. A right frontal craniotomy and transcortical approach through the right middle frontal gyrus was performed. The tumour cyst was visualised at a depth of 1.5 cm. Xanthochromic fluid was aspirated from the cyst cavity. At the depth of the cystic cavity, the solid component of the lesion was visualised as a reddish-brown, mulberry-like, lobulated structure (Fig. 3B). Complete excision of the lesion was performed (including the peripheral hemosiderin ring) which had reached the depth into the subarachnoid space but was not attached to the optic nerve. A post-operative CT scan the next day also showed complete excision of the solid component (Fig. 3C).
Histopathological examination confirmed the diagnosis of a CCM (Fig. 3D). Post-operatively, she recovered well.
At a follow-up of 2 months, the patient was completely free of her pre-operative symptoms. At follow-up of 4 months, the patient remained symptom-free and an MRI was done which showed residual gliosis and haemoglobin degradation products in the right antero-inferior frontal region, with complete resolution of hydrocephalus and midline shift (Fig. 3E).
Discussion
CCMs are one of the most common intracranial vascular malformations, having been variously called cryptic angioma, cavernous angioma, cavernous haemangioma, cavernoma, and ‘angiographically occult vascular malformation’ [1]. However, as per the current ISSVA classification, CCMs are classified as simple vascular malformations type III—venous malformations [35].
The definition of a giant cavernous malformation has been arbitrary. Lawton et al. mentioned it to be 6 cm [13], whereas Kan et al. mentioned it to be 4 cm [14]. We believe this value of 4 cm may be more apt for CCMs in children and should be used as a standard for reporting in the future as it is a commonly used threshold for most brain tumours as well.
Though the peak age of presentation of CCMs has been reported to be in the third and fourth decades [2, 3, 5], about one-fourth of CCMs have been reported in paediatric patients [3, 27, 36, 37]. Giant CCMs are rare [3, 5, 11, 12, 23, 25]. However, when they do occur, they seem to be more common in the paediatric age group than in adults [16, 26]. Acciari et al. (2009), mentioned that cavernous malformations rarely appear clinically before the first year of life [27]. However, in our literature review of giant CCMs in the paediatric age group, we found that 14 of the 53 patients presented before the age of 1 year, and 21 of them before the age of 2 years (Table 2); the youngest being a 1-day-old neonate [14]. Their occurrence in this group of patients likely implies that they arise due to failure of normal embryonic vascular development [26].
Our review of literature for paediatric giant CCMs also revealed that 22/53 (41.51%) patients were female, and 31/53 (58.49%%) patients were male suggesting slight male preponderance. However, there is a discordance in literature since both male [29, 33] and female [8, 14, 26, 38] preponderance has been reported. Wang et al. described a male preponderance in children and a female preponderance in adults [16].
Familial occurrence in CCMs has been reported to occur in about 20–50% of patients. Multiple CCMs have been reported to occur in up to 30% of sporadic and up to 84% of familial cases. However, neither familial nor multiple, giant CCMs have been reported [8, 10, 26, 38].
CCMs are most commonly located in the supratentorial region (in up to 87.5% cases) as reported in the literature [3,4,5, 10, 17, 27]. The remaining occur in the infratentorial compartment, with the majority being in the brainstem [1, 39]. In our literature review of giant CCMs in the paediatric age group, most of them (83.02%) did occur in the supratentorial region (44/53 patients); whereas infratentorial location was seen in 17.31% of patients (9/53 patients). In the supratentorial region, the giant CCMs in the paediatric group were in the deep brain areas (paraventricular region, basal ganglia, or thalamus) in 8 patients, and in an intraventricular location in 4 patients. In the infratentorial region, however, giant CCMs in the paediatric age group occurred more commonly in the cerebellum (8 patients) than in the brainstem (1 patient). We believe this is likely due to the eloquence of the brainstem, whereby a CCM located here would be more likely to cause symptoms and come to clinical attention before it could reach a ‘giant’ size.
Histologically, CCMs consist of sinusoid like capillary vessels lying adjacent to one another, with little or no intervening brain parenchyma [1]. Their growth has been postulated to be due to various mechanisms such as ectasia of vascular channels, new hemosiderin deposition with reactive gliosis, and recurrent macro and micro haemorrhages; followed by the organization of the clot, pseudo-capsule formation, and secondary expansion [2, 3, 8, 10, 11, 16, 23, 31]. However, it has also been reported that they can show evidence of expansile growth or infiltration without haemorrhage, thus mimicking a neoplasm [8, 13, 16, 25, 29, 31, 40]. The largest reported lesion in our review, by size (in terms of maximum dimension in any one axis), was 14 cm (reported by de Andrade et al. in 2002) [21].
Zabramski et al. in 1994 proposed a classification of CCMs, based on their MRI appearance which depends in turn on the age of haemorrhage [41]. Mottolese et al. found that this classification was not particularly appropriate for paediatric patients, and they proposed their classification for the paediatric age group based on the MR signal characteristics of CCMs, their morphology, and clinical picture [36]. However, so far, no radiological classification system has incorporated the morphological variant of giant CCMs.
Giant CCMs can often have a heterogenous presentation on CT scan, and can therefore cause diagnostic dilemmas. They can be misdiagnosed as oligodendroglioma due to the presence of calcifications, as anaplastic or pilocytic astrocytoma in cases of isodense appearance with poor contrast enhancement, or as ependymoma or even metastatic melanoma with intra-tumoral haemorrhage. Thrombosed AVMs, toxoplasmosis, and cysticercosis are the other differentials [3, 5, 11, 22, 37].
On MRI, the appearance of a giant CCM is similar to that of a smaller cavernous angioma which is a heterogeneous ‘popcorn-like’ mass (with or without cysts) that reflects various stages of blood breakdown products, surrounded by the compressed, gliotic, and haemosiderin-stained brain [13, 17]. Gradient echo sequences and SWI are the most sensitive imaging methods for the detection of the peripheral haemosiderin ring [4, 16]. Contrast enhancement is generally not significant, but may sometimes be marked, and is another cause for diagnostic dilemmas with neoplasms; especially when associated with mass effect and perilesional oedema [10, 12, 25, 29, 38]. This was the case in one of our patients as well (Case 3) where the differential diagnoses considered were ependymoma and craniopharyngioma. Cavernoma was not suspected until surgery revealed the typical solid-cystic appearance of a nodular mulberry coloured lesion along with xanthochromic fluid in the surrounding cyst. Angiographically, cavernous malformations are occult, but an associated developmental venous anomaly may often be seen; although this is rarely, if ever, the cause of haemorrhage [1, 9].
CCMs in the paediatric age group present clinically, most commonly with seizures, which may be focal or generalised [3, 27, 36]. Other modes of presentation are due to focal neurological deficit (which occurs due to pressure on the underlying brain) and haemorrhage (which may be recurrent and mild, or overt) [1]. Slow-growing lesions can often present with symptoms of raised intracranial pressure due to obstructive hydrocephalus or the mass itself occupying significant intracranial space [38]. In Wang’s paper, the common initial symptom in paediatric patients with giant CCMs included seizures in 43.5%, focal neurological deficits in 39.1%, and macrocephaly in 34.8% [16]. Haemorrhage is more likely to occur in children, than in adults [3, 16, 27, 36, 37].
In our literature review of giant CCMs in the paediatric age group, we found that seizures were present in 47.17% (25/53 patients) of patients at presentation. However, with giant CCMs, features of raised intracranial pressure (37.74%) (20/53 patients) and local mass effect i.e., focal neurological deficit (seen in 18/53 or 33.96% patients) (Table 2), are seen more frequently than in smaller sized cavernomas, which seems obvious considering their sheer size. Headache was present in 22.65% of patients (12/53) at presentation and vomiting in 18.87% of patients (10/53) with giant CCMs in the paediatric age group. Macrocephaly was also a common mode of presentation seen (16.98% patients or 9/53 patients) in younger children with giant CCMs (up to the age of 7 years). Symptomatology at presentation was not reported for 2 patients.
In children as in adults, the therapeutic management of CCMs includes watchful waiting, antiepileptic therapy, surgical removal and rarely, even radiosurgery [27]. Giant CCMs are generally not asymptomatic; hence, the wait and watch strategy is often not employed in their management. Radiosurgery is also not employed for the management of giant CCMs. Surgery is indicated in case of acute haemorrhage, focal neurological deficits, seizures, and symptoms of mass effect or raised intracranial pressure. It is also especially recommended for infratentorial cavernous angiomas, even if they may be clinically silent, due to the higher associated risk of bleeding and poor outcome should it occur [27]. Complete microsurgical removal is the mainstay of treatment and is considered curative [27, 39, 42,43,44]. Excision of the peripheral haemosiderin ring is recommended in hemispheric lesions with epilepsy [45]. However, this may not be possible while working in deep brain or eloquent areas to better preserve neurological function, as in our first case.
The shortest direct trajectory to the cavernoma is always the preferred approach for surgical excision [34]. Our experience also supports a direct approach. We also rely on intraoperative neuronavigation to guide the surgical trajectory and the extent of surgical resection. Venous anatomy of the surgical trajectory is carefully studied in preoperative imaging to avoid venous injury. Adjuvants like intra-operative neuromonitoring and preoperative diffusion tensor imaging are always used for lesions in the basal ganglia and the brainstem; endoscope assistance and awake craniotomy may also be useful based on the location of the cavernoma. We prefer an excision by piecemeal method to avoid injury, especially in the eloquent areas [39].
In our literature review of paediatric giant CCMs, surgical intervention was performed in 52 patients. Gross total resection was achieved in 39 patients. One patient underwent near-total resection. Subtotal or partial excision was done in 6 patients. The extent of surgical resection was not reported in 6 patients (Table 1). Subtotal or partial excision of giant cavernomas may relieve symptoms of mass effect in the short-term, but long-term outcome is often not reported for these patients in terms of their neurological status, as well as lesion recurrence on imaging. Our second patient with the large CPA cavernoma presented to us with progressive growth in the lesion after 2 prior surgical attempts in which the lesion was incompletely excised. In the experience of the senior author, of the 104 patients with CCMs who have been operated on, 5 patients (who underwent incomplete resections for various reasons) have had recurrent bleeds for which redo operation was required.
Although giant CCMs may be associated with a worse prognosis than their smaller sized counterparts, the predominant factor driving prognosis is their location rather than their size [11]. In our review of paediatric giant CCMs, 37 patients improved with surgical treatment (in terms of their symptomatology at presentation), either immediately after surgery or on long term follow up, and 9 patients (from one series) [10] were reported to have an uneventful postoperative outcome. Even those patients who underwent subtotal or partial resection improved postoperatively [4, 26, 30]. Focal neurological deficit worsened or persisted in 2 patients after surgery [2, 22]. Death was reported in 2 patients who had presented in-extremis, surgery was performed for one of them (4-year-old female child) [24], and support was withdrawn for the other (1-day-old male infant) [14]. The long-term outcome of these after excision is generally favourable and complete surgical removal leads to cure [8, 23, 31].
Conclusion
Giant CCMs are rare and may differ from their smaller counterparts in clinical presentation and imaging, often presenting with a diagnostic challenge. Early treatment may be necessary for most of them. Complete microsurgical removal remains the mainstay of treatment. Though transient neurological deficits may be encountered, surgery for gross total resection leads to a favourable outcome for reduction of mass effect, as well as other well-known benefits of preventing further re-bleeds and achieving long-term seizure control.
Data availability
All data generated or analysed during this study are included in this manuscript.
Abbreviations
- CCM:
-
Cerebral cavernous malformation
- MRI:
-
Magnetic resonance imaging
- CT:
-
Computed tomography
- ISSVA:
-
International Society for the Study of Vascular Anomalies
- SWI:
-
Susceptibility weighted imaging
- AVM:
-
Arteriovenous malformation
- GTR:
-
Gross total resection
- STR:
-
Subtotal resection
- NR:
-
Not reported
- CPA:
-
Cerebellopontine angle
References
Deopujari CE (2012) Cavernomas of the Brain. In: Tandon PN, Ramamurthi R. Ramamurthi and Tandon’s textbook of neurosurgery, 3rd edn. Jaypee Brothers Medical Publishers (P) Ltd, New Delhi, pp 1109–1115.
Braga BP, Costa LB Jr, Lemos S, Vilela MD (2006) Cavernous malformations of the brainstem in infants. Report of two cases and review of the literature. J Neurosurg 104(6 Suppl):429–33. https://doi.org/10.3171/ped.2006.104.6.429
Gezen F, Karatas A, Is M, Yildirim U, Aytekin H (2008) Giant cavernous haemangioma in an infant. Br J Neurosurg 22(6):787–789. https://doi.org/10.1080/02688690802108780
Jurkiewicz E, Marcinska B, Malczyk K, Grajkowska W, Daszkiewicz P, Roszkowski M (2013) Giant cerebellar cavernous malformation in 4-month-old boy. Case report and review of the literature. Neurol Neurochir Pol 47(6):596–600. https://doi.org/10.5114/ninp.2013.39078
Avci E, Oztürk A, Baba F, Karabağ H, Cakir A (2007) Huge cavernoma with massive intracerebral hemorrhage in a child. Turk Neurosurg 17(1):23–26
Agrawal A, Banode P, Shukla S (2012) Giant cavernous hemangiomas of the brain. Asian J Neurosurg 7(4):220–222. https://doi.org/10.4103/1793-5482.106660
Kim DS, Park YG, Choi JU, Chung SS, Lee KC (1997) An analysis of the natural history of cavernous malformations. Surg Neurol 48(1):9–17; discussion 17–8. https://doi.org/10.1016/s0090-3019(96)00425-9
Son DW, Lee SW, Choi CH (2008) Giant cavernous malformation: a case report and review of the literature. J Korean Neurosurg Soc 43(4):198–200. https://doi.org/10.3340/jkns.2008.43.4.198
Parizel MR, Menovsky T, Van Marck V, Lammens M, Parizel PM (2014) Giant cavernous malformations in young adults: report of two cases, radiological findings and surgical consequences. JBR-BTR 97(5):274–278. https://doi.org/10.5334/jbr-btr.1327
Ozgen B, Senocak E, Oguz KK, Soylemezoglu F, Akalan N (2011) Radiological features of childhood giant cavernous malformations. Neuroradiology 53(4):283–289. https://doi.org/10.1007/s00234-010-0783-5
Grujić J, Jovanović V, Tasić G, Savić A, Stojiljković A, Matić S, Lepić M, Rotim K, Rasulić L (2020 ) GIANT CAVERNOUS MALFORMATION WITH UNUSUALLY AGGRESSIVE CLINICAL COURSE: A CASE REPORT. Acta Clin Croat 59(1):183–187. https://doi.org/10.20471/acc.2020.59.01.24
Ozsoy KM, Oktay K, Gezercan Y, Cetinalp NE, Olguner SK, Erman T (2017) Giant cavernous malformations in childhood: a case report and review of the literature. Pediatr Neurosurg 52(1):30–35. https://doi.org/10.1159/000447407
Lawton MT, Vates GE, Quinones-Hinojosa A, McDonald WC, Marchuk DA, Young WL (2004) Giant infiltrative cavernous malformation: clinical presentation, intervention, and genetic analysis: case report. Neurosurgery 55(4):979–980. https://doi.org/10.1227/01.neu.0000137277.08281.48
Kan P, Tubay M, Osborn A, Blaser S, Couldwell WT (2008) Radiographic features of tumefactive giant cavernous angiomas. Acta Neurochir (Wien) 150(1):49–55; discussion 55. https://doi.org/10.1007/s00701-007-1455-z
Khosla VK, Banerjee AK, Mathuriya SN, Mehta S (1984) Giant cystic cavernoma in a child. Case report J Neurosurg 60(6):1297–1299. https://doi.org/10.3171/jns.1984.60.6.1297
Wang C, Zhao M, Wang J, Wang S, Zhang D, Zhao J (2018) Giant cavernous malformations: a single center experience and literature review. J Clin Neurosci 56:108–113. https://doi.org/10.1016/j.jocn.2018.06.042
Hassani FD, Karekezi C, El Abbadi N (2020) Rare case of giant pediatric cavernous angioma of the temporal lobe: a case report and review of the literature. Surg Neurol Int 11:7. https://doi.org/10.25259/SNI_468_2019
Hayashi T, Fukui M, Shyojima K, Utsunomiya H, Kawasaki K (1985) Giant cerebellar hemangioma in an infant. Childs Nerv Syst 1(4):230–233. https://doi.org/10.1007/BF00270768
Kawagishi J, Suzuki M, Kayama T, Yoshimoto T (1993) Huge multilobular cavernous angioma in an infant: case report. Neurosurgery 32(6):1028–30; discussion 1030–1. https://doi.org/10.1227/00006123-199306000-00026
Reyns N, Assaker R, Louis E, Lejeune JP (1999) Intraventricular cavernomas: three cases and review of the literature. Neurosurgery 44:648–654
de Andrade GC, Prandini MN, Braga FM (2002) Cavernoma gigante: relato de dois casos [Giant cavernous angioma: report of two cases]. Arq Neuropsiquiatr 60(2-B):481–6.
Chicani CF, Miller NR, Tamargo RJ (2003) Giant cavernous malformation of the occipital lobe. J Neuroophthalmol 23(2):151–153. https://doi.org/10.1097/00041327-200306000-00010
Muzumdar DP, Bhatjiwale MG, Goel A (2003) Giant cerebral cavernous haemangioma: a case report and review of literature. J Clin Neurosci 10(3):348–351. https://doi.org/10.1016/s0967-5868(03)00012-2
Corapçioğlu F, Akansel G, Gönüllü E, Yildiz K, Etuş V (2006) Fatal giant pediatric intracranial cavernous angioma. Turk J Pediatr 48(1):89–92
Kim YJ, Kim JE, Kim NR, Kim HS (2007) Imaging findings of giant cavernous malformation with a focal infiltrative pattern. Pediatr Radiol 37(10):1039–42. https://doi.org/10.1007/s00247-007-0553-7
van Lindert EJ, Tan TC, Grotenhuis JA, Wesseling P (2007) Giant cavernous hemangiomas: report of three cases. Neurosurg Rev 30(1):83–92; discussion 92. https://doi.org/10.1007/s10143-006-0042-8
Acciarri N, Galassi E, Giulioni M, Pozzati E, Grasso V, Palandri G, Badaloni F, Zucchelli M, Calbucci F (2009) Cavernous malformations of the central nervous system in the pediatric age group. Pediatr Neurosurg 45(2):81–104. https://doi.org/10.1159/000209283
Li M, Li Y, Xu G, Mei J (2010) Giant intracranial cavernous haemangioma presented with enlarged head circumference in a child. J Paediatr Child Health 46(12):788–789. https://doi.org/10.1111/j.1440-1754.2010.01919.x
Thakar S, Furtado SV, Ghosal N, Hegde AS (2010) A peri-trigonal giant tumefactive cavernous malformation: case report and review of literature. Childs Nerv Syst 26(12):1819–23. https://doi.org/10.1007/s00381-010-1237-4
Lew SM (2010) Giant posterior fossa cavernous malformations in 2 infants with familial cerebral cavernomatosis: the case for early screening. Neurosurg Focus 29(3):E18. https://doi.org/10.3171/2010.5.FOCUS10119
Mohindra S, Sodhi HS, Rane S (2013) Tumefactive presentation of a supratentorial cavernous hemangioma: a report of two cases. J Pediatr Neurosci 8(3):232–234. https://doi.org/10.4103/1817-1745.123689
Villaseñor-Ledezma J, Budke M, Alvarez-Salgado JA, Cañizares MA, Moreno L, Villarejo F (2017) Pediatric cerebellar giant cavernous malformation: case report and review of literature. Childs Nerv Syst 33(12):2187–2191. https://doi.org/10.1007/s00381-017-3550-7
Hirata K, Ihara S, Sato M, Matsumaru Y, Yamamoto T (2017) Hyper-vascular giant cavernous malformation in a child: a case report and review. Childs Nerv Syst 33(2):375–379. https://doi.org/10.1007/s00381-016-3234-8
Rangnekar RD, Vilanilam GC, Krishnakumar K, Abraham M (2021) Giant cavernomas: gigantic propositions for a lilliputian problem? Neurol India 69(1):153–156. https://doi.org/10.4103/0028-3886.310114
ISSVA Classification of Vascular Anomalies (2018) International Society for the Study of Vascular Anomalies. Available at https://issva.org/classification. Accessed 30th Jun 2021
Mottolese C, Hermier M, Stan H, Jouvet A, Saint-Pierre G, Froment JC, Bret P, Lapras C (2001) Central nervous system cavernomas in the pediatric age group. Neurosurg Rev 24(2–3):55–71; discussion 72–3. https://doi.org/10.1007/pl00014581
Maraire JN, Awad IA (1995) Intracranial cavernous malformations: lesion behavior and management strategies. Neurosurgery 37(4):591–605. https://doi.org/10.1227/00006123-199510000-00001
Kim IC, Kwon KY, Rhee JJ, Lee JW, Hur JW, Lee HK (2013) Giant cystic cerebral cavernous malformation with multiple calcification—case report. J Cerebrovasc Endovasc Neurosurg 15(3):255–259. https://doi.org/10.7461/jcen.2013.15.3.255
Deopujari CE, Jacob T, Karmarkar V (2016) Surgical approaches to deep seated cavernous malformations. J Neurosurg Imaging Techniques 1(2):52–65
Siddiqui AA, Jooma R (2001) Neoplastic growth of cerebral cavernous malformation presenting with impending cerebral herniation: a case report and review of the literature on de novo growth of cavernomas. Surg Neurol 56(1):42–45. https://doi.org/10.1016/s0090-3019(01)00505-5
Zabramski JM, Wascher TM, Spetzler RF, Johnson B, Golfinos J, Drayer BP, Brown B, Rigamonti D, Brown G (1994) The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg 80(3):422–432. https://doi.org/10.3171/jns.1994.80.3.0422
Bertalanffy H, Gilsbach JM, Eggert HR, Seeger W (1991) Microsurgery of deep-seated cavernous angiomas: report of 26 cases. Acta Neurochir (Wien) 108(3–4):91–99. https://doi.org/10.1007/BF01418515
Cenzato M, Stefini R, Ambrosi C, Giovanelli M (2008) Post-operative remnants of brainstem cavernomas: incidence, risk factors and management. Acta Neurochir (Wien) 150(9):879–86; discussion 887. https://doi.org/10.1007/s00701-008-0008-4
Steinberg GK, Chang SD, Gewirtz RJ, Lopez JR (2000) Microsurgical resection of brainstem, thalamic, and basal ganglia angiographically occult vascular malformations. Neurosurgery 46(2):260–70; discussion 270–1. https://doi.org/10.1097/00006123-200002000-00003
Ruan D, Yu XB, Shrestha S, Wang L, Chen G (2015) The role of hemosiderin excision in seizure outcome in cerebral cavernous malformation surgery: a systematic review and meta-analysis. PLoS One 10(8):e0136619. https://doi.org/10.1371/journal.pone.0136619
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
As this is a retrospective study and there was no deviation from standard of care that was provided to the patients, ethics approval was not required from the Institutional Ethics Committee.
Consent for publication
Parents of the patients included in the study were informed that their patient’s clinical data and imaging photographs may be used for educational purposes such as presentation in conferences/journals, and consent was obtained. No personal identifying information has been submitted in this manuscript or in Figs. 1, 2, and 3.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shroff, K., Deopujari, C., Karmarkar, V. et al. Paediatric giant cavernomas: report of three cases with a review of the literature. Childs Nerv Syst 37, 3835–3845 (2021). https://doi.org/10.1007/s00381-021-05286-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-021-05286-6